

Summary
Human and murine succinic semialdehyde dehydrogenase (SSADH; gamma-hydroxybutyric (GHB) aciduria) deficiency represents an epileptic disorder associated with hyperGABA- and hyperGHB-ergic states. Despite significant neurotransmitters alterations, well-defined single-cell electrophysiological studies, aimed to provide insight into regional neuropathology, have been lacking. In this study, we characterized the effect of residual SSADH enzyme function/increased GABA levels on single-cell hippocampal electrophysiology in SSADH+/+ (wild-type; WT), SSADH+/− (heterozygous; HET), and SSADH−/− (knock-out; KO) mice. Tonic extrasynaptic GABAA receptor (GABAAR)-mediated currents were elevated in HET and KO mice, whereas phasic synaptic GABAAR currents were unaltered in dentate gyrus granule cells. Similarly, tonic GABAAR-mediated currents were increased in dentate gyrus interneurons of KO animals, while phasic GABAergic neurotransmission was unaffected in the same cells. Our results indicate global disruption of cortical networks in SSADH KO mice, affecting both excitatory and inhibitory neurons. Our findings provide new clues concerning seizure evolution in the murine model (absence→tonic-clonic→status epilepticus), and extend pathophysiological insight into human SSADH deficiency.
Keywords: Dentate gyrus, Granule cell, Interneuron, GABA, GABAA receptor, Mouse
Introduction
Succinic semialdehyde dehydrogenase (SSADH) deficiency is an autosomal-recessively inherited disorder of γ-aminobutyric acid (GABA) catabolism, which shows a high prevalence of delayed neurological development, ataxia, sleep disturbances, and epileptic seizures (Pearl et al., 2009). SSADH deficient patients display supraphysiological levels of GABA and γ-hydroxybutyric acid (GHB) in the brain, and it is hypothesized that the excessive levels of these neurotransmitters cause detrimental effects on brain development and functioning (Novotny et al., 2003). Accordingly, it is believed that an overactivation of the receptors for GABA (GABAA and GABAB receptors) or for GHB (GABAB and putative GHB receptors) plays a crucial role in the development of pathology in SSADH deficiency, for which little or no efficient treatment exists.
In order to study the pathophysiology of SSADH deficiency, SSADH knock-out (KO) mice were recently generated, which mimic the severe form of the human disorder (Hogema et al., 2001). Indeed, SSADH KO mice show elevated GABA and GHB in brain and urine, which is associated with ataxia and absence seizures at postnatal days 14–18, that progress into lethal tonic-clonic seizures at P18–20. Recent studies have shown that GABA is elevated in SSADH KO mouse brains during the earliest stages of brain development (Jansen et al., 2008). Single-cell electrophysiological investigations have also demonstrated that the principal neurons of the neocortex (Drasbek et al., 2008) and the hippocampus (Nylen et al., 2008) are detrimentally affected by complete abrogation of SSADH activity in knock-out mice.
Several important questions remain with respect to the impact of SSADH deficiency on neuronal functioning. Firstly, it is important to determine how the SSADH gene dosage impacts single-neuron physiology, since this might offer insight into how SSADH regulates the extracellular GABA levels in physiological and pathophysiological situations. Secondly, to understand better the disease mechanisms, it is of interest to determine how elevated GABA levels induce signaling in different neuron populations in the brain, particularly excitatory glutamatergic neurons versus inhibitory GABAergic interneurons.
Here, we have studied brain slices from mice that harbor 100, 50 or 0% of full SSADH gene dosage. We focused on GABAA receptor currents induced by the ambient extracellular levels of GABA in the three genotypes, and studied how tonic GABA currents and GABAergic synaptic activity correlate with reduced SSADH gene dosage. Examining two distinct neuron populations in the dentate gyrus of the mouse hippocampal formation, our studies serve to expand our understanding of gene dosage in SSADH deficiency, and its impact on different neuron populations in cortical circuits.
Methods
Mouse breeding
All studies were conducted in accordance with Danish and European legislation regarding laboratory animals. All efforts were made to minimize animal suffering and to reduce the number of mice used. Wild-type (WT), SSADH heterozygous (HET), and SSADH knockout (KO) littermates were generated from heterozygous breeding. Genotyping of the mice were carried out using PCR (Hogema et al., 2001) (Fig. 1B). Mice were kept in a university animal facility with a 12/12-h light/dark cycle with unrestricted access to food and water. The mice did not display overt seizures upon handling, or preparation for anesthesia.
Figure 1.
