

Abstract
Haemophagocytic lymphohistiocytosis (HLH) is a life-threatening immunodeficiency characterized by severe systemic hyper-inflammatory responses to infectious or other triggers of the immune system. In many patients, the underlying cause of HLH is a genetic defect leading to defective CD8+ T-cell and natural killer cell granule-mediated cytotoxicity. The treatment of HLH consists principally of immune suppression followed by allogeneic haematopoietic cell transplantation (HCT) to cure the underlying defect and prevent relapse of HLH. Initial treatment regimens consist of steroids coupled with either etoposide or antithymocyte globulin, +/- cyclosporine. Complete responses are observed in only 50-75% of patients and even after a complete response, relapse and death still occur. The only definitive, long-term cure for patients with genetic forms of HLH is allogeneic HCT. Unfortunately, allogeneic HCT for patients with HLH is often complicated by critical illness, extensive organ involvement, active infections, or refractory HLH. For these reasons, patients are unusually prone to developing transplant-related toxicities and complications. In recent years, great strides have been made with regard to the care and transplantation of patients with HLH. Here we review the current state of the treatment of patients with HLH with allogeneic HCT, highlighting the important steps forward that have been made with reduced-intensity conditioning.
Keywords: transplantation, immunodeficiency, haemophagocytic syndrome, BMT
Introduction
Haemophagocytic lymphohistiocytosis (HLH) is a rare, life-threatening immunodeficiency characterized by severe systemic hyper-inflammatory responses to infectious or other (known or unknown) triggers to the immune system. In many patients, the underlying cause of HLH is a defect in one of several genes whose protein products participate in processes leading to effective CD8+ cytotoxic T-cell and NK cell granule-mediated cytotoxicity. Several of the genetic conditions that cause HLH are grouped together as Familial HLH (FHLH). These include mutations in PRF1 (Stepp, et al 1999), UNC13D (Feldmann, et al 2003), STX11 (zur Stadt, et al 2005) and STXBP2(Cote, et al 2009, zur Stadt et al, 2009). Abnormalities of XIAP (BIRC4) are also associated with FHLH phenotypes, though patients are more commonly classified as having the related disorder, X-linked lymphoproliferative disease (XLP) (Marsh, et al 2010a, Rigaud, et al 2006). XLP is classically caused by mutations in SH2D1A.(Coffey, et al 1998, Nichols, et al 1998, Sayos, et al 1998) Mutations in RAB27A(Menasche, et al 2000) and LYST(Barbosa, et al 1996, Nagle, et al 1996) also cause distinct genetic syndromes which prominently include HLH: Griscelli syndrome, type 2, and Chediak-Higashi syndrome, respectively.
Despite the growing number of genetic defects that are known to cause HLH, the underlying pathophysiology of HLH appears to be similar among most patients. All of the known genes, except for XIAP, have been shown to be essential for normal granule-mediated cytotoxicity. Because the genetic defects result in decreased or absent cytotoxicity, a viral or other antigenic trigger can lead to inappropriate hyper-inflammatory responses. These responses may be driven by heightened and protracted immune stimulation in the absence of normal antigen clearance, with resultant prolonged activation and hyperproliferation of T cells, hypercytokinaemia, and loss of the homeostatic measures that would normally ensure down-regulation of the immune response. The resulting inflammatory milieu results in host organ damage that is ultimately fatal in the absence of appropriate therapy.
The treatment of HLH consists principally of immune suppression, coupled with treatment of the underlying trigger if possible. Treatment regimens usually consist of steroids coupled with either etoposide or antithymocyte globulin (ATG).(Henter, et al 2002, Mahlaoui, et al 2007) Cyclosporine is also often used. Published reports indicate that complete responses are observed in only 50-75% of patients. However, even after a complete response, relapse and death may still occur. Currently, a trial evaluating the efficacy of a “hybrid” immunotherapy approach using dexamethasone, ATG, and etoposide is underway in North America (http://clinicaltrials.gov/ct2/show/NCT01104025). However, the only definitive long-term cure for patients with genetic forms of HLH remains allogeneic haematopoietic cell transplantation (HCT). The same holds true for cases that progress while on established therapies or experience relapse of HLH after initial remission.
Navigating a successful allogeneic HCT for patients with HLH is often complicated. Many patients are very ill prior to HCT due to extensive organ involvement with HLH. Many patients have one or more infections. Active HLH itself is associated with profound intrinsic depression of many innate and adaptive immune responses (Sumegi, et al 2011), which may be further crippled by the immune suppressants used for therapy of the disease. Additionally, patients may have frankly active or smoldering HLH at the time of transplantation. For these reasons, patients are unusually prone to developing transplant-related toxicities, infectious complications, and recurrent manifestations of HLH during the initial post-transplant period.
Despite these challenges, great strides have been made in the care and transplantation of patients with HLH. In order to summarize the experience with allogeneic HCT of patients with HLH, we performed a review of the literature using combinations of the terms haemophagocytic lymphohistiocytosis, erythrophagocytic lymphohistiocytosis, X-linked lymphoproliferative disease, haematopoietic cell transplantation, stem cell transplantation, bone marrow transplantation, reduced-intensity conditioning (RIC), Chediak-Higashi syndrome, Griscelli syndrome, alemtuzumab, and treatment. Here we review the current state of the treatment of patients with HLH with allogeneic HCT, