

Abstract
An efficient enantioselective approach to form trans-γ-lactams in up to 99% yield, 93% ee and >20/1 dr using unactivated imines has been developed. The cyclohexyl-substituted azolium and the weak base sodium o-chlorobenzoate are most suitable for this transformation. Notably, the process involves cooperative catalysis by N-heterocyclic carbene and Brønsted acid.
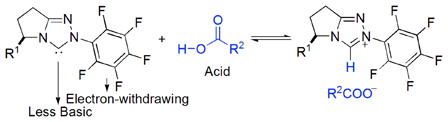
Umpolung reactions catalyzed by N-heterocyclic carbenes (NHCs) have proven a quickly growing field in the past decade.1 Since 2000, our group has focused on the design of carbene precursors and the exploration of new asymmetric reactions by NHC catalysis. We have developed two families of catalysts (morpholine-based and pyrrolidine-based triazolium salts), and successfully applied the azolium salts as precatalysts for Stetter, redox and cascade reactions efficiently affording versatile products in good enantioselectivity.2–4 Of these catalysts, the triazolium precatalysts with an N- pentaflurophenyl group installed to tune the electronic and steric nature exhibit excellent catalytic activity. The corresponding carbenes derived from this scaffold are far less basic and thus could be compatible with weak acids. This is in agreement with our previous observation that sodium acetate can deprotonate the pentafluorophenyl triazolium salts to give the free carbenes and acetic acid.4a Interestingly, the presence of the Brønsted acid does not interfere with the action of the carbenes.
 |
(1) |
We hypothesized that, if an acid with low pKa value does not neutralize a carbene (eq 1),5 the carbene and the acid could play different roles in a reaction system leading to new types of reactions. Based on this hypothesis, we were interested in exploring a NHC/Brønsted acid cooperative strategy in catalysis. Although the concept of cooperative catalysis, such as metal/Brønsted acid,6 NHC/Ag,7 and NHC/Mg or Ti,8 has emerged in organic synthesis, a NHC/Brønsted acid cooperative catalysis has not been demonstrated.9
Homoenolates generated by the reaction of NHC with enals are vinylogous acyl anions, and their reactivity was first reported independently by the Glorius and Bode groups in 2004.10 Since then, investigations have documented their utility to prepare cis-γ-lactams,8a,11 γ-butyrolactones,12 cyclopentenes8b–c,13 and heterocycles,14 among others.15 With few exceptions,11d,12d,13b,15c the enantioselective control of homoenolates still remains challenging. Keeping this in mind, we turned our attention to homoenolate chemistry and the design of new types of chiral azolium salts to achieve products in high enantioselectivity.
We imagined that the conjugate acid of the base used to generate the carbene could be induced to activate a basic functionality such as an imine with the further prospect that a chiral base would potentially lead to a second means of controlling stereochemistry in the bond-forming event. This combination may also conceivably give access to an inaccessible diastereomer and prove complementary to existing methods. Thus, combining homoenolates and acid activated imines,8,16 is expected to generate valuable γ- lactams17 (Scheme 1). Herein, we report an NHC/Brønsted acid-mediated reaction of enals with unactivated imines affording trans-γ-lactams in good yields, high enantioselectivities and good diastereoselectivities.
Scheme 1.