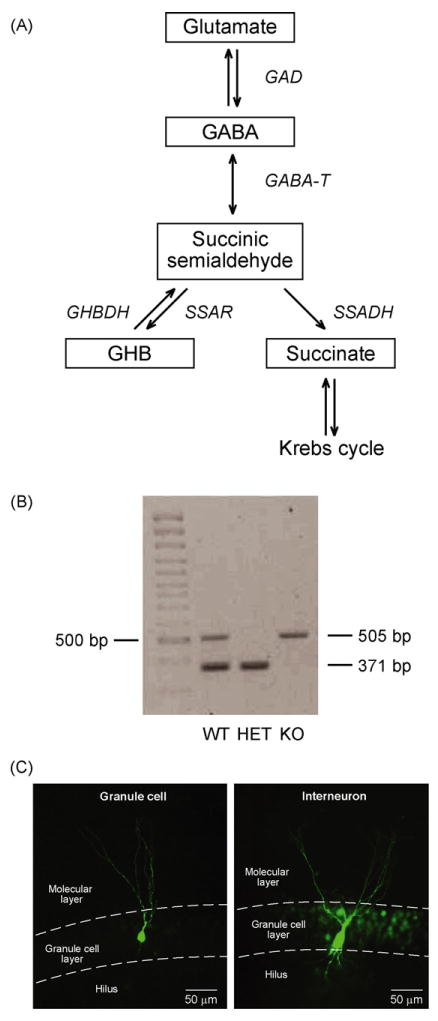
(A) The GABA metabolic pathway. GABA is synthetized from glutamate by GAD (glutamic acid decarboxylase). During GABA metabolism, GABA-T (GABA transaminase) removes an amino group from GABA, producing SSA (succinic semialdehyde). Mediated by SSADH (succinic semialdehyde dehydrogenase), SSA is converted to succinate which enters the Krebs cycle. SSA may also be converted into an alternative by-product, GHB (γ-hydroxybutyrate) catalyzed by succinic semialdehyde reductase (SSAR). SSADH deficiency leads to accumulation of GABA and GHB. GHBDH (GHB dehydrogenase). (B) Genotyping of SSADH knock-out mice. DNA was isolated from mouse tails, and PCR genotyping results were obtained for three offspring of heterozygous parents. Displayed are the results for WT, HET, and KO mice. (C) Photomicrograph of a biocytin injected granule cell and interneuron in mouse dentate gyrus.
Brain slice preparation
Postnatal days 10–18 (P10–18) mice of either sex were anesthetized deeply with isoflurane and decapitated. The brain was dissected out, and placed in ice-cold artificial cerebrospinal fluid (ACSF) composed of (in mM): 126 NaCl, 2.5 Kcl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaCO3, 10 d-glucose (osmolarity 305–315 mosmol/kg), pH 7.4 when bubbled with carbogen (5% CO2–95% O2). 350-μm-Thick coronal slices were prepared with Vibratome 3000 Plus (Vibratome Company, St Louis, MO) in ice-cold bubbled ACSF. Slices rested in a holding chamber at least 1 h in bubbled ACSF at room temperature (22–25 °C). To improve slice quality, 2 mM kynurenic acid, 0.2 mM ascorbic acid, and 0.2 mM pyruvic acid were added to the ACSF during slicing and storage.
Brain slice electrophysiology
For whole-cell patch-clamp recordings, slices were transferred into a recording chamber that was perfused with 33 ± 1°C bubbled ACSF at 2–3 ml/min. Recordings were carried out using a MultiClamp 700B amplifier (Axon Instruments, Union City, CA). Granule cells and interneurons were visualized using a custom-built infrared microscope (Versascope, E. Marton Electronics, Cacoga Park, CA) equipped with a 40× water-immersion objective (Olympus, Ballerup, Denmark) and CCD100 camera (DAGE-MTI, Michigan City, IN). Interneurons were identified by the 3–4-fold larger cell-body compared to granule cells, localized on the boundary of the granule cell layer and the hilus. Patch-pipettes were pulled from borosilicate glass (OD = 1.5 mm, ID = 0.8 mm; Garner Glass Company, Claremont, CA) using a DMZ Universal Puller (Zeitz Instruments, Munich, Germany). After filling the pipettes with intracellular solution containing (in mM): 140 CsCl, 2 MgCl2, 0.05 EGTA, 10 HEPES, adjusted to pH 7.2 with CsOH (280–290 mosmol/kg), their resistances were 3–5MΩ. Giga seals (>1 GΩ) were always obtained before break in. Cells were held in voltage-clamp mode at a holding potential (Vhold) of −70 mV, while resistance was compensated by 70% (lag 10 μs). During the experiment, the whole-cell capacitance and series resistance were monitored, and recording was terminated if series resistance increased by >50% or exceeded 20 MΩ. The mean capacitance was 7.1 ± 0.3 pF in granule cells and 31.4 ± 1.6 pF in interneurons, which was used to verify the cell type. Furthermore, a subset of neurons was injected with biocytin to visualize their typical granule cell or interneuron appearance (Fig. 1C).
