

Abstract
The interaction of amyloid-β (Aβ) peptide with Cu(II) appears to play an important role in the etiology of Alzheimer’s disease. At physiological pH, the Cu(II) coordination in Aβ is heterogeneous, and there exist at least two binding modes in which Cu(II) is coordinated by histidine residues. Electron spin resonance studies have revealed a picture of the Cu(II) binding at a higher or lower pH, where only one of the two binding modes is almost exclusively present. We describe a procedure to directly examine the coordination of Cu(II) to each histidine residue in the dominant binding mode at physiological pH. We use nonlabeled and residue-specifically 15N-labeled Aβ(1–16). For quantitative analysis, the intensities of three-pulse electron spin-echo envelope modulation (ESEEM) spectra are analyzed. Spectral simulations show that ESEEM intensities provide information about the contribution of each histidine residue. Indeed, the ESEEM experiments at pH 6.0 confirm the dominant contribution of His6 to the Cu(II) coordination as expected from the work of other researchers. Interestingly, however, the ESEEM data obtained at pH 7.4 reveal that the contributions of the three residues to the Cu(II) coordination are in the order of His14 ≈ His6 > His13 in the dominant binding mode. The order indicates a significant contribution from the simultaneous coordination by His13 and His14 at physiological pH, which has been underappreciated. These findings are supported by hyperfine sublevel correlation spectroscopy experiments. The simultaneous coordination by the two adjacent residues is likely to be present in a non–β-sheet structure. The coexistence of different secondary structures is possibly the molecular origin for the formation of amorphous aggregates rather than fibrils at relatively high concentrations of Cu(II). Through our approach, precise and useful information about Cu(II) binding in Aβ(1–16) at physiological pH is obtained without any side-chain modification, amino acid residue replacement, or pH change, each of which might lead to an alteration in the peptide structure or the coordination environment.
Introduction
Amyloid-β (Aβ), a naturally occurring polypeptide with 39–43 amino acid residues, is implicated in the pathogenesis of Alzheimer’s disease. The aggregation of Aβ, which is normally soluble in body fluids, into insoluble deposits is a crucial pathological event in the disease. Several studies have suggested that the intermediates in the aggregation process may account for the neurotoxicity of the disease.1–5 The aggregation of the polypeptide is influenced by other chemical species such as serum proteins, apolipoproteins, phospholipids, and transition metal ions.6 In particular, some transition metal ions such as Cu(II) and Zn(II) affect the aggregation by direct coordination with the polypeptide.7,8
Recent research shows that Cu(II) and Zn(II) are found to be co-localized with aggregated Aβ in the brain of patients with Alzheimer’s disease.9 Also, the transition metal ions are deeply associated with the change in the secondary structure and morphology of Aβ peptide.10 In particular, in vitro research has revealed that with increasing Cu(II) concentrations, amorphous aggregates become dominant over ordered fibrils.11 While the effect of the Zn(II) binding on the aggregation and toxicity is controversial,12–15 the coordination of Cu(II) is generally believed to increase the toxicity.10,16,17 The role of Cu(II) as an etiological factor is, therefore, of biomedical interest.
From a molecular point of view, there has been considerable interest in determining the binding site(s) of Cu(II) in Aβ. A number of scientific approaches have revealed that histidine residues are involved in the Cu(II) coordination. Since Aβ(1–40) and Aβ(1–42), the two major forms found in the amyloid deposits, have three histidine residues, His6, His13, and His14, it has been an important issue to determine which histidine residues coordinate to Cu(II). The work performed by Karr et al.18–20 suggests that Aβ(1–40) and Aβ(1–42) have essentially the same Cu(II)-binding site as shorter Aβ peptides, Aβ(1–28) and Aβ(1–16). Also, the presence of a primary Cu(II)-binding site near the N-terminus region containing the three histidine residues is accepted by researchers21–29 even though the work of Sarell et al.29 suggests that the two shorter Aβ peptides have a relative weak second Cu(II)-binding site. Thus, the two shorter peptides serve as good model systems to investigate the Cu(II) coordination in Aβ especially when an equimolar or subequimolar amount of Cu(II) is present with the peptide.
Despite contradictory results from some groups,19,21 it appears to be consensual that all the three histidine residues, His6, His13, and His14, coordinate to Cu(II) in the equimolar Cu(II)–Aβ(1–16) complex.22–28 A number of electron spin resonance (ESR) studies have, however, suggested that there exists an equilibrium between at least two different Cu(II)-coordination environments in the equimolar complex at physiological pH.22,24,26 Syme et al.22 have reported that two sets of continuous-wave (CW) ESR signals typical of type II Cu(II) complexes are detected from the mixture of Cu(II) and either Aβ(1–28) or Aβ(1–16) at a pH of 7.4. They have also shown that the component with a g|| value of 2.26, Component I, is prevalent at a lower pH, whereas the component with a g|| value of 2.22, Component II, is dominant at a higher pH. Drew et al.24 have suggested Cu(II)-binding modes based on their CW-ESR and hyperfine sublevel correlation (HYSCORE) results. Their experimental data and simulations indicate that His6 coordinates to Cu(II) in Component I and Component II, whereas His13 and His14 simultaneously coordinate to Cu(II) in Component II, but not in Component I. The HYSCORE results of Dorlet et al.26 have also suggested similar Cu(II)-binding modes for Component I. On the other hand, Sarell et al.29 have argued that in both Component I and Component II, two histidine residues, one of which is His6, equatorially coordinate to Cu(II) while the other axially coordinates to Cu(II).
While most researchers propose that two histidine residues simultaneously coordinate to Cu(II) on the equatorial plane in Component I, the number of simultaneously coordinating histidine residues in Component II is controversial. Nevertheless, the binding modes suggested by these groups are consistent with one another in that His6 plays a more pivotal role in the Cu(II) coordination. However, their results may not be fully reflective of the binding modes at physiological pH because most of the experiments have been performed at a lower or higher pH.
Our group has already reported that all of the three residues are involved in the Cu(II) coordination at physiological pH, and their overall contributions are not significantly different.23 Nevertheless, our pervious work was performed under conditions where both components contribute to the signal. Thus, our previous results only showed the average representation of Cu(II) coordination in Aβ(1–16), and the nature of the equilibrium was not truly appreciated.
In this context, we carefully reexamine the contribution of each histidine residue to the Cu(II) coordination in Aβ(1–16), particularly in Component I, at a pH of 7.4 by employing pulsed ESR spectroscopy and exploiting a ternary complex system. The difference between the contributions of His6, His13, and His14 to the Cu(II) coordination in Component I, the dominant component at physiological pH, is our primary interest. We are also interested in the significance of the simultaneous coordination by His13 and His14, which has been suggested to be unlikely in Component I.24,26 To obtain information about the Cu(II) coordination in Component I, we have performed three-pulse electron spin-echo envelope modulation (ESEEM) experiments on equimolar mixtures of Cu(II) and either Aβ(1–16) or its 15N-labeled version at a relatively low magnetic field where Component II is negligible.
Intriguingly, our ESEEM results suggest that the contributions of the three histidine residues to the Cu(II) coordination in Component I are in the order of His14 ≈ His6 > His13, which can only be explained by a significant contribution of the simultaneous Cu(II)-coordination by the two imidazole rings of His13 and His14 in Component I. The observation at physiological pH is informative in that the coordination by the two adjacent residues is likely to lead to the formation of a non–β-sheet structure, which is believed to be associated with toxicity. The biochemical significance of the Cu(II) coordination by the His13–His14 dyad is further discussed in this article.
To assess the relative Cu(II)-binding affinity for each histidine residue, we have introduced a Cu(II)–diethylenetriamine (dien) complex, which is in turn mixed with nonlabeled or 15N-labeled Aβ(1–16) to form a ternary complex. The ESEEM spectra of the ternary complexes indicate that the Cu(II)-binding affinities for the three histidine residues are in the order of His14 > His6 ≈ His13 at a pH of 7.4. The order may account for the unexpected high contribution of His14 to the Cu(II) coordination in Component I at physiological pH.
Also, a broader interpretation of our results leads to the suggestion that the contributions of the three histidine residues to the Cu(II) coordination in Component II are in the order of His13 > His6 ≈ His14, and intermolecular bridges may form by sharing of His13 by two Cu(II) ions in Component II. All in all, our experiments are meaningful in that the interaction between Cu(II) and each histidine residue at physiological pH is traced via a simple model system.