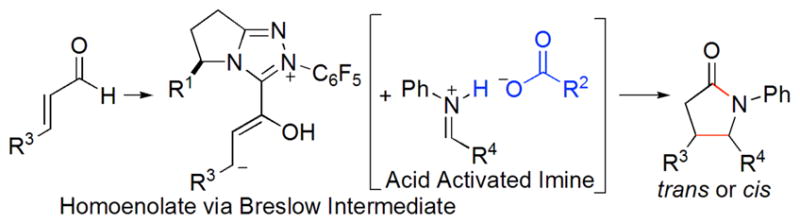
Proposed NHC/Brønsted Acid Cooperative Catalysis
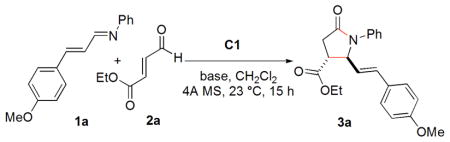
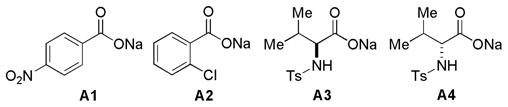
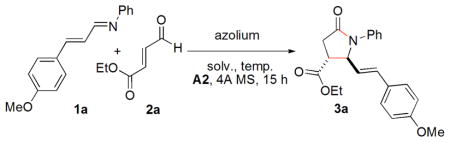
We initiated our study by the reaction of the sterically unhindered and unactivated N-(4-methoxycinnamylidene)aniline (1a) with strongly electrophilic and commercially available ethyl trans-4-oxo-2- butenoate (2a). This reaction was carried out in CH2Cl2 at room temperature in the presence of base18 and 4A MS for 15 h using benzyl-substituted triazolium salt C1 as a catalyst. When strong bases such as KHMDS or Et3N are used, the product lactam is generated in poor yield and selectivity. Under these conditions, enal 2a competitively hydrates or oxidizes with adventitious water or oxygen forming a carboxylic acid which may serve as the activator to the imine. Indeed, when Et3N is used as a stoichiometric base, this pathway is eliminated (entry 2, Table 1). The use of carboxylate bases, on the other hand, is expected to generate stronger conjugate acids capable of activating aldimine 1a. With NaOAc, the reaction occurs to afford the annulation product lactam 3a in 62% yield.19 Surprisingly, the trans- lactam (42% ee) is formed in preference to the cis-isomer (entry 4, Table 1). This is the first NHC-catalyzed synthesis of trans-γ-lactam as the major product.8a When the slightly weaker base PhCOONa is employed, 3a is formed in 72% yield and 50% ee (entry 5). Following this trend, the much weaker bases A1 and A2 (conjugate acids: p-NO2C6H4COOH, pKa[H2O] = 3.1 and o-ClC6H4COOH, pKa[H2O] = 2.9) result in the formation of 3a in higher yields and higher ee’s (entries 6 and 7). When the bases A3 and A4 derived from chiral amino acids are used, 3a is formed in lower enantioselectivity but with a difference between the two entries indicative of a match and mismatch (entries 8 and 9). It is noteworthy that weak bases A3 and A4 are still capable of deprotonating the azolium to generate active catalyst.
Table 1.
Base Screena
 | ||||
|---|---|---|---|---|
| entry | base | yield (%)b | drc | ee (%)d |
| 1 | Et3N | 39 | 9:1 | 14 |
| 2e | Et3N | trace | --- | --- |
| 3 | KHMDS | 25 | 5/1 | 23 |
| 4 | NaOAc | 62 | 5/1 | 42 |
| 5 | PhCOONa | 72 | 6/1 | 50 |
| 6 | A1 | 94 | 5/1 | 57 |
| 7 | A2 | 94 | 5/1 | 59 |
| 8 | A3 | 76 | 6/1 | 33 |
| 9 | A4 | 80 | 6/1 | 41 |
| ||||
Conditions: 2a, 0.2 mmol; 1a, 0.1 mmol; base, 20 mol%; C1 (structure in Table 2), 20 mol%; CH2Cl2, 1 mL; 4A MS under Ar.
NMR yields of trans and cis-diastereomers (internal standard).
The ratio of trans/cis determined by 1H NMR.
Ee of trans-3a determined by chiral HPLC.
1.2 eq of Et3N was used.