Data acquisition and analysis
Currents were low-pass filtered (8-pole Bessel) at 3 kHz before digitizing at 20 kHz using a DA converter (BNC-2110), a PCI acquisition board (PCI-6014, National Instruments, Austin, TX), and a custom-written LabView 6.1 (National Instruments) based software containing an acquisition interface and analysis module (EVAN v. 1.4, courtesy of Istvan Mody). This software was also used to detect and analyze sIPSCs with amplitude detection threshold of 6–8 pA. All events were visually inspected before making an average of a minimum of 50 events. Event amplitude, 10–90% rise time, and frequency were measured, whereas the IPSC weighted decay time constant (τw) was calculated using double-exponential fits. Tonic GABAA receptor-mediated currents were defined by their sensitivity to a high concentration of the selective GABAA receptor antagonist SR95531. The SR95531 sensitive current was observed as an outward shift in the holding current. For quantification, 5 ms long samples were taken from the recording every 100 ms and plotted against time, removing baseline points falling onto the decay of IPSCs (Drasbek and Jensen, 2006). Mean currents were calculated in 4 s long segments at three time points (denoted a, b and c): just before SR95531 application (b), 20 s before (a) and after (c). The tonic current was c–b, while the variation of the baseline (b–a), defining the stability of the recording, was allowed to be maximally 6 pA.
Statistical evaluation
Differences in the sIPSC parameters and mean currents densities between control, HET and KO mice were tested unpaired two-sample Student's t-tests. Data were expressed as means ± SEM, where n indicates the number of neurons.
Exponential (gene-)dose-response relationships for dentate gyrus granule cell and interneuron were evaluated using by testing the hypothesis of linear regression of logarithmically transformed current density values on the reductions in gene dosage (control 0%, HET 50% and KO 100%) using analysis of variance, and differences in slope and intercept between granule cells and interneurons were tested using a two-sample t-test.
Morphological identification of granule cells and interneurons
Intracellular labeling was performed as described previously (Holm et al., 2009). Briefly, injections were carried out by including 0.5% biocytin in the internal pipette solution. Slices were transferred to a solution of 4% paraformaldehyde at 4°C overnight, washed with PBS (0.1 M, pH7.4), and incubated overnight with FITC-conjugated avidin-d (2 μl/ml, Vector Laboratories) in PBS and 0.3% Triton X-100. Slices were then washed with PBS and embedded with fluorescent mounting medium (Dako) and labeled neurons visualized by epifluorescence on a Leica AF6000 LX workstation using a 10× objective.
Drugs
Kynurenic acid, ascorbic acid, biocytin and SR95531 were obtained from Sigma (St Louis, MO), while pyruvic acid was purchased from MP Biomedicals (Irvine, CA). SR95531 (5 mM) was dissolved in 50% DMSO (dimethyl sulfoxyde) and 50% dH2O (distilled water). All drugs were stored at −20°C until use.
Results
To investigate the effect of SSADH gene dosage on single-cell physiology and GABAergic inhibition in the mouse dentate gyrus, we electrophysiologically analyzed brain slices from WT, SSADH HET and SSADH KO mice. Whole-cell patch-clamp recordings were performed from granule cells and interneurons, and GABAergic currents were recorded in the presence of the ionotropic glutamate receptor antagonist kynurenic acid (2 mM).
Tonic extrasynaptic GABAA receptor-mediated currents in dentate gyrus granule cells are increased in SSADH deficient mice
The activation of extrasynaptic GABAA receptors by the ambient GABA in the brain slices can be quantified by applying the GABAA antagonist SR95531 to the slice chamber (Jensen et al., 2003). Thus, if present, GABAA receptor-mediated currents appear as an outward shift in the holding current upon SR95531 application (Fig. 2A). In WT slices, voltage-clamp recordings at a Vhold of −70 mV were carried out from dentate gyrus granule cells. Injection of SR95531 (∼100 μM) into the recording chamber revealed a tonic GABAA current in these cells, which was 0.9 ± 0.2 pA/pF (n = 13) when normalized to the cell capacitance. The current density was significantly larger both in granule cells in SSADH HET slices (2.2 ± 0.5 pA/pF, n = 12, P < 0.05) and in SSADH KO slices (6.3 ± 1.5 pA/pF, n = 7, P < 0.01). Additionally, the current density in SSADH KO mice was also significantly larger than in SSADH HET mice (P < 0.05). Fig. 2B suggests that the tonic GABA current increased non-linearly with a decreased SSADH gene dosage, which will be analyzed in more detail below.
Figure 2.