Experimental Section
Peptide Synthesis and Cu(II) Complex Preparation
Isotopically enriched [G-15N]-Nα-Fmoc-Nτ-trityl-L-histidine, in which all nitrogen atoms are enriched with 15N, was purchased from Cambridge Isotope Laboratory (Andover, MA). Three different analogues of Aβ(1–16) peptide, DAEFRHDSGYEVHHQK, with an 15N-labeled histidine residue at either position 6, 13, or 14 were synthesized at the Peptide Synthesis Facility in University of Pittsburgh via the conventional solid-phase fluorenylmethoxycarbonyl chemistry.30,31 The three 15N-labeled Aβ(1–16) analogues were termed Aβ(1–16)H6[15N], Aβ(1–16)H13[15N], and Aβ(1–16)H14[15N], respectively. Each of the analogues was characterized by high performance liquid chromatography and mass spectrometry. Nonlabeled Aβ(1–16) peptide was purchased from rPeptide (Bogart, GA). Isotopically enriched [63Cu]Cl2 was purchased from Cambridge Isotope Laboratory. Diethylenetriamine and N-ethylmorpholine (NEM) were purchased from Sigma–Aldrich (St. Louis, MO).
A 100 mM NEM buffer with a pH of 7.4 was prepared by mixing NEM and hydrochloric acid in 50% glycerol. Another NEM buffer with a pH of 6.0 was prepared in the same fashion. Then, 2.5 mM solutions of Aβ(1–16), Aβ(1–16)H6[15N], Aβ(1–16)H13[15N], and Aβ(1–16)H14[15N] were prepared in the pH 7.4 NEM buffer as well as in the pH 6.0 NEM buffer. Separately, 10 mM Cu(II) stock solutions were prepared in the two buffers. For each of the Aβ(1–16) analogues, an equimolar Cu(II)–peptide mixture was prepared with a final concentration of 1.25 mM. A 6 mM solution of dien was also prepared in the pH 7.4 NEM buffer. Then, a 1.2:1 molar ratio mixture of dien and Cu(II) was prepared with a final concentration of 2.5 mM in Cu(II). For each of the Aβ(1–16) analogues, a 1.2:1:1 dien–Cu(II)–peptide mixture was prepared with a final concentration of 1.25 mM in the peptide.
Electron Spin Resonance Spectroscopy
Cu(II)–peptide mixtures dissolved in the pH 7.4 buffer or in the pH 6.0 buffer and dien–Cu(II)–peptide mixtures dissolved in the pH 7.4 buffer were used for ESR experiments. A 200 μL aliquot of each Cu(II)–peptide or dien–Cu(II)–peptide mixture solution was transferred into a quartz tube with an inner diameter of 3 mm. All ESR experiments were performed on a Bruker ElexSys E580 FT/CW X-band Spectrometer equipped with a Bruker ER 4118X-MD5 dielectric ring resonator. The temperature was adjusted with an Oxford ITC503 temperature controller and an Oxford CF935 dynamic continuous-flow cryostat connected to an Oxford LLT650 low-loss transfer tube.
Continuous-wave ESR experiments were carried out on the sample solutions. All CW-ESR signals were collected at 80 K with a microwave frequency of approximately 9.69 GHz. The magnetic field was generally swept from 2600 G to 3600 G for a total of 1024 data points. Other instrumental parameters include a time constant of 40.96 ms, a conversion time of 81.92 ms, a modulation amplitude of 4 G, a modulation frequency of 100 kHz, and a microwave power of 0.1993 mW. The experimentally obtained spectra were compared with the corresponding simulated spectra in Symphonia and WINEPR provided by Bruker.
Field-swept echo experiments were performed on the sample solutions at 20 K. Then, three-pulse ESEEM experiments were performed on the same sample solutions at 20 K with a conventional stimulated-echo pulse sequence of π/2 – τ – π/2 – T – π/2 – τ – echo. The first pulse separation, τ, was set at either 144 ns, 192 ns, or 200 ns, and the second pulse separation, T, was varied from either 288 ns or 400 ns with a step size of 16 ns for a total of 1024 points. The pulse length was 16 ns, and the magnetic field strength was fixed at one of 2800 G, 2950 G, 3100 G, 3226 G, 3350 G, 3360 G, and 3375 G; Component I of the Cu(II)–Aβ(1–16) mixtures, whether 15N-labeled or not, is almost exclusively present at 2800 G at physiological pH; 2950 G, 3100 G, and 3226 G are close to the g|| position of the dien–Cu(II)–peptide mixtures; and 3350 G, 3360 G, and 3375 G correspond to the g⊥ positions where the echo intensity is a maximum. In addition, a four-step phase cycle was employed to eliminate unwanted signals.32,33 The real parts of the collected raw data were baseline-corrected and fast Fourier-transformed. Then, the final spectra were obtained as the magnitude of the Fourier transforms. Several ESR parameters for some of the experimentally obtained spectra were determined by spectral simulations based upon the methods provided by Lee et al.34 and Stoll and Britt.35
Four-pulse HYSCORE experiments were performed at 20 K with a pulse sequence of π/2 – τ – π/2 – t1 – π – t2 – π/2 –τ – echo. The first pulse separation, τ, was set at either 144 ns or 200 ns, and both the second pulse separation, t1, and the third pulse separation, t2, were varied from either 144 ns or 200 ns with a step size of 16 ns for a total of either 256 or 512 points. The pulse lengths were 16 ns and 32 ns for π/2 and π pulses, respectively, and the magnetic field strength was fixed at approximately 3360 G or 3375 G, where the echo intensity is a maximum. In addition, a four-step phase cycle was employed to eliminate unwanted signals. The real parts of the collected two-dimensional data were baseline-corrected and apodized with a Hamming window in both dimensions. Then, the processed data were zero-filled to 1024 points in both dimensions before being fast Fourier-transformed. The final spectra were obtained as the contour plots of the magnitude of the two-dimensional Fourier transforms.
Results
A series of CW-ESR and pulsed ESR experiments were conducted on the nonlabeled and 15N-labeled Cu(II)–Aβ(1–16) mixtures and dien–Cu(II)–Aβ(1–16) mixtures. Each of the 15N-labeled Aβ(1–16) peptide analogues used here contains only one 15N-enriched hisitidine residue at either His6, His13, or His14. These 15N-labeled analogues are termed Aβ(1–16)H6[15N], Aβ(1–16)H13[15N], and Aβ(1–16)H14[15N], respectively.
Two Components Present in the Mixture of Cu(II) and Aβ(1–16) at Physiological pH
Continuous-wave ESR experiments were carried out on the equimolar mixture of Cu(II) and the nonlabeled Aβ(1–16) dissolved in the 100 mM NEM buffer at a pH of 7.4. Figure 1 shows the experimentally obtained CW-ESR spectra of the equimolar mixture and spectral simulation results. It is clear that two components are present in the mixture. The g|| value and A|| value of the major component (Component I) are determined to be 2.27 ± 0.005 and 171 ± 1 G, respectively, by spectral simulations. The values are consistent with a square-planar Cu(II)-coordination geometry with three nitrogen donors and one oxygen donor on the equatorial plane.36 Similarly, the g|| value and g|| value of the minor component (Component II) are determined to be 2.23 ± 0.005 and 157 ± 1 G, respectively and the values are consistent with three nitrogen donors and one oxygen donor or four nitrogen donors on the equatorial plane.36 The simulation results also show that the minor component accounts for 25–30% of the Cu(II)–peptide complex. The comparison between experimental and simulated spectra is provided in Supporting Information Figure S1 and Table S1.
Figure 1.