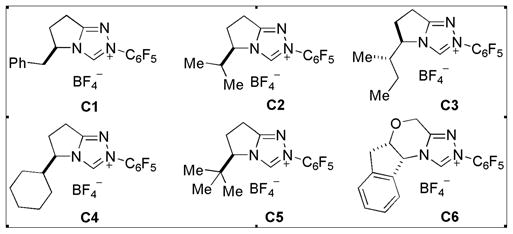
We envisioned that moderately increased steric hindrance on the azolium would provide better chiral environment since the nucleophilic carbanion on the intermediate homoenolate is distant from the chiral center. When isopropyl-substituted triazolium salt C2 is employed as a catalyst with A2, 3a is generated in 66% ee (entry 1, Table 2). The azoliums C3 and C4 with slightly bulkier groups compared to C2 were prepared from the corresponding amino acids, delivering higher enantioselectivities (71% ee and 76% ee, respectively, entries 2 and 3). The much bulkier azolium C5 is ineffective (entry 4). Triazolium salt C6 reveals moderate catalytic activity affording 3a in low ee (46%) (entry 5). Next, the reaction temperature and solvent were investigated using the best catalyst C4 and base A2. When the temperature is lowered to 0 °C, yield, dr and ee are improved (entry 6). Even lower temperature (−10 °C) affects enantioselectivity slightly (entry 7). Common solvents such as toluene, CHCl3 and ClCH2CH2Cl are less effective compared to CH2Cl2. The use of other solvents such as THF, ether or alcohols delivers 3a in only trace amount. However, the use of acetonitrile as a solvent 3a provides 3a in 91% ee, but with moderate yield (entry 8). Satisfyingly, when inexpensive acrylonitrile is utilized as a solvent instead of CH3CN, 3a is generated in excellent yield (93%), good ee (90%) and good diastereoselectivity (11/1) (entry 10).
Table 2.
Optimization of Azolium, Temperature and Solventa
 | ||||||
|---|---|---|---|---|---|---|
| entry | cat. | solvent | temp (°C) | yieldb (%) | drc | eed (%) |
| 1 | C2 | CH2Cl2 | 23 | 97 | 6/1 | 66 |
| 2 | C3 | CH2Cl2 | 23 | 96 | 6/1 | 71 |
| 3 | C4 | CH2Cl2 | 23 | 93 | 6/1 | 76 |
| 4 | C5 | CH2Cl2 | 23 | trace | --- | --- |
| 5 | C6 | CH2Cl2 | 23 | 72 | 5/1 | 46 |
| 6 | C4 | CH2Cl2 | 0 | 98(99) | 9/1 | 87 |
| 7 | C4 | CH2Cl2 | −10 | 97 | 9/1 | 88 |
| 8 | C4 | CH3CN | 0 | 76 | 10/1 | 91 |
| 9 | C4 | CH3CH2CN | 0 | 66 | 12/1 | 87 |
| 10 | C4 | H2C=CHCN | 0 | 96(93) | 11/1 | 91 |
| ||||||
See Table 1 (isolated yield in parentheses).
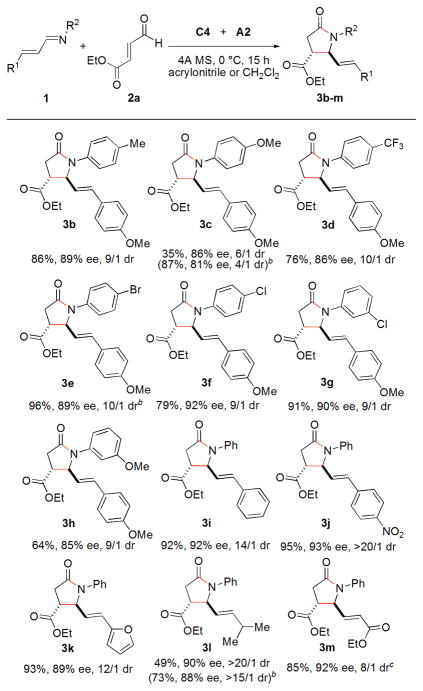
Under these optimized conditions, the substrate scope of α, β-unsaturated imines was evaluated and the results are shown in Table 3.20 The electronic nature of the imines was investigated first. When an electron-neutral group (–Me) and electron-deficient groups (i.e., –CF3, –Br and –Cl) are at the para or meta position of the phenyl group of aldimines, the cyclization is slightly affected to afford the products 3b, 3d, 3e, 3f and 3g in good yields, good dr, and high enantioselectivities (86–92% ee’s). In contrast, the reaction is more efficient in CH2Cl2 using para or meta-methoxy-substituted imines (i.e., 3c, 35% yield, 86% ee, 6/1 dr in acrylonitrile and 87% yield, 81% ee, 4/1 dr in CH2Cl2). Using the aldimines derived from cinnamaldehyde, para-nitrocinnamaldehyde and 3-(2-furyl)acrylaldehyde, the products 3i–3k are obtained in excellent yields and good enantioselectivities (92%, 93% and 89% ee’s, respectively). The imine from (E)-4-methylpent-2-enal undergoes cyclization to give 3l in low yield in acrylonitrile due to decomposition of the imine.21 The reaction proceeds well in CH2Cl2 to yield 3l in 73% yield and 88% ee. However, the in situ generated imine from enal and aniline gives 3m in 85% yield, 92% ee and 8/1 diastereoselectivity.