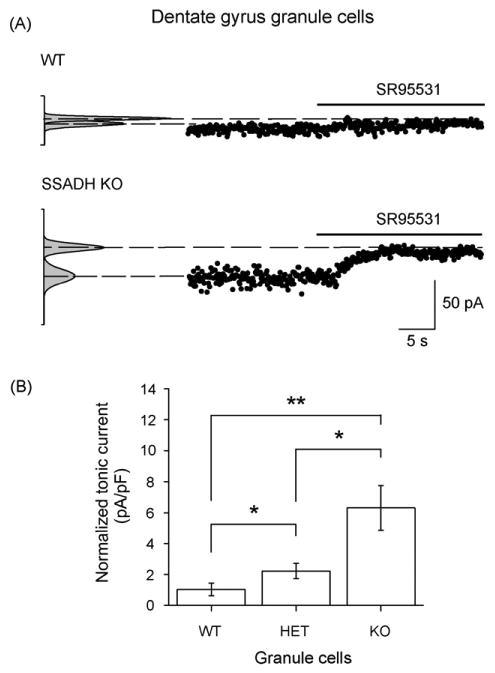
Tonic GABAergic inhibition in dentate gyrus granule cells depends on SSADH gene dosage. (A) The holding current was plotted against time and used to estimate the tonic current sensitive to the GABAA receptor antagonist SR95531 (∼100 μM). In this representative WT granule cell, the SR95531 sensitive tonic GABAA current was 5.8 pA. In the granule cell from SSADH KO mouse, the tonic GABAA current was larger and reached 34.0 pA. Cells were clamped at a Vhold of −70 mV. The Gaussian curves (left) represent all-points histograms of the holding current. Medians represent the mean currents, while the deviation correlates with the noise level. After SR95531 application, there is a decrease in the noise level. (B) Histogram showing that the average tonic GABAA receptor-mediated current densities in granule cells depended upon the SSADH gene dosage. The number of cells included were WT (n = 13), HET (n = 12), and KO (n = 7). *P < 0.05; **P < 0.01.
Phasic synaptic GABAA receptor-mediated currents in dentate gyrus granule cells are unaltered in SSADH deficiency
Activity of GABAergic synapses onto granule cells gives rise to spontaneous inhibitory postsynaptic currents (sIPSCs). Voltage-clamp recordings in granule cells were again carried out, and in all genotypes, sIPSCs appeared as fast inward currents (Fig. 3A and B). In WT granule cells, sIPSCs demonstrated mean amplitudes, weighted decay time constants, rise times, and mean frequencies that were comparable between WT, HET and KO mice (Fig. 3C). The sIPSC activity showed no significant differences between the 3 genotypes (P > 0.05), indicating that, in excitatory granule cells, the GABAergic synaptic function is comparable to WT and HET animals in SSADH KO mice.
Figure 3.
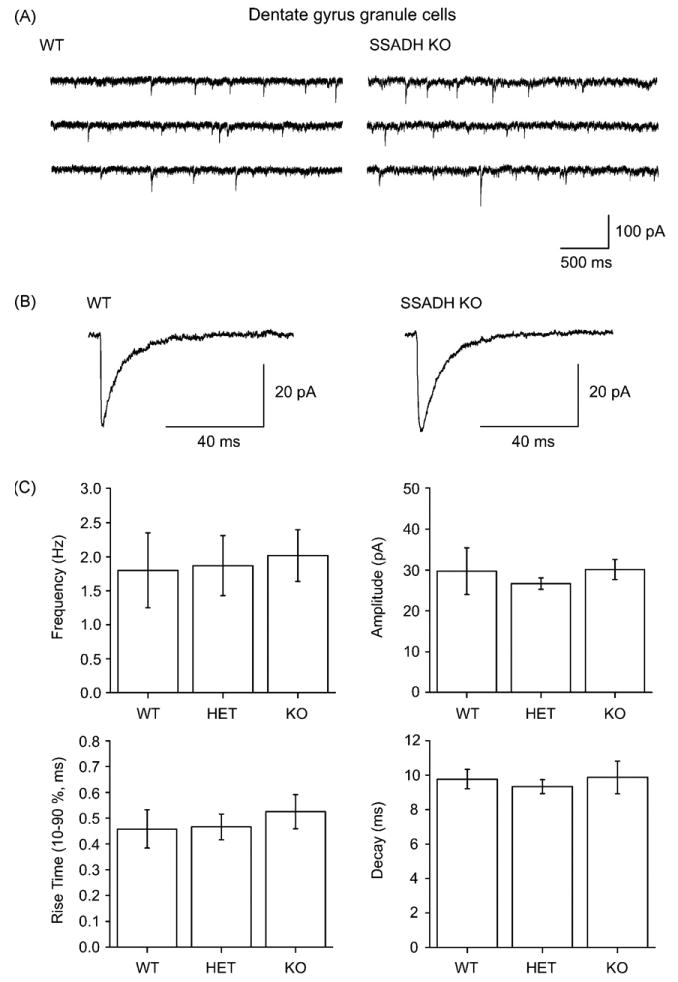
Phasic GABAergic inhibition in dentate gyrus granule cells is not affected in SSADH KO mice. (A) Sweeps of spontaneous inhibitory postsynaptic currents (sIPSCs) recorded from granule cells show no differences in WT and SSADH KO mice. (B) Averages of sIPSCs from a WT and a KO cell showing similar waveforms. (C) Histograms summarizing the average frequencies, amplitudes, rise times (10–90%), and the decay time constants of sIPSCs in granule cells recorded from WT, HET, and KO mice. The kinetics of sIPSCs were similar for all groups, showing non-significant differences between the genotypes (P > 0.05).