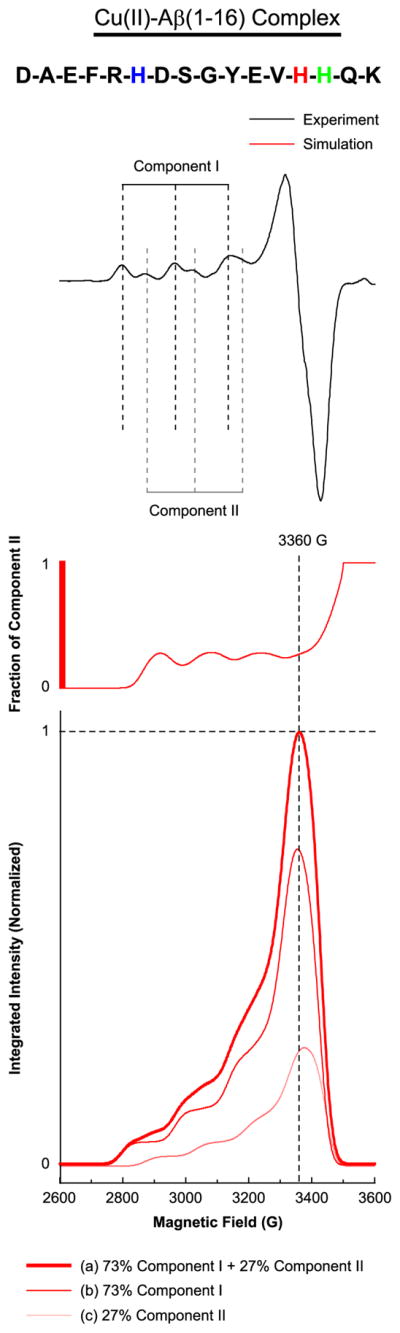
Multiple components present in the Cu(II)–Aβ(1–16) complex suggested by the experimental and simulated CW-ESR spectrum of Aβ(1–16) mixed with an equimolar amount of Cu(II). The amino acid sequence of Aβ(1–16) is illustrated with His6, His13, and His14 indicated in blue, red, and green, respectively. The experimentally obtained CW-ESR spectrum, which contains two clearly distinguished components, Component I and Component II, is shown at the top. The fraction of Component II as a function of the magnetic field is shown at the middle. At the bottom, the curves corresponding to 73% Component I, 27% Component II, and the mixture thereof are shown in red, pink, and bold red, respectively. Component II accounts for approximately a quarter of the Cu(II)–Aβ(1–16) complex at 3360 G while there is practically no contribution of Component II below 2810 G.
Contribution of Each Histidine Residue to the Cu(II) Coordination in Component I at Two Different pHs of 7.4 and 6.0
In our previous work, we had suggested that all of the three histidine residues in Aβ(1–16) contribute to a similar extent to the Cu(II) coordination at physiological pH.23 In that work, however, three-pulse ESEEM experiments were carried out at a magnetic field of approximately 3360 G, where the ESR signal intensity reflects both Component I and Component II. Thus, our previous ESEEM results represent the average aspect of the Cu(II)–Aβ(1–16) complex at physiological pH. Figure 1 also shows the contribution of Component II to the overall signal intensity based on our CW-ESR simulations; Component II accounts for approximately 25% of the total signal at 3360 G. In order to evaluate the contribution of each histidine residue to the Cu(II) coordination exclusively in Component I without changing pH, we took advantage of a magnetic field where the contribution of Component II is negligible, as suggested by Santagelo et al.37,38 on a Cu(II)–DNA complex. As illustrated in Figure 1, Component II gives practically no ESR signal below 2810 G. We performed three-pulse ESEEM experiments on the equimolar mixtures of Cu(II) and the Aβ(1–16) analogues including Aβ(1–16), Aβ(1–16)H6[15N], Aβ(1–16)H13[15N], and Aβ(1–16)H14[15N] at 2800 G.
Figure 2 shows the ESEEM spectrum of the nonlabeled Aβ(1–16) peptide mixed with Cu(II) at 2800 G at a pH of 7.4. The spectrum has three peaks at or around 0.55, 1.01, and 1.54 MHz. The three ESEEM frequencies, ν0, ν−, and ν+ for the NQI transitions are given by:39
Figure 2.
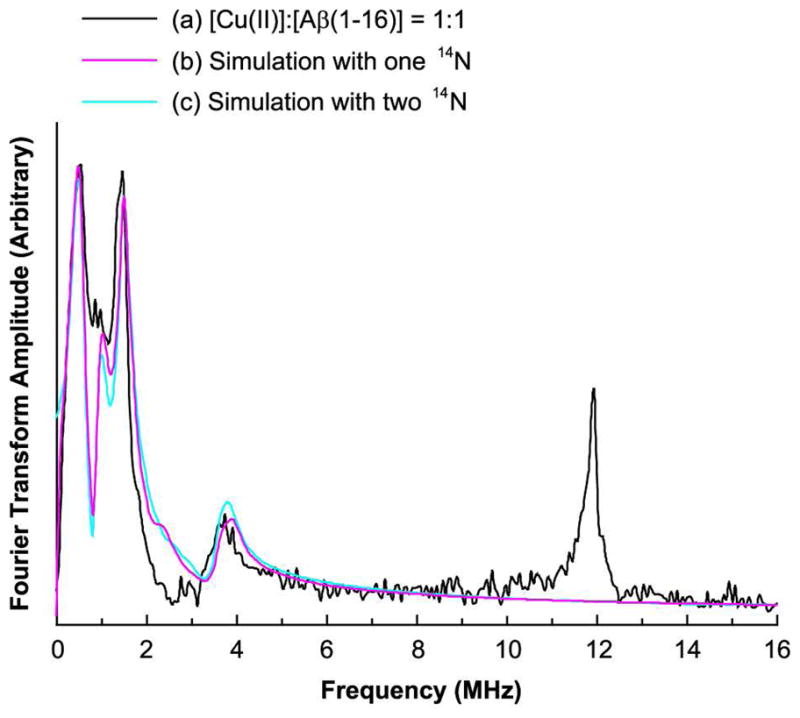
Experimentally obtained and simulated three-pulse ESEEM spectra of the isotopically nonlabeled Aβ(1–16) peptide mixed with an equimolar amount of Cu(II) at 2800 G at pH 7.4. The Simulated spectra with two ESEEM-active 14N nuclei are in good agreement with the experimental result.
| [1] |
where e is the electron charge, q is the z-component of the electric field gradient across the nucleus, Q is the 14N nuclear quadrupole moment, eegr; is the asymmetry parameter, and h is Planck’s constant. With these frequencies in eq [1], the nuclear quadrupole parameters, e2qQ/h and η, are determined to be 1.70 ± 0.03 MHz and 0.65 ± 0.02, respectively, which are comparable to those for Cu(II) complexes of histidine imidazole in proteins that were determined by McCracken et al.40–44 In addition to the three peaks, each spectrum also has a broad peak around 3.8 MHz. Based on the analysis of the NQI frequencies and the three-pulse ESEEM and HYSCORE data obtained at 3360 G, we have concluded that the broad peak around 3.8 MHz is due to the double-quantum transition in the other electron spin manifold.
Figure 2 also shows the corresponding simulated spectra overlaid with the experimentally obtained spectrum. Since two histidine residues are likely to simultaneously coordinate to Cu(II) on the equatorial plane, two ESEEM-active 14N nuclei are assumed to yield the best fit. Aside from the peaks due to 14N nuclei, another peak appears around 11.9 MHz, which is almost the Larmor frequency of 1H at 2800 G. The peak is attributed to the presence of hydrogen atom(s) that weakly interact with the electron spin of Cu(II).
In our work, we analyze the integrated intensity, typically between 0 and 8 MHz, in the three-pulse ESEEM spectra of the nonlabeled and 15N-labeled Cu(II)–Aβ(1–16) complexes to elucidate the contributions of the three histidine residues to the Cu(II) coordination.23 The corresponding two-pulse ESEEM experiments would potentially lead to similar results. However, the peaks at the sum and difference frequencies also appear in two-pulse ESEEM spectra. These peaks are not present in threepulse spectra, which simplifies analysis. Also, the linewidths of two-pulse ESEEM spectra are broader than those of three-pulse spectra. Thus, three-pulse experiments have advantages over two-pulse counterparts for comparing peak intensities and areas under the curve. The modulation depths of the frequencies due to ESEEM-active 14N nuclei decrease when some of the 14N nuclei are replaced with 15N because the single-quantum transition of 15N nuclei does not substantially contribute to the ESEEM signal.45–50 However, it is difficult to obtain the modulation depth of each frequency because there are several different frequencies attributed to 14N in an ESEEM time-domain curve. Thus, instead of comparing the modulation depths, we compare the integrated intensity of the ESEEM spectrum of the nonlabeled version with that of the 15N-labeled ones. Since the modulation depth of the 1H-ESEEM frequency is not significantly affected by a replacement of 14N with 15N, the integrated intensity of the 1H-ESEEM is utilized to normalize the integrated intensity of the 14N-ESEEM region. Detailed calculations show that this normalized 14N-ESEEM intensity is a monotonic function of the fraction of 14N that is replaced with 15N. The relevant equations include eqs [S-40], [S-49], and [S-59] provided in Supporting Information Appendix (4)–(6). Also, simulations show that the normalized 14N-ESEEM intensity increases by a factor of approximately 1.8 when the number of coupled ESEEM-active 14N nuclei increases from one to two provided that the two nuclear spins are equivalent. Detailed explanations about the method used here are provided in Supporting Information Appendix (4)–(8).