Table 3.
Evaluation of Imine Substrate Scope
|
Conditions: 2a, 0.2 mmol; imine 1, 0.1 mmol; A2, 20 mol%; C4, 20 mol%; solv., 0.8 mL; all reactions were carried out under Ar in the presence of 4A MS at 0° C for 15 h. The trans/cis ration is determined by 1H NMR prior to purification. The ee’s are determined by chiral HPLC.
CH2Cl2 as a solvent instead of acrylonitrile.
The starting material imine is formed in situ.
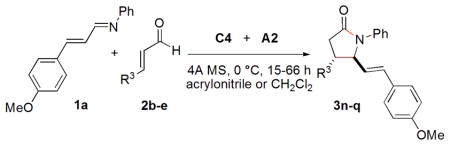
A variety of enals as suitable substrates were explored (Table 4). By using a ketone as a substituent on the enal, the desired lactam 3n is produced in 62% yield and >15/1 dr, but only in 66% ee (entry 1). Less nucleophilic para-nitrocinnamaldehyde is fully converted to 3o in 93% ee and 14/1 dr. In contrast, much less nucleophilic para-bromocinnamaldehyde and cinnamaldehyde undergo cyclization less efficiently to give 3p and 3q in 56% and 48% yields, respectively, but in good enantioselectivities and dr.
Table 4.
Exploration of Various Enalsa
 | |||||
|---|---|---|---|---|---|
| entry | R3 | product | yield (%) | dr | ee (%) |
| 1b |
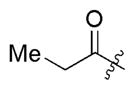 2b 2b
|
3n | 62 | 11/1 | 66 |
| 2c | 4-NO2-C6H4 2c | 3o | 99 | 14/1 | 93 |
| 3d | 4-Br-C6H4 2d | 3p | 58 | 17/1 | 91 |
| 4d | Ph 2e | 3q | 48 | 20/1 | 90 |
Conditions: aldehyde 2, 0.2 mmol; 1a, 0.1 mmol; A2, 20 mol%; C4; 20 mol%; solv., 0.8 mL; all reactions were carried out under Ar in the presence of 4A MS at 0 °C. The trans/cis ratio is determined by 1H NMR prior to purification. The ee’s are determined by chiral HPLC.
15 h in acrylonitrile.
36 h in CH2Cl2.
66 h in CH2Cl2.
 |
(2) |
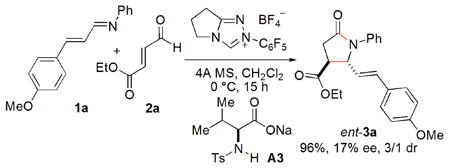
In order to probe the role of acid in our system, the reaction of the α,β-unsaturated imines 1a with the enal 2a was examined using achiral carbene and chiral, enantioenriched amino acids. We found that amino acids bearing stronger electron-withdrawing groups on nitrogen result in higher enantioselectivity. Product ent-3a is obtained in 96% isolated yield, 17% ee, and 3/1 dr using A3 as the base (eq. 2).22 Although the enantioselectivity is low, the result suggests that hydrogen-bonding exists in the transformation and indicates that it is possible to develop an approach with achiral carbenes and chiral acids16 to deliver products with high enantioselectivity.
A few possible reaction pathways involving homoenolate equivalents have been proposed in the literature.11,14 A plausible mechanism for our reaction is related as shown in Scheme 2. The enal 2 first reacts with carbene to generate Breslow intermediate. This homoenolate equivalent attacks acid activated imine perhaps via a hydrogen-bonding intermediate 4. Steric hindrance leads to R3 and alkenyl in the anti position. Proton transfer then results in the formation of acyl carboxylate 5. The nitrogen species replaces carbene to afford the product 3 and free carbene.