Tonic GABAA receptor-mediated currents in dentate gyrus interneurons are increased in SSADH deficient mice
Interneurons of the dentate gyrus were identified by their 3–4 times larger cell bodies in the infrared videomicroscope, and their 4.4-fold larger cell capacitance, the latter reflecting the electrically clamped membrane area (Glykys and Mody, 2007). Interneurons in WT slices showed a tonic GABAA current density of 1.1 ± 0.2 pA/pF (Fig. 4A, n = 5). HET slices showed no significant difference in current density (1.3 ± 0.4 pA/pF, n = 8, P > 0.05) from WT, however, in KO slices the current density was significantly larger than WT (2.4 ± 0.4 pA/pF, n = 8, P < 0.05) but not significantly larger than HET (P = 0.15, Fig. 4B). Again, sIPSCs were not different in interneurons across the three genotypes with comparable amplitudes, decay time constants, rise times and frequencies between WT, HET and KO slices (Fig. 5A–C). Although more variability was observed for sIPSCs in interneurons compared to granule cells, the sIPSC activity showed no significant differences between the 3 genotypes (P > 0.05, Fig. 5).
Figure 4.
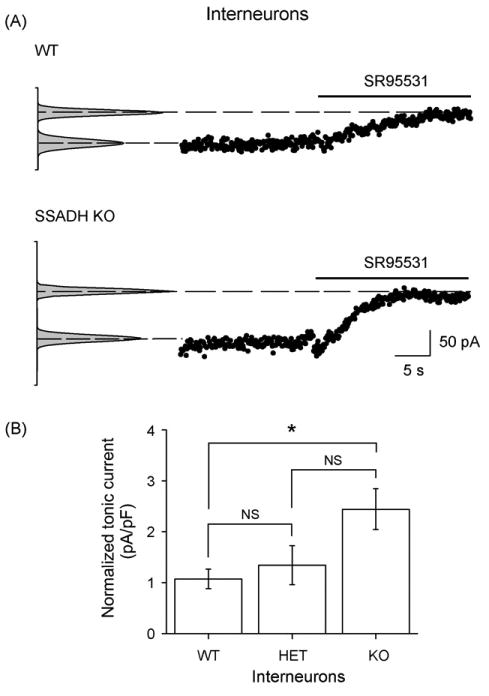
SSADH KO mice show increased SR95531 sensitive tonic GABAA currents in dentate gyrus interneurons. (A) Holding current values showing the SR95531 sensitive GABAA receptor-mediated current. The WT interneuron displayed a tonic current of 55.9 pA, while the KO interneuron reached 97.3 pA. (B) Histogram showing that the average tonic GABAA receptor-mediated current densities in interneurons dependent upon the SSADH gene dosage. These were significantly upregulated in SSADH KO, but not in SSADH HET mice. The number of cells included were WT (n = 5), HET (n = 8) and KO (n = 8). *P < 0.05; NS: non-significant.
Figure 5.
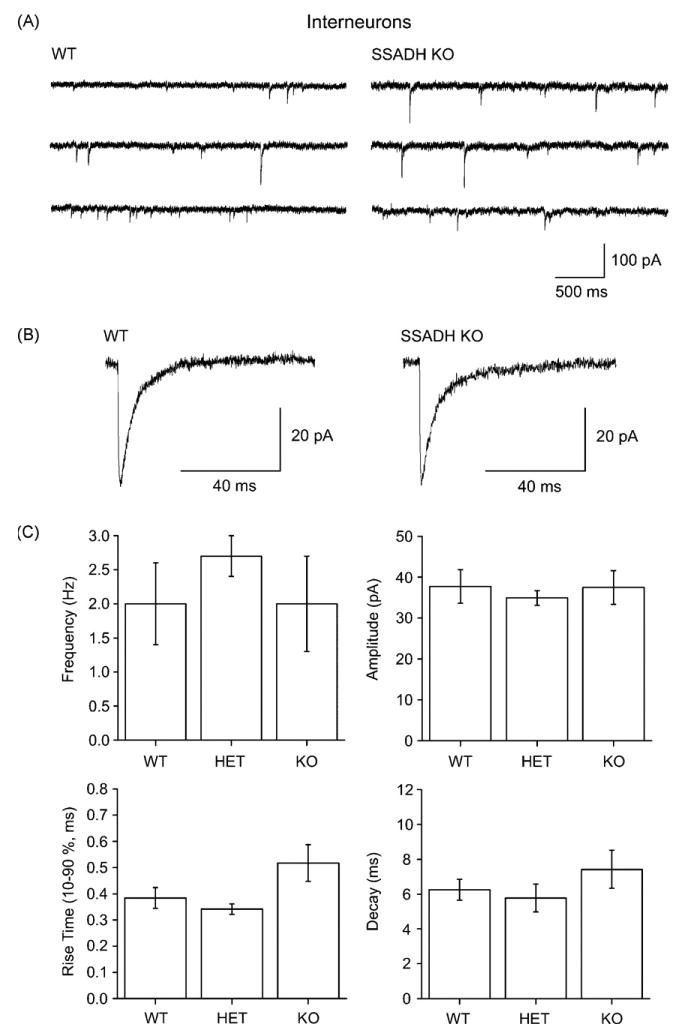
Phasic GABAergic inhibition in dentate gyrus interneurons is not affected in SSADH KO mice. (A) Sweeps of spontaneous inhibitory postsynaptic currents (sIPSCs) recorded from interneurons show no differences in WT and SSADH KO mice. (B) Averages of sIPSCs from a WT and a KO cell showing similar waveforms. (C) Histograms showing the average frequencies, amplitudes, rise times (10–90%), and the decay time constants of sIPSCs in interneurons recorded from WT, HE, and KO mice. The kinetics of the sIPSCs showed no significant differences between the 3 genotypes (P < 0.05).