This analysis has been applied to the ESEEM data obtained in this work. Figure 4 shows the comparison of the ESEEM spectra of the nonlabeled and 15N-labeled Aβ(1–16) mixed with an equimolar amount of Cu(II) at pH 7.4. The four spectra have almost identical spectral shapes including peak positions in the 14N-ESEEM region below 8 MHz. The similarity in the peak shapes shown in Figure 3 implies that the ESEEM-active 14N of each histidine residue has almost identical nuclear transition frequencies.43 Even with the almost identical frequencies, however, possible difference in the orientation of each histidine residue might also lead to a different normalized 14N-ESEEM intensity. Nevertheless, simulations show that the change in the normalized intensity by such orientational effects is within 5% of the average value for the case of a single electron spin interacting with one 14N nucleus. Detailed explanations are provided in Supporting Information Appendix (8).
Figure 4.

Three-pulse ESEEM and field-swept echo detected spectra of the nonlabeled and 15N-labeled Aβ(1–16) analogues mixed with an equimolar amount of Cu(II) at pH 6.0. The decrease in the 14N-ESEEM intensity below 8 MHz is more prominent when His6 is enriched with 15N. On the other hand, the 1H-ESEEM intensity of each spectrum is essentially identical.
Figure 3.

Three-pulse ESEEM and field-swept echo detected spectra of the nonlabeled and 15N-labeled Aβ(1–16) analogues mixed with an equimolar amount of Cu(II) at 2800 G at pH 7.4. The decrease in the 14N-ESEEM intensity below 8 MHz is more prominent when His6 or His14 is enriched with 15N. On the other hand, the 1H-ESEEM intensity of each spectrum is essentially identical.
As shown in Figure 3, it is clear that the three ESEEM spectra of the 15N-labeled versions have considerably lower intensities in the 14N-ESEEM region below 8 MHz whereas all of the four spectra display nearly identical 1H-ESEEM intensities at or around 11.9 MHz. At a pH of 7.4, the normalized 14N-ESEEM intensity decreases by approximately 40%, 20%, and 40% for the Cu(II) complexes of Aβ(1–16)H6[15N], Aβ(1–16)H13[15N], and Aβ(1–16)H14[15N], respectively, compared with the intensity of the nonlabeled version. The ESEEM spectra at 2800 G indicate that the contribution of His6 or His14 to the Cu(II) coordination in Component I is distinguishably higher than that of His13 at physiological pH. On the other hand, with the uncertainty due to the orientational effects and the error due to the baseline drift in the ESEEM spectra, it is expected that the difference between His6 and His14 may not be significant. The integrated intensity of each spectrum and the estimated relative contribution of each histidine residue to the 14N-ESEEM signal are provided in Supporting Information Table S3.
Similarly, Figure 4 illustrates the comparison of the ESEEM spectra of the nonlabeled and 15N-labeled Aβ(1–16) mixed with an equimolar amount of Cu(II) at a pH of 6.0. The field-swept echo detected spectra reveal that the echo is a maximum at or around 3350 G. Also, it is clear from the first derivative of the field-swept echo detected spectrum of the nonlabeled version that Component I is almost exclusively present. The four ESEEM spectra obtained at 3350 G have almost identical spectral shapes including peak positions in the 14N-ESEEM region below 8 MHz. The ESEEM spectra of the 15N-labeled versions have considerably lower intensities in the 14N-ESEEM region below 8 MHz whereas all of the four spectra display nearly identical 1H-ESEEM intensities at or around 14.3 MHz. The normalized 14N-ESEEM intensity decreases by approximately 45%, 30%, and 30% for the Cu(II) complexes of Aβ(1–16)H6[15N], Aβ(1–16)H13[15N], and Aβ(1–16)H14[15N], respectively, compared with the intensity of the nonlabeled version. Two other sets of ESEEM spectra obtained with either different magnetic field or different pulse separation display a similar trend. Detailed information is provided in Supporting Information Table S4.
Affinity of Each Histidine Residue for Cu(II) Traced with the Dien–Cu(II)–Aβ(1–16) Ternary Complex
In order to assess the propensity of Cu(II) to bind to each of His6, His13, and His14, we introduced a Cu(II)–dien complex, which normally forms a stable ternary complex with a monodentate ligand.43,51
First, we performed CW-ESR and field-swept echo experiments on the nonlabeled and 15N-labeled Aβ(1–16) analogues mixed with an equimolar amount of the Cu(II)–dien complex. Figure 5 shows the CW-ESR spectrum of the mixture of the Cu(II)–dien complex and the nonlabeled Aβ(1–16) peptide. The g|| and A|| values determined by spectral simulations are 2.22 and 191 G, respectively, which are of a typical dien–Cu(II)–imidazole derivative complex with a square-planar geometry, as reported by several researchers.43,51,52 Thus, it is certain that Cu(II) is equatorially coordinated by dien, a tridendate ligand, and one histidine residue, a monodendate ligand. Also, the presence of only one component suggests that the Cu(II) coordination by each histidine residue leads to almost identical coordination geometry. The comparison between experimental and simulated spectra is provided in Supporting Information Figure S2 and Table S2.
Figure 5.

Three-pulse ESEEM and field-swept echo detected spectra of the nonlabeled and 15N-labeled Aβ(1–16) analogues mixed with an equimolar amount of the Cu(II)–dien complex at pH 7.4. The CW-ESR spectrum of the nonlabeled version at pH 7.4 is also presented. The CW-ESR spectrum and the field-swept echo detected spectra display characteristic features of dien–Cu(II)–imidazole ternary complexes. The decrease in the 14N-ESEEM intensity below 8 MHz is most prominent when His14 is enriched with 15N. On the other hand, the 1H-ESEEM intensity is not significantly affected by the replacement of 14N with 15N.
Next, we performed three-pulse ESEEM experiments on the four mixtures at four magnetic fields: 2950 G, 3100 G, and 3226 G, which are close to the g|| region, and 3375 G, at which the echo intensity is a maximum. Figure 5 also shows the ESEEM spectra of the four mixtures obtained at the four different magnetic fields. Each of the four spectra obtained at 2950 G has three relatively narrow peaks at or around 0.55, 1.03, and 1.55 MHz and a broad peak around 4.0 MHz. The three narrow peaks appear at almost the same frequencies as in the corresponding spectra obtained at the other magnetic fields while the frequency of the broad peak changes with the magnetic field. Employing the same analysis as in the case of the Cu(II)–Aβ(1–16) complexes, we have concluded that the three peaks below 2 MHz are mainly due to the NQI transitions of 14N, and the broad peak around 4.0 MHz is due to the double-quantum transition. The nuclear quadrupole parameters, e2qQ/h and eegr;, are determined to be 1.72 ± 0.03 MHz and 0.64 ± 0.02, respectively. The ESEEM spectra reveal that Cu(II) is coordinated by histidine in the four mixtures. Also, another peak appears at around 12.6 MHz, which is almost the Larmor frequency of 1H at 2950 G. The peak is attributed to the presence of hydrogen atom(s) that weakly interact with the electron spin of Cu(II).
It is noticeable that the four spectra obtained at 2950 G have almost identical spectral shapes including peak positions even though the intensities of some peaks are different. Like the case of the Cu(II)–Aβ(1–16) complexes, the similarity in the spectral shape signifies that the ESEEM signals in the spectra are almost independent of the histidine residue that coordinates to Cu(II). However, the three ESEEM spectra of the 15N-labeled versions have considerably lower intensities in the 14N-ESEEM region below 8 MHz whereas all of the four spectra display nearly identical 1H-ESEEM intensities around 12.6 MHz. The normalized 14N-ESEEM intensity decreases by approximately 30%, 30%, and 40% for the Cu(II)–dien complexes of Aβ(1–16)H6[15N], Aβ(1–16)H13[15N], and Aβ(1–16)H14[15N], respectively, compared with the intensity of the nonlabeled version. The three-pulse ESEEM experiments performed at 3100 G, 3226 G, and 3375 G on the same samples show essentially the same trend, as illustrated in Figure 5. The integrated intensity of each spectrum and the estimated relative contribution of each histidine residue to the 14N-ESEEM signal is provided in Supporting Information Table S5. The contribution of each histidine residue to the 14N-ESEEM, which is dependent on the affinity of each histidine residue for Cu(II), is in the order of His14 > His6 ≈ His13, where the distinction between the latter two is not as clear as that between the first two. This order may explain the high contribution of His14 to the Cu(II) coordination in Component I of the Cu(II)–Aβ(1–16) complex. Therefore, it is inferred that at physiological pH, His14 plays an important role in the major component, Component I, due to its higher affinity for Cu(II).