Scheme 2.

Proposed Pathway for trans-γ-Lactam Formation
In conclusion, we have developed an efficient NHC-catalyzed asymmetric approach to synthesize trans-γ-lactams in high yields, high enantioselectivities and >20/1 dr with unactivated imines. It is noteworthy that electron-rich carbenes have historically been used to catalyze homoenolate chemistry.1b Our findings are promising in the application of homoenolates using electron-deficient carbene catalysis. This method involves cooperative NHC/Brønsted acid catalysis. In the transformation, cyclohexyl-substituted carbene and o-chlorobenzoic acid are most efficient. The application of cooperative catalysis in other reactions and the utility of chiral carbenes for asymmetric homoenolate transformation are in progress.
Supplementary Material
Acknowledgments
We thank NIGMS (GM72586), Amgen, and Roche for support. DAD is grateful to Roche for an Excellence in Chemistry award. We thank Kevin M. Oberg (CSU) for solving the crystal structure to determine absolute configuration of products.
Footnotes
Supporting Information. Experimental procedures, characterization data, absolute configuration determination of product, NMR and HPLC spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For selected reviews, see: Enders D, Niemeier O, Hensler A. Chem Rev. 2007;107:5606. doi: 10.1021/cr068372z.Nair V, Vellalath S, Babu BP. Chem Soc Rev. 2008;37:2691. doi: 10.1039/b719083m.Read de Alaniz J, Rovis T. Synlett. 2009:1189. doi: 10.1055/s-0029-1216654.Phillips EM, Chan A, Scheidt KA. Aldrichimica Acta. 2009;42:55.Moore JL, Rovis T. Top Curr Chem. 2010;291:77. doi: 10.1007/978-3-642-02815-1_18.Vora HU, Rovis T. Aldrichimica Acta. 2011;44:3.
- 2.For asymmetric Stetter, see: Kerr MS, Read de Alaniz J, Rovis T. J Am Chem Soc. 2002;124:10298. doi: 10.1021/ja027411v.Kerr MS, Rovis T. J Am Chem Soc. 2004;126:8876. doi: 10.1021/ja047644h.Read de Alaniz J, Rovis T. J Am Chem Soc. 2005;127:6284. doi: 10.1021/ja0425132.Liu Q, Rovis T. J Am Chem Soc. 2006;128:2552. doi: 10.1021/ja058337u.Cullen SC, Rovis T. Org Lett. 2008;10:3141. doi: 10.1021/ol801047k.Liu Q, Perreault S, Rovis T. J Am Chem Soc. 2008;130:14066. doi: 10.1021/ja805680z.Liu Q, Rovis T. Org Lett. 2009;11:2856. doi: 10.1021/ol901081a.DiRocco DA, Oberg KM, Dalton DM, Rovis T. J Am Chem Soc. 2009;131:10872. doi: 10.1021/ja904375q.DiRocco DA, Rovis T. J Am Chem Soc. 2011;133:10402. doi: 10.1021/ja203810b.
- 3.For redox reactions, see: Reynolds NT, Read de Alaniz J, Rovis T. J Am Chem Soc. 2004;126:9518. doi: 10.1021/ja046991o.Reynolds NT, Rovis T. J Am Chem Soc. 2005;127:16406. doi: 10.1021/ja055918a.Vora HU, Rovis T. J Am Chem Soc. 2007;129:13796. doi: 10.1021/ja0764052.Vora HU, Moncecchi JR, Epstein O, Rovis T. J Org Chem. 2008;73:9727. doi: 10.1021/jo8020055.Vora HU, Rovis T. J Am Chem Soc. 2010;132:2860. doi: 10.1021/ja910281s.
- 4.For cascade reactions, see: Lathrop SP, Rovis T. J Am Chem Soc. 2009;131:13628. doi: 10.1021/ja905342e.Filloux CM, Lathrop SP, Rovis T. Proc Natl Acad Sci USA. 2010;107:20666. doi: 10.1073/pnas.1002830107.Ozboya KE, Rovis T. Chem Sci. 2011 doi: 10.1039/C1SC00175B.