Increases in tonic GABAA currents in SSADH deficiency are larger in granule cells than in interneurons
From the results on tonic currents (Figs. 2 and 4), it appeared that the increase in tonic currents with reduced gene dosage was non-linear, and relatively larger in granule cells. To statistically evaluate this, we first tested the hypothesis of an exponential relationship, on the basis of analysis of variance for linear regression on logarithmically (ln) transformed values. This was accepted for both dentate gyrus granule cells and interneurons, yielding P = 0.56 and P = 0.28, respectively (see “Methods”). The slopes of the regression lines were 0.021 ± 0.0045 and 0.0084 ± 0.0036, and intercepts were 1.6 ± 0.37 and 0.68 ± 0.25 for granule cells and interneurons, respectively (Fig. 6). These values were significantly different between the two cell types (two-sample t-test, P < 0.0001 for both slope and intercept). Thus, we conclude that the increase in tonic current was non-linear, and larger in granule cells.
Figure 6.
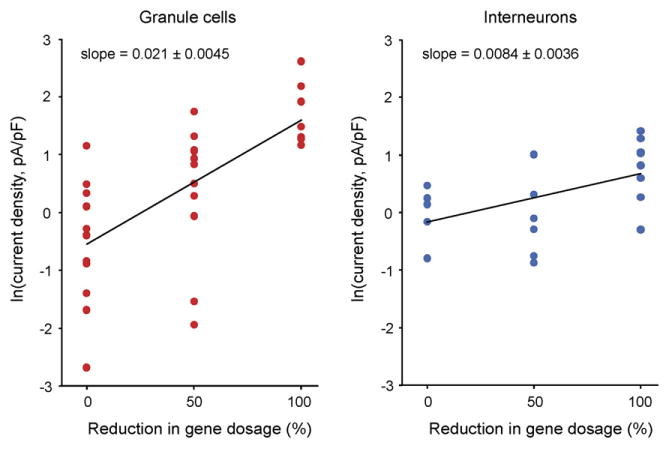
Linear regression of logarithmically transformed current density values in granule cells and interneurons. Linear regression (of the ln transformed data) is accepted on the basis of analysis of variance of granule cells (P = 0.55), and interneurons (P = 0.28). The slope and intercept of regression lines were significantly different between the two types of neurons (2-sample t-test, P < 0.0001, for slope and intercept).
Discussion
In this study, we found that a decreased SSADH gene dosage caused a non-linear increase in the tonic GABAA receptor-mediated current in the dentate gyrus. This increase was electrophysiologically observed both in principal cells and interneurons, demonstrating a strengthened tonic inhibition when SSADH function is impaired. Reducing the SSADH gene dosage to 50% had a minor, but statistically significant, impact on the electrophysiological responses in granule cells, but not in interneurons, while a total SSADH gene knock-out led to substantial increases in extrasynaptic GABAA receptor activation. Since this increase occurred in both cell types, we conclude that augmented GABAA receptor activation is a hallmark of the hyperGABAergic state in two major neuronal types in the hippocampal formation, i.e. principal cells and interneurons. This might be an important aspect of understanding the neurological symptoms in human SSADH deficiency, which likely involves several distinct cellular subtypes in cortical networks.
SSADH enzyme activity correlates with GABA levels in SSADH deficient mice
It was previously found that the SSADH gene dosage biochemically correlates with SSADH enzyme activities in the brain. Thus, SSADH HET mice show 52% of the brain enzyme activity compared with WT, while SSADH KOs show 0% activity (Hogema et al., 2001). Analyzing contents in different tissues and fluids, GABA is increased 8.7- (urine) and 2.8-(total brain) fold in SSADH KOs, respectively, with no discernable changes in HET animals. Based on these observations, we analyzed how decreasing SSADH activities translated into tonic GABA currents measured by patch-clamping two major cell types in the dentate gyrus. These experiments showed that GABA receptor activation increased by 2.4- and 6.9-fold in granule cells in SSADH HET and KO mice, while the corresponding numbers for interneurons were 1.3- and 2.3-fold. In agreement, regression analysis showed that the non-linear increase in tonic currents was significantly larger in granule cells compared with interneurons.