Simultaneous Coordination of Any Two Histidine Residues in the Cu(II)–Aβ(1–16) Complex Suggested by HYSCORE
Figure 6 shows the 14N- and 15N-ESEEM regions of the HYSCORE spectra of the equimolar mixtures of Cu(II) and the nonlabeled and 15N-labeled Aβ(1–16) analogues. Each of the four spectra obtained at approximately 3360 G has a cross-peak around (1.6 MHz, 8.0 MHz). No such cross-peak is observed in the HYSCORE spectra of the dien–Cu(II)–Aβ(1–16) complexes. The HYSCORE spectra of the dien–Cu(II)–Aβ(1–16) complexes are provided in Supporting Information Figure S8. Since the frequencies corresponding to the double-quantum transition are approximately 4.0 MHz, the cross-peak is likely to be due to the existence of two or more 14N nuclei coupled with the electron spin of Cu(II). The possible appearance of a cross-peak from the correlation between a fundamental frequency at one electron spin manifold and a combination frequency at the other manifold in HYSCORE has also been proposed.53,54 Given that the NQI and double-quantum frequencies are almost identical irrespective of the coordinated histidine residue(s), the simultaneous contributions of two 14N nuclei from different histidine residues to the same electron spin system can explain the presence of the cross-peak in all of the four HYSCORE spectra.
Figure 6.

14N- and 15N-ESEEM regions of the HYSCORE spectra of the nonlabeled and 15N-labeled Aβ(1–16) analogues mixed with an equimolar amount of Cu(II) at 3360 G at pH 7.4. Each of the four spectra has a cross-peak around (1.6 MHz, 8.0 MHz), which indicates multiple histidine coordination.
Discussion
Contribution of Each Histidine Residue to the Cu(II) Coordination in Component I at Two Different pHs of 7.4 and 6.0
Our ESEEM experiments performed at a pH of 6.0 show that the contributions of the three histidine residues to the Cu(II) coordination in Component I are in the order of His6 > His13 ≈ His14. The results at pH 6.0 are consistent with those of several other researchers including Drew et al.24 and Dorlet et al.26 and thus validate our approach. However, our ESEEM data obtained at a pH of 7.4 suggest that the contributions of the three histidine residues to the Cu(II) coordination in Component I are in the order of His14 ≈ His6 > His13. The results at physiological pH are significant because very few researchers have emphasized the importance of His14 over His6 or His13, in the Cu(II)–Aβ(1–16) complex, and indeed several groups have suggested a dominant role of His6 in the complex.21,22,24,26,28,55
In fact, the CW-ESR results of Drew et al.24 and the HYSCORE results of Dorlet et al.26 suggest that the contribution of His6 to the Cu(II) coordination in Component I is twice as much as that of His13 or His14. To investigate Component I, however, Drew et al.24 adjusted the pH to 6.3 or 6.9 while Dorlet et al.26 used samples prepared at a pH of 6.5 for the same purpose. The Cu(II)-binding site(s) and involved amino acid residues for Component I are unlikely to be significantly altered by a change in the pH from 6.3 to 7.4 because the ESR parameters including the g|| and A|| values of Component I are found to remain almost unchanged over a wide range of pH values including 7.4.22,26,29 Nevertheless, it has already been revealed that the pH dependence of the equilibrium between Component I and Component II in the Cu(II)–Aβ(1–28) complex is perturbed by a replacement of one histidine residue with alanine, and the extent of the perturbation is dependent on which residue is replaced.29 Also, Ma et al.56 have shown that the pKa values of the three histidine residues are slightly different from one another and dependent on the solution conditions. Therefore, it is also probable that the Cu(II)-binding affinities of the three histidine residues change with pH, and the change is more pronounced in one residue than in another. Furthermore, the pKa values reported by Ma et al.56 are in the range of 5.9 to 7.8, which indicates that both protonated and deprotonated forms are significant, and the ratio between the two forms is sensitive to the pH value in a pH range where Component I is dominant. The lower pKa value of His6 might explain its dominant contribution to the Cu(II) coordination at pH 6.0. Such considerations are compelling arguments for the examination of Cu(II) coordination at physiological pH.
Taken together, the contributions of the three histidine residues to the Cu(II) coordination suggested by our results are in the order of His14 ≈ His6 > His13 in Component I at physiological pH. Our results, therefore, support the contention that the three histidine residues do not equally contribute to the Cu(II) coordination in Component I, and coordinated histidine residues may be in exchange with noncoordinated histidine residues. Our ESEEM data suggest that in Component I, all of the three residues are involved in the Cu(II) coordination, and His14 is at least as significant a contributor as His6 at physiological pH while the contribution of His6 becomes more significant as the pH decreases.
Relationship between Cu(II)-Binding Affinity and Contribution to the Cu(II) Coordination in Component I
Our ESEEM results on the Cu(II)–Aβ(1–16) complexes at 2800 G show that His6 has a significantly higher contribution in Component I than His13. On the other hand, the difference between the two histidine residues is less prominent in the dien–Cu(II)–Aβ(1–16) complex. In order to rationalize the disparity, we propose that the conformation of Component I might be more stable with His6 coordinated to Cu(II) than His13 coordinated to Cu(II) even though the affinities of the two residues for Cu(II) are similar to each other. The proposition is supported by the work of Sarell et al.29 who have shown that a replacement of His6 with alanine reduces the Cu(II)-binding affinity of Aβ(1–28) more substantially than a replacement of His13 or His14. The ITC data of Hong et al.55 also indicate that the Cu(II)–His6 coordination is entropically more favored than the Cu(II) coordination by His13 or His14 due to the proximity of His6 to the N-terminus, which is also likely to be involved in the Cu(II) coordination in Aβ(1–16).
On the other hand, our ESEEM results on the dien–Cu(II)–Aβ(1–16) complexes also reveal that His14 has the highest Cu(II)-binding affinity at a pH of 7.4. These findings are consistent with the work performed by Furlan et al.57,58 who have suggested that Asp1 and His14 interact with each other by electrostatic attraction, which leads to the high metal binding affinity of His14. In fact, the indirect involvement of the carboxylate group of Asp1 in the Cu(II) coordination of Aβ peptide has also been proposed by Karr et al.59 They have suggested that the carboxylate group of Asp1 participates in a hydrogen-bond that stabilizes Component I at physiological pH and the hydrogen-bond is sensitive to a pH change. Considering that pKa values of histidine residues are reported to be in the range of 5.9 to 7.8, it is possible that His14 is hydrogen-bonded to Asp1. Whether the interaction between Asp1 and His14 is electrostatic or hydrogen-bonding-like, it is capable of affecting the Cu(II)-binding affinity of His14. Together, it is probable that the high affinity of His14 for Cu(II) explains the high contribution of the residue to the Cu(II) coordination in Component I while the relatively high contribution of His6 in Component I is due to the overall structure of the complex.
Researchers have suggested that two histidine residues simultaneously and intramolecularly coordinate to Cu(II) on the equatorial plane in Component I.24–26,29 Interestingly, their suggestion is consistent with our ESEEM results. In the spectra obtained at 3375 G in Figure 4 and Figure 5, the double quantum-peak is more prominent in the nonlabeled Cu(II)–Aβ(1–16) complex at pH 6.0 than in the dien–Cu(II)–Aβ(1–16) complex, where only one histidine residue equatorially coordinates to Cu(II) at the same time. These results indicate that more than one histidine residue simultaneously coordinate to Cu(II) in at least a fraction of Component I in the Cu(II)–Aβ(1–16) complex.42 Also, calculations show that the normalized 14N-ESEEM intensity of the Cu(II)–Aβ(1–16) complex is approximately 1.7 times as much as that of the complex with dien, which indicates that approximately two histidine residues simultaneously coordinate to Cu(II) in Component I. These results are further supported by spectral simulations. More detailed information is provided in Supporting Information Figure S7, Figure S10, Table S6, and Table S8.
Figure 7 illustrates the suggested Cu(II)-binding modes of Aβ(1–16) at pH 7.4 and pH 6.0. We propose that at physiological pH, Component I is in turn made up of three different subcomponents, Subcomponent IA, IB, and IC, in each of which two histidine residues simultaneously coordinate to Cu(II) through the imidazole rings. The relative contributions of the three histidine residues are approximately 40%, 20%, and 40% from His6, His13, and His14, respectively. These values lead to the estimation that the subcomponent with the simultaneous coordination by His6 and His14, Subcomponent IB, accounts for approximately 60% of Component I. Each of the other two subcomponents explains about 20%. At physiological pH, Subcomponent IB accounts for 45% of overall Cu(II) complexes including Component I and Component II, while other species are also significant.