- 5.An equilibrium certainly exists between the azolium/base and carbene/conjugate acid. However, through the use of an acidic azolium with a weak base, we hoped to generate the NHC and conjugate acid in significant enough quantities to engage in productive cooperative catalysis. For a report involving the use of another very weak base in NHC catalysis, see: Kaeobamrung J, Mahatthananchai J, Zheng P, Bode JW. J Am Chem Soc. 2010;132:8810. doi: 10.1021/ja103631u.
- 6.For selected examples, see: Hu W, Xu X, Zhou J, Liu WJ, Huang H, Hu J, Yiang L, Gong LZ. J Am Chem Soc. 2008;130:7782. doi: 10.1021/ja801755z.Li C, Villa-Marcos B, Xiao J. J Am Chem Soc. 2009;131:6967. doi: 10.1021/ja9021683.Lu Y, Johnstone TC, Arndtsen BA. J Am Chem Soc. 2009;131:11284. doi: 10.1021/ja904185b.Zhou S, Fleischer S, Junge K, Beller M. Angew Chem Int Ed. 2011;50:5120. doi: 10.1002/anie.201100878.
- 7.Chen Z, Yu X, Wu J. Chem Commun. 2010;46:6356. doi: 10.1039/c0cc01207f.For compatible catalysis of NHC/Pd, see: Nemoto T, Fukuda T, Hamada Y. Tetrahedron Lett. 2006;47:4365.Lebeuf R, Hirano K, Glorius F. Org Lett. 2008;10:4243. doi: 10.1021/ol801644f.He J, Tang S, Liu J, Sun Y, Pan X, She X. Tetrahedron Lett. 2009;50:430.
- 8.(a) Raup DEA, Cardinal-David B, Holte D, Scheidt KA. Nat Chem. 2010;2:766. doi: 10.1038/nchem.727. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cardinal-David B, Raup DEA, Scheidt KA. J Am Chem Soc. 2010;132:5345. doi: 10.1021/ja910666n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cohen D, Cardinal-David B, Scheidt KA. Angew Chem Int Ed. 2011;50:1678. doi: 10.1002/anie.201005908. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cohen D, Cardinal-David B, Roberts JM, Sarjeant AA, Scheidt KA. Org Lett. 2011;13:1068. doi: 10.1021/ol103112v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hydrogen-bonding to improve enantioselectivity in NHC catalysis has been reported; see: Mennen SM, Blank JT, Tran-Dube MB, Imbriglio JE, Miller SJ. Chem Commun. 2005:195. doi: 10.1039/b414574g.He L, Zhang YR, Huang XL, Ye S. Synthesis. 2008:2825.O’Toole SE, Connon SJ. Org Biomol Chem. 2009;7:3584. doi: 10.1039/b908517c.
- 10.(a) Burstein C, Glorius F. Angew Chem Int Ed. 2004;43:6205. doi: 10.1002/anie.200461572. [DOI] [PubMed] [Google Scholar]; (b) Sohn SS, Rosen EL, Bode JW. J Am Chem Soc. 2004;126:14370. doi: 10.1021/ja044714b. [DOI] [PubMed] [Google Scholar]
- 11.(a) He M, Bode JW. Org Lett. 2005;7:3131. doi: 10.1021/ol051234w. [DOI] [PubMed] [Google Scholar]; (b) Rommel M, Fukuzumi T, Bode JW. J Am Chem Soc. 2008;130:17266. doi: 10.1021/ja807937m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nair V, Varghese V, Babu BP, Sinu CR, Suresh E. Org Biomol Chem. 2010;8:761. doi: 10.1039/b922981g. [DOI] [PubMed] [Google Scholar]; (d) Zheng P, Gondo CA, Bode JW. Chem Asian J. 2011;6:614. doi: 10.1002/asia.201000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Sohn SS, Rosen EL, Bode JW. J Am Chem Soc. 2004;126:14370. doi: 10.1021/ja044714b. [DOI] [PubMed] [Google Scholar]; (b) Burstein C, Tschan S, Xie X, Glorius F. Synthesis. 2006:2418. [Google Scholar]; (c) Nair V, Vellalath S, Poonoth M, Mohan R, Suresh E. Org Lett. 2006;8:507. doi: 10.1021/ol052926n. [DOI] [PubMed] [Google Scholar]; (d) Li Y, Zhao Z-A, He H, You S-L. Adv Synth Catal. 2008;350:1885. [Google Scholar]