Increased extracellular GABA levels induce tonic GABAA receptor activity
A number of previous studies have demonstrated that elevated ambient GABA in the brain leads to tonic, and presumed, extrasynaptic GABA receptor activation. It was found that rat granule cells showed increased GABAA receptor currents, when the extracellular GABA was increased by inhibiting GABA uptake with NO-711, with little or no change in the GABAergic synaptic function (Nusser and Mody, 2002). Similar findings were obtained in GABA transporter 1 (GAT1) KO mice, where extracellular GABA levels are elevated, and hippocampal pyramidal cells show a similar increase in tonic extrasynaptic GABAA responses (Jensen et al., 2003). It remains possible that intracellular accumulation of GABA in SSADH deficient cells may in fact inhibit GABA uptake via GAT1, since GAT function depends on low cytosolic GABA levels (Wu et al., 2001). This hypothesis remains untested, however, although inhibition of GABA uptake, and even reversal of the GABA transporter, occurs when cytosolic GABA is increased by blocking GABA transaminase with vigabatrin (Wu et al., 2001).
GABAA and GABAB mediated mechanisms in the mouse model of SSADH deficiency
Other electrophysiological alterations in GABAA and GABAB mediated functions have also been described in the neocortex and hippocampus of SSADH KO mice. Firstly, Drasbek et al. (2008) showed that neocortical layer 2/3 pyramidal cells displayed similar evoked and spontaneous IPSCs in SSADH KOs, but exhibited increased tonic currents. In an independent study, electrically evoked GABAA receptor-mediated synaptic transmission was downregulated, which was associated with a decreased GABAA receptor ligand binding in the CA1 region (Wu et al., 2006). Additionally, the spontaneous GABA release in the CA1 region was reported to be downregulated, which can be rescued by a ketogenic diet (Nylen et al., 2008). Thus, our findings somewhat contrast those from CA1, although this could be due to brain-region or cell type specific changes induced by the SSADH deficiency.
Finally, reduced slow GABAB receptor-mediated responses were observed in the hippocampus (Buzzi et al., 2006). The latter finding points to the hypothesis that G-protein coupled GABAB receptors are desensitized by high levels of the agonists, GABA and GHB. In addition, downstream effectors such as GIRK channels could be downregulated as a result of the high activation of GABAB receptors in SSADH KO mice. Whether downregulation of cortical GABAB receptor function plays a role in the conversion from absence to tonic-clonic seizures will await further studies.
Conclusions and functional aspects
We conclude that SSADH deficiency increases GABAA mediated tonic inhibition that is possibly caused by increased extracellular GABA concentrations globally in the brain. Indeed, the cell-specific changes in GABA responses in SSADH KO mice point to distinct effects of GABA on major types of neurons, including excitatory and inhibitory cells. This is particularly relevant when the effects on the neural network are assessed, and how it translates into in vivo findings that SSADH deficient mice display, including electrophysiological and behavioral absence seizures (Cortez et al., 2004). It was recently found that increased GABA can synchronize thalamocortical circuits, which may underlie typical absence seizures (Cope et al., 2009). On the other hand, the progression into convulsive status epilepticus might involve other mechanisms, as discussed above.
Despite the plethora of discrete structural and biochemical alterations in SSADH deficient mice (Gupta et al., 2004), our results indicate that excitatory and inhibitory neurons of the dentate gyrus are exposed to elevated GABA levels in a gene dosage dependent manner. This could tip the balance between excitation and inhibition in the dentate gyrus in this model of perturbed GABA metabolism, and possibly be linked to the symptoms related to the limbic system, including emotional and cognitive disturbances in human SSADH deficiency.
Acknowledgments
We thank Lene Wind Steffensen and Lone Overgaard for excellent technical assistance. This study was supported by grants from the Lundbeck Foundation (Denmark), Velux Foundation (Denmark), Gangsted Foundation (Denmark), the Danish Medical Research Council, and NIH HD 58553-02/HD 58553-02S2 from the United States Public Health Service.