Figure 7.

Difference in Cu(II)-binding modes of Aβ(1–16) between pH 7.4 and pH 6.0 suggested by ESR spectroscopy. While His6 accounts for approximately 50% of the Cu(II)–histidine coordination in Component I at pH 6.0, the contribution of His14 is at least as significant as that of His6 at pH 7.4. At pH 7.4, Component I is composed of three subcomponents, Subcomponent IA, IB, and IC, in each of which two imidazole rings from two different histidine residues coordinate to Cu(II).
Interestingly, Furlan et al.57,58 have proposed that the simultaneous coordination by His6 and His14 is favored over that by His6 and His13 in the Zn(II)–Aβ(1–16) and Cu(I)–Aβ(1–16) complex. Presumably, the dominance of Subcomponent IB is due to both the high affinity of His14 and the stability provided by the Cu(II)–His6 coordination. Also, it is probable that Subcomponent IA is less favored because of the relatively lower Cu(II)-binding affinity of His13. More importantly, it is strongly suggested that Subcomponent IC is less dominant than Subcomponent IB due to the entropic penalty, but the high Cu(II)-binding affinity of His14 causes the subcomponent to be as significant as Subcomponent IA.
Simultaneous Coordination by His13 and His14 in Component I at Physiological pH and Its Biochemical Significance
As previously mentioned, our ESEEM results suggest that the contribution of the three histidine residues to the Cu(II) coordination in Component I are in the order of His14 ≈ His6 > His13 at physiological pH. These findings have led to the conclusion that three subcomponents, Subcomponent IA, IB, and IC, are present at physiological pH. The conclusion is also supported by the presence of the cross-peak around (1.6 MHz, 8.0 MHz) in the HYSCORE spectra of the nonlabeled and 15N-labeled Cu(II)–Aβ(1–16) complexes, as illustrated in Figure 6. In particular, the cross-peak around (1.6 MHz, 8.0 MHz) in the HYSCORE spectrum of the Cu(II)–Aβ(1–16)H6[15N] complex strongly suggests that His13 and His14 coordinate to a Cu(II) ion at the same time.
These findings are significant because the simultaneous coordination by His13 and His14 in the equatorial plane in Component I has not been appreciated by many researchers.24,26,55 The simultaneous Cu(II)-coordination by His13 and His14 through the two imidazole rings is not expected for a β-sheet structure because the two adjacent residues would be forced to be on opposite sides of the β-sheet.19,60 Thus, the simultaneous coordination by the two residues signifies that the peptide structure is not a β-sheet near the residues in the presence of Cu(II). Recently, Ahmed et al.61 have reported that relatively small oligomers of Aβ(1–42) with a non–β-sheet secondary structure are neurotoxic while their conversion to protofibrils or fibrils having a β-sheet structure reduces toxicity. It is possible that the expected neurotoxicity of Cu(II) is due to inhibition of the formation of a β-sheet structure by binding to His13 and His14.
Our group has previously revealed that the in vitro morphology of Aβ peptide depends on the Cu(II) concentration: as the Cu(II) concentration increases, the fraction of amorphous aggregates increases while the formation of fibrils is retarded.11 Especially, a thioflavin-T fluorescence assay has suggested that the amount of Aβ fibrils formed in the presence of two equivalents of Cu(II) is less than half that formed at an equimolar level of Cu(II). Herein, we propose that the existence of a significant amount of Subcomponent IC leads to the dominance of amorphous aggregates in the morphology of Aβ peptide. Figure 8 illustrates the formation of fibrils and amorphous aggregates and the suggested role of Subcomponent IC. With no Cu(II), soluble Aβ peptide molecules normally undergo changes in the secondary structure from the random coil to the β-sheet to form fibrils. When a substantial amount of Subcomponent IC is present with other species, however, at least some of the Aβ peptide molecules have a non–β-sheet structure. As a consequence, the formation of fibrils is inhibited, and amorphous aggregates become more significant. It is probable that while several factors may affect the amount of Subcomponent IC, the Cu(II)-to-peptide ratio is one of them. Also, since His6 is not equatorially involved in Subcomponent IC, its overall structure is likely to be different from that of Subcomponent IA or IB. The difference might be another factor that leads to the preference of amorphous aggregates, which are intrinsically less ordered than fibrils. Without Subcomponent IC, the fast equilibrium between Subcomponent IA and IB, whose overall structures are expected to be similar to each other, would lead to the formation of ordered fibrils.
Figure 8.
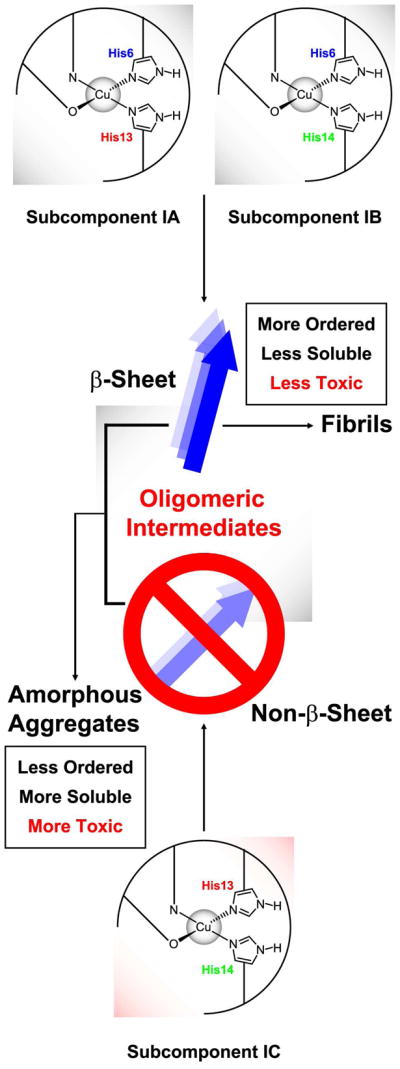
Formation of fibrils and amorphous aggregates of Aβ peptide in the presence of Cu(II) and the suggested role of the simultaneous intramolecular coordination by two imidazole rings of His13 and His14. With a significant amount of Subcomponent IC, the β-sheet and non–β-sheet structures coexist, which leads to the preference of amorphous aggregates over fibrils.
Recently, Dong et al.62 have shown that in the presence of an equimolar amount of Cu(II), the fibril formation of Aβ(13–21)K16A, a mutant with intact His13 and His14, is inhibited. On the other hand, fibrils are observed with Ac-Aβ(13–21)H14A, a mutant with only one histidine residue. More intriguingly, they have also suggested that in the major component of the Cu(II)–Aβ(13–21)K16A complex, only one imidazole ring from one peptide is involved in the Cu(II) coordination on the equatorial plane.62 Their findings are consistent with our proposition in that a small but significant amount of a Cu(II) complex with two equatorially and intramolecularly bound imidazole rings can inhibit the formation of fibrils.
Difference in the Cu(II)–Histidine Coordination between Component I and Component II
Our previous work suggests that when the two components are considered together, the contribution of each histidine residue is not significantly different from one another at physiological pH.23 On the other hand, the ESEEM spectra obtained at 2800 G indicate different contributions of the three histidine residues in Component I. We surmise that the histidine residues that are less involved in Component I at a certain instance are more likely to serve as ligands in Component II. Since the contributions of the three histidine residues in Component I are in the order of His14 ≈ His6 > His13, the contributions of those residues in Component II are likely to be in the order of His13 > His6 ≈ His14 at physiological pH. Given that Component II only accounts for 25–30% of the overall Cu(II)–Aβ(1–16) complex, the contribution of His13 in Component II is expected to be much higher than that of His6 or His14 in Component I. Interestingly, the very high contribution of His13 in Component II occurs despite the relatively low propensity of Cu(II) to bind to His13 as shown in Figure 5. In fact, the amount of His13 in Component II is higher than that expected if Component II was solely intramolecular in nature, and His13 was present in all complexes of Component II. We, therefore, hypothesize that the two nitrogen atoms of the imidazole ring of His13 may coordinate to different Cu(II) ions to form intermolecular bridges in a small fraction of Component II. The possibility of bridges might explain the ESR parameters such as g|| and A|| values that fall into the border-line between three equatorial nitrogen donors and four equatorial nitrogen donors. In addition, the possible presence of bridges might contribute, at least to some extent, to the controversy in the Cu(II) coordination of Component II.