- 13.(a) Nair V, Vellalath S, Poonoth M, Suresh E. J Am Chem Soc. 2006;128:8736. doi: 10.1021/ja0625677. [DOI] [PubMed] [Google Scholar]; (b) Chiang PC, Kaeobamrung J, Bode JW. J Am Chem Soc. 2007;129:3520. doi: 10.1021/ja0705543. [DOI] [PubMed] [Google Scholar]; (c) Chiang PC, Rommel M, Bode JW. J Am Chem Soc. 2009;131:8714. doi: 10.1021/ja902143w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) He M, Struble JR, Bode JW. J Am Chem Soc. 2006;128:8418. doi: 10.1021/ja062707c. [DOI] [PubMed] [Google Scholar]; (b) Chan A, Scheidt KA. J Am Chem Soc. 2007;129:5334. doi: 10.1021/ja0709167. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) He M, Bode JW. J Am Chem Soc. 2008;130:418. doi: 10.1021/ja0778592. [DOI] [PubMed] [Google Scholar]; (d) Phillips EM, Reynolds TE, Scheidt KA. J Am Chem Soc. 2008;130:2416. doi: 10.1021/ja710521m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chan A, Scheidt KA. J Am Chem Soc. 2008;130:2740. doi: 10.1021/ja711130p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Seayad J, Patra PK, Zhang Y, Ying JY. Org Lett. 2008;10:953. doi: 10.1021/ol800003n. [DOI] [PubMed] [Google Scholar]
- 15.(a) Nair V, Babu BP, Vellalath S, Varghese V, Raveendran AE, Suresh E. Org Lett. 2009;11:2507. doi: 10.1021/ol900571x. [DOI] [PubMed] [Google Scholar]; (b) Nair V, Sinu CR, Babu BP, Varghese V, Jose A, Suresh E. Org Lett. 2009;11:5570. doi: 10.1021/ol901918x. [DOI] [PubMed] [Google Scholar]; (c) Fang X, Jiang K, Xing C, Hao L, Chi YR. Angew Chem Int Ed. 2011;50:1910. doi: 10.1002/anie.201007144. [DOI] [PubMed] [Google Scholar]
- 16.For selected examples and reviews, see: Akiyama T. Chem Rev. 2007;107:5744. doi: 10.1021/cr068374j.You SL, Cai Q, Zeng M. Chem Soc Rev. 2009;38:2190. doi: 10.1039/b817310a.Hashimoto T, Kimura H, Maruoka K. Angew Chem Int Ed. 2010;49:6844. doi: 10.1002/anie.201003600.
- 17.Sun PP, Chang MY, Chiang MY, Chang NC. Org Lett. 2003;5:1761. doi: 10.1021/ol0344164.Roberson CW, Woerpel KA. J Org Chem. 1999;64:1434. doi: 10.1021/jo982375x.Pohmakotr M, Yotapan N, Tuchinda P, Kuhakarn C, Reutrakul V. J Org Chem. 2007;72:5016. doi: 10.1021/jo070533r.For examples of trans-γ-lactams as cores of natural products, see: Barrett AGM, Head J, Smith ML, Stock NS, White AJP, Williams DJ. J Org Chem. 1999;64:600.Feeling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Angew Chem Int Ed. 2003;42:355. doi: 10.1002/anie.200390115.
- 18.When the reaction is carried out in the absence of a base, trace desired product is generated.
- 19.Under these conditions, 3m was also formed (7%), the residual product derived from in situ imine exchange.
- 20.Other imines: N-Bn and N-acyl α,β-unsaturated imines afford trace amounts of the desired lactams. N-Ts imine does not produce the lactam. The N-phenyl aldimine derived from para-bromobenzaldehyde delivers the lactam in modest yield and selectivity under these conditions (~50% yield, 2:1 trans/cis, 62/58 % ee).
- 21.No lactam product resulting from (E)-4-methylpent-2- enal as a homoenolate was observed.
- 22.When Boc-protected L-valine is utilized, 3a was formed in lower enantioselectivity.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.