References
- Buzzi A, Wu Y, Frantseva MV, Perez Velazquez JL, Cortez MA, Liu CC, Shen LQ, Gibson KM, Snead OC., 3rd Succinic semialdehyde dehydrogenase deficiency: GABA(B) receptor-mediated function. Brain Res. 2006;1090:15–22. doi: 10.1016/j.brainres.2006.02.131. [DOI] [PubMed] [Google Scholar]
- Cope DW, Di Giovanni G, Fyson SJ, Orbán G, Errington AC, Lőrincz ML, Gould TM, Carter DA, Crunelli V. Enhanced tonic GABA(A) inhibition in typical absence epilepsy. Nat Med. 2009;15:1392–1398. doi: 10.1038/nm.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez MA, Wu Y, Gibson KM, Snead OC., 3rd Absence seizures in succinic semialdehyde dehydrogenase deficient mice: a model of juvenile absence epilepsy. Pharmacol Biochem Behav. 2004;79:547–553. doi: 10.1016/j.pbb.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Drasbek KR, Jensen K. THIP, a hypnotic and antinociceptive drug, enhances an extrasynaptic GABA(A) receptor-mediated conductance in mouse neocortex. Cereb Cortex. 2006;16:1134–1141. doi: 10.1093/cercor/bhj055. [DOI] [PubMed] [Google Scholar]
- Drasbek KR, Vardya I, Delenclos M, Gibson KM, Jensen K. SSADH deficiency leads to elevated extracellular GABA levels and increased GABAergic neurotransmission in the mouse cerebral cortex. J Inherit Metab Dis. 2008;31:662–668. doi: 10.1007/s10545-008-0941-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Mody I. Activation of GABA(A) receptors: views from outside the synaptic cleft. Neuron. 2007;56:763–770. doi: 10.1016/j.neuron.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Gupta M, Polinsky M, Senephansiri H, Snead OC, Jansen EE, Jakobs C, Gibson KM. Seizure evolution and amino acid imbalances in murine succinate semialdehyde dehydrogenase (SSADH) deficiency. Neurobiol Dis. 2004;16:556–562. doi: 10.1016/j.nbd.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Hogema BM, Gupta M, Senephansiri H, Burlingame TG, Taylor M, Jakobs C, Schutgens RB, Froestl W, Snead OC, Diaz-Arrastia R, Bottiglieri T, Grompe M, Gibson KM. Pharmacologic rescue of lethal seizures in mice deficient in succinate semialdehyde dehydrogenase. Nat Genet. 2001;29:212–216. doi: 10.1038/ng727. [DOI] [PubMed] [Google Scholar]
- Holm MM, Nieto-Gonzalez JL, Vardya I, Vaegter CB, Nykjaer A, Jensen K. Mature BDNF, but not proBDNF, reduces excitability of fast-spiking interneurons in mouse dentate gyrus. J Neurosci. 2009;29:12412–12418. doi: 10.1523/JNEUROSCI.2978-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen EE, Struys E, Jakobs C, Hager E, Snead OC, Gibson KM. Neurotransmitter alterations in embryonic succinate semialdehyde dehydrogenase (SSADH) deficiency suggest a heightened excitatory state during development. BMC Dev Biol. 2008;8:112. doi: 10.1186/1471-213X-8-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K, Chiu CS, Sokolova I, Lester HA, Mody I. GABA transporter-1 (GAT1)-deficient mice: differential tonic activation of GABA(A) versus GABA(B) receptors in the hippocampus. J Neurophysiol. 2003;90:2690–2701. doi: 10.1152/jn.00240.2003. [DOI] [PubMed] [Google Scholar]
- Novotny EJ, Jr, Fulbright RK, Pearl PL, Gibson KM, Rothman DL. Magnetic resonance spectroscopy of neurotransmitters in human brain. Ann Neurol. 2003;54(Suppl 6):S25–S31. doi: 10.1002/ana.10697. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Mody I. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J Neurophysiol. 2002;87:2624–2628. doi: 10.1152/jn.2002.87.5.2624. [DOI] [PubMed] [Google Scholar]
- Nylen K, Velazquez JL, Likhodii SS, Cortez MA, Shen L, Leshchenko Y, Adeli K, Gibson KM, Burnham WM, Snead OC., 3rd A ketogenic diet rescues the murine succinic semialdehyde dehydrogenase deficient phenotype. Exp Neurol. 2008;210:449–457. doi: 10.1016/j.expneurol.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl PL, Gibson KM, Cortez MA, Wu Y, Carter Snead O, 3rd, Knerr I, Forester K, Pettiford JM, Jakobs C, Theodore WH. Succinic semialdehyde dehydrogenase deficiency: lessons from mice and men. J Inherit Metab Dis. 2009;32:343–352. doi: 10.1007/s10545-009-1034-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Buzzi A, Frantseva M, Velazquez JP, Cortez M, Liu C, Shen L, Gibson KM, Snead OC., 3rd Status epilepticus in mice deficient for succinate semialdehyde dehydrogenase: GABA(A) receptor-mediated mechanisms. Ann Neurol. 2006;59:42–52. doi: 10.1002/ana.20686. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wang W, Richerson GB. GABA transaminase inhibition induces spontaneous and enhances depolarization-evoked GABA efflux via reversal of the GABA transporter. J Neurosci. 2001;21:2630–2639. doi: 10.1523/JNEUROSCI.21-08-02630.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]