If all of the three imidazole rings of the three histidine residues simultaneously and intramolecularly coordinate to Cu(II) in Component II, the His13–His14 dyad provides two imidazole rings. Then, Component II would play a role similar to that of Subcomponent IC in the inhibition of the fibril formation. In fact, the cross-peak around (1.6 MHz, 8.0 MHz) in our HYSCORE spectra might be partially due to the simultaneous coordination by two histidine residues in Component II. However, more investigation is needed to obtain more precise information about the Cu(II)-coordination environment in Component II.
Regarding the overall contribution of each histidine residue to the Cu(II) coordination in the Cu(II)–Aβ(1–16) complex, some values have already been reported by other groups.24,26,29,55 Drew et al.24 have argued that His6, His13, and His14 account for approximately 50%, 25%, and 25% of the overall Cu(II)-histidine coordination at a pH of 6.3, respectively, and the contribution of each histidine residue at a pH of 8.0 is almost identical. With their data, one can expect the contributions of His6, His13, and His14 at a pH of 7.4 to be approximately 39%, 31%, and 31%, respectively, which is not significantly different from our previous results showing almost equal contributions of the three residues.
However, the overall contribution of each histidine residue at physiological pH expected based on the HYSCORE results of Dorlet et al.26 appears to be inconsistent with our results. As they suggest that only one histidine residue coordinate to Cu(II) at the same time in Component II, it is anticipated that the overall contribution of His6 is still significantly higher than that of His13 or His14 at pH 7.4, where Component I with simultaneous Cu(II) coordination by two histidine residues is dominant. We suspect that the contribution of His6 in Component I might be overestimated in their HYSCORE spectra because a cross-peak due to 15N is normalized by a cross-peak due to 14N, which is supposed to decrease with the increase in the contribution of 15N. Nevertheless, their results are consistent with our results and those of Drew et al.24 in that the contribution of His6 is higher than that of His13 in Component I.
Summary
In order to obtain more precise information about Component I and Component II, both of which comprise the Cu(II)–Aβ(1–16) complex at physiological pH, we have performed three-pulse ESEEM experiments at 2800 G where only Component I is exclusively present. The ESEEM spectra of the equimolar mixtures of Cu(II) and the nonlabeled and 15N-labeled Aβ(1–16) analogues show that the contribution of His14 to the Cu(II) coordination in Component I is at least as much as that of His6 while His13 contributes significantly less than the other two residues at physiological pH. These findings are in contrast with the expectation of His6 playing a dominant role in the Cu(II) coordination in Component I at physiological pH.
The ESEEM spectra of the mixtures of the Cu(II)–dien complex and the Aβ(1–16) analogues suggest that His14 has a better Cu(II)-binding affinity than the other two residues, which may account for the higher contribution of His14 to the Cu(II) coordination in Component I. On the other hand, the contribution of His6 to the Cu(II) coordination in Component I appears to be higher than expected from its Cu(II)-binding affinity. On the basis of our results and the data from other groups, we have suggested that there exist three different subcomponents in Component I at physiological pH. In each of the three subcomponents, two histidine residues simultaneously and intramolecularly coordinate to Cu(II) on the equatorial plane. While the Cu(II)–His6 coordination leads to a stable overall structure of Subcomponent IA (His6–His13) and IB (His6–His14), the presence of Subcomponent IC (His13–His14) may be explained by the high Cu(II)-binding affinity of His14. The existence of the simultaneous coordination by two imidazole rings is also confirmed by the appearance of the cross-peak around (1.6 MHz, 8.0 MHz) in our HYSCORE spectra.
The presence of Subcomponent IC at pH 7.4 may explain the retarded growth of fibrils and the formation of amorphous aggregates in the presence of a significant amount of Cu(II). We have proposed that the non–β-sheet nature of Subcomponent IC inhibits the formation of fibrils, and the distinct structural difference between Subcomponent IC and the other two may lead to the formation of less ordered aggregates. The proposition might explain the role of Cu(II) in the toxicity associated with Aβ peptide, based on the consensus that small oligomers with a non–β-sheet secondary structure are neurotoxic.
In addition, we have conjectured that His13 contributes more to the Cu(II) coordination in Component II than in Component I, which would explain the negligible difference in the contribution of the three histidine residues to the Cu(II) coordination in the Aβ(1–16) complex made up of both components. Also, we suggest, from the interpretation of our results, that intermolecular bridges may form through His13 in a small fraction of Component II.
In brief, our findings are important in that the pH-dependence of the contribution of each histidine to the Cu(II) coordination is revealed by ESEEM spectroscopy. All of our results help find more precise Cu(II)-binding modes by providing critical information obtained at physiological pH. Also, we provide an atomic-level insight into the formation of Aβ aggregates in the presence of Cu(II), which is believed to be an essential event in Alzheimer’s disease.
Supplementary Material
Acknowledgments
This work was supported by a National Institutes of Health grant (5R01NS053788). We are grateful to the Peptide Synthesis Facility of University of Pittsburgh for the peptide preparation. We also thank Dr. Sharon Ruthstein for helpful discussions.
Footnotes
Supporting Information Available: CW-ESR simulation (Figure S1, Table S1, Figure S2, and Table S2), detailed information about the three-pulse ESEEM spectra of the nonlabeled and 15N-labeled Cu(II)–Aβ(1–16) complexes (Figure S3), time-domain curves of the nonlabeled and 15N-labeled Cu(II)–Aβ(1–16) and dien–Cu(II)–Aβ(1–16) complexes (Figure S4, Figure S5, and Figure S6), intensities of the 14N-ESEEM and 1H-ESEEM regions in the three-pulse ESEEM spectra (Table S3, Table S4, and Table S5), number of histidine residues that simultaneously coordinate to Cu(II) in Component I (Figure S7, Table S6), HYSCORE spectra of the nonlabeled and 15N-labeled dien–Cu(II)–Aβ(1–16) complexes (Figure S8), three-pulse ESEEM simulation (Figure S9 and Table S7), and detailed information about the change in modulation depths of the 14N frequencies by a replacement of 14N with 15N (Appendix (1)–(8) including Figure S10, Figure S11, Table S8, and Table S9). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hardy J, Selkoe DJ. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Kirkitadze MD, Bitan G, Teplow DB. J Neurosci Res. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- 3.Carrotta R, Manno M, Bulone D, Martorana V, Biagio PLS. J Biol Chem. 2005;280:30001–30008. doi: 10.1074/jbc.M500052200. [DOI] [PubMed] [Google Scholar]
- 4.Haass C, Selkoe DJ. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 5.Crouch PJ, Harding SME, White AR, Camakaris J, Bush AI, Masters CL. Int J Biochem Cell Biol. 2008;40:181–198. doi: 10.1016/j.biocel.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 6.McLaurin J, Yang DS, Yip CM, Fraser PE. J Struct Biol. 2000;130:259–270. doi: 10.1006/jsbi.2000.4289. [DOI] [PubMed] [Google Scholar]
- 7.Bush AI. Trends Neurosci. 2003;26:207–214. doi: 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 8.Talmard C, Guilloreau L, Coppel Y, Mazarguil H, Faller P. ChemBioChem. 2007;8:163– 165. doi: 10.1002/cbic.200600319. [DOI] [PubMed] [Google Scholar]
- 9.Miller LM, Wang Q, Telivala TP, Smith RJ, Lanzirotti A, Miklossy J. J Struct Biol. 2006;155:30–37. doi: 10.1016/j.jsb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 10.Faller P. ChemBioChem. 2009;10:2837–2845. doi: 10.1002/cbic.200900321. [DOI] [PubMed] [Google Scholar]
- 11.Jun S, Saxena S. Angew Chem, Int Ed. 2007;46:3959–3961. doi: 10.1002/anie.200700318. [DOI] [PubMed] [Google Scholar]
- 12.Cuajungco MP, Fagét KY. Brain Res Rev. 2003;41:44–56. doi: 10.1016/s0165-0173(02)00219-9. [DOI] [PubMed] [Google Scholar]
- 13.Garai K, Sahoo B, Kaushalya SK, Desai R, Maiti S. Biochemistry. 2007;46:10655–10663. doi: 10.1021/bi700798b. [DOI] [PubMed] [Google Scholar]
- 14.Talmard C, Leuma Yona R, Faller P. J Biol Inorg Chem. 2009;14:449–455. doi: 10.1007/s00775-008-0461-9. [DOI] [PubMed] [Google Scholar]
- 15.Miller Y, Ma B, Nussinov R. Proc Natl Acad Sci USA. 2010;107:9490–9495. doi: 10.1073/pnas.0913114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, Cuajungco MP, Gray DN, Lim J, Moir RD, Tanzi RE, Bush AI. Biochemistry. 1999;38:7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 17.Dai X, Sun Y, Gao Z, Jiang Z. J Mol Neurosci. 2010;41:66–73. doi: 10.1007/s12031-009-9282-8. [DOI] [PubMed] [Google Scholar]
- 18.Karr JW, Kaupp LJ, Szalai VA. J Am Chem Soc. 2004;126:13534–13538. doi: 10.1021/ja0488028. [DOI] [PubMed] [Google Scholar]
- 19.Karr JW, Akintoye H, Kaupp LJ, Szalai VA. Biochemistry. 2005;44:5478–5487. doi: 10.1021/bi047611e. [DOI] [PubMed] [Google Scholar]
- 20.Karr JW, Szalai VA. Biochemistry. 2008;47:5006–5016. doi: 10.1021/bi702423h. [DOI] [PubMed] [Google Scholar]
- 21.Kowalik-Jankowska T, Ruta M, Wisniewska K, Lankiewicz L. J Inorg Biochem. 2003;95:270–282. doi: 10.1016/s0162-0134(03)00128-4. [DOI] [PubMed] [Google Scholar]
- 22.Syme CD, Nadal RC, Rigby SEJ, Viles JH. J Biol Chem. 2004;279:18169–18177. doi: 10.1074/jbc.M313572200. [DOI] [PubMed] [Google Scholar]
- 23.Shin B-k, Saxena S. Biochemistry. 2008;47:9117–9123. doi: 10.1021/bi801014x. [DOI] [PubMed] [Google Scholar]
- 24.Drew SC, Noble CJ, Masters CL, Hanson GR, Barnham KJ. J Am Chem Soc. 2009;131:1195–1207. doi: 10.1021/ja808073b. [DOI] [PubMed] [Google Scholar]
- 25.Drew SC, Masters CL, Barnham KJ. J Am Chem Soc. 2009;131:8760–8761. doi: 10.1021/ja903669a. [DOI] [PubMed] [Google Scholar]
- 26.Dorlet P, Gambarelli S, Faller P, Hureau C. Angew Chem Int Ed. 2009;48:9273–9276. doi: 10.1002/anie.200904567. [DOI] [PubMed] [Google Scholar]
- 27.Hureau C, Coppel Y, Dorlet P, Solari PL, Sayen S, Guillon E, Sabater L, Faller P. Angew Chem Int Ed. 2009;48:9522–9525. doi: 10.1002/anie.200904512. [DOI] [PubMed] [Google Scholar]
- 28.Faller P, Hureau C. Dalton Trans. 2009:1080–1094. doi: 10.1039/b813398k. [DOI] [PubMed] [Google Scholar]
- 29.Sarell CJ, Syme CD, Rigby SEJ, Viles JH. Biochemistry. 2009;48:4388–4402. doi: 10.1021/bi900254n. [DOI] [PubMed] [Google Scholar]
- 30.Merrifield RB. J Am Chem Soc. 1963;85:2149–2154. [Google Scholar]
- 31.Fields GB, Nobel RL. Int J Pept Protein Res. 1990;35:161–214. doi: 10.1111/j.1399-3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]
- 32.Fauth JM, Schweiger A, Braunschweiler L, Forrer J, Ernst RR. J Magn Reson. 1986;66:74–85. [Google Scholar]
- 33.Gemperle C, Aebli G, Schweiger A, Ernst RR. J Magn Reson. 1990;88:241–256. [Google Scholar]
- 34.Lee HI, Doan PE, Hoffman BM. J Magn Reson. 1999;140:91–107. doi: 10.1006/jmre.1999.1803. [DOI] [PubMed] [Google Scholar]
- 35.Stoll S, Britt RD. Phys Chem Chem Phys. 2009;11:6614–6625. doi: 10.1039/b907277b. [DOI] [PubMed] [Google Scholar]
- 36.Peisach J, Blumberg WE. Arch Biochem Biophys. 1974;165:691–708. doi: 10.1016/0003-9861(74)90298-7. [DOI] [PubMed] [Google Scholar]
- 37.Santangelo M, Medina-Molner A, Schweiger A, Mitrikas G, Spingler B. J Biol Inorg Chem. 2007;12:767–775. doi: 10.1007/s00775-007-0230-1. [DOI] [PubMed] [Google Scholar]
- 38.Santangelo MG, Antoni PM, Spingler B, Jeschke G. ChemPhysChem. 2010;11:599–606. doi: 10.1002/cphc.200900672. [DOI] [PubMed] [Google Scholar]
- 39.Dikanov SA, Tsvetkov YD, Bowman MK, Astashkin AV. Chem Phys Lett. 1982;90:149–153. [Google Scholar]
- 40.McCracken J, Peisach J, Dooley DM. J Am Chem Soc. 1987;109:4064–4072. [Google Scholar]
- 41.McCracken J, Desai PR, Papadopoulos NJ, Villafranca JJ, Peisach J. Biochemistry. 1988;27:4133–4137. doi: 10.1021/bi00411a034. [DOI] [PubMed] [Google Scholar]
- 42.McCracken J, Pember S, Benkovic SJ, Villafranca JJ, Miller RJ, Peisach J. J Am Chem Soc. 1988;110:1069–1074. [Google Scholar]
- 43.Jiang F, McCracken J, Peisach J. J Am Chem Soc. 1990;112:9035–9044. [Google Scholar]
- 44.Magliozzo RS, Bubacco L, McCracken J, Jiang F, Beltramini M, Salvato B, Peisach J. Biochemistry. 1995;34:1513–1523. doi: 10.1021/bi00005a007. [DOI] [PubMed] [Google Scholar]
- 45.Mims WB. Phys Rev B. 1972;5:2409–2419. [Google Scholar]
- 46.Lai A, Flanagan HL, Singel DJ. J Chem Phys. 1988;89:7161–7166. [Google Scholar]
- 47.McCracken J, Peisach J, Cote CE, McGuirl MA, Dooley DM. J Am Chem Soc. 1992;114:3715–3720. [Google Scholar]
- 48.Tang XS, Diner BA, Larsen BS, Gilchrist ML, Lorigan GA, Britt RD. Proc Natl Acad Sci USA. 1994;91:704–708. doi: 10.1073/pnas.91.2.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh V, Zhu Z, Davidson VL, McCracken J. J Am Chem Soc. 2000;122:931–938. [Google Scholar]
- 50.Stoll S, Calle C, Mitrikas G, Schweiger A. J Magn Reson. 2005;177:93–101. doi: 10.1016/j.jmr.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 51.Mims WB, Peisach J. J Chem Phys. 1978;69:4921–4930. [Google Scholar]
- 52.Gerfen GJ, Singel DJ. J Chem Phys. 1994;100:4127–4137. [Google Scholar]
- 53.Hubrich M, Jeschke G, Schweiger A. J Chem Phys. 1996;104:2172–2184. [Google Scholar]
- 54.Jeschke G, Rakhmatullin R, Schweiger A. J Magn Reson. 1998;131:261–271. doi: 10.1006/jmre.1998.1367. [DOI] [PubMed] [Google Scholar]
- 55.Hong L, Carducci TM, Bush WD, Dudzik CG, Millhauser GL, Simon JD. J Phys Chem B. 2010;114:11261–11271. doi: 10.1021/jp103272v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma K, Clancy EL, Zhang Y, Ray DG, Wollenberg K, Zagorski MG. J Am Chem Soc. 1999;121:8698–8706. [Google Scholar]
- 57.Furlan S, La Penna G. Phys Chem Chem Phys. 2009;11:6468–6481. doi: 10.1039/b822771c. [DOI] [PubMed] [Google Scholar]
- 58.Furlan S, Hureau C, Faller P, La Penna G. J Phys Chem B. 2010;114:15119–15133. doi: 10.1021/jp102928h. [DOI] [PubMed] [Google Scholar]
- 59.Karr JW, Szalai VA. J Am Chem Soc. 2007;129:3796–3797. doi: 10.1021/ja068952d. [DOI] [PubMed] [Google Scholar]
- 60.Morgan DM, Dong J, Jacob J, Lu K, Apkarian RP, Thiyagarajan P, Lynn DG. J Am Chem Soc. 2002;124:12644–12645. doi: 10.1021/ja0273086. [DOI] [PubMed] [Google Scholar]
- 61.Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE, Smith SO. Nat Struct Mol Biol. 2010;17:561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dong J, Canfield JM, Mehta AK, Shokes JE, Tian B, Childers WS, Simmons JA, Mao Z, Scott RA, Warncke K, Lynn DG. Proc Natl Acad Sci USA. 2007;104:13313–13318. doi: 10.1073/pnas.0702669104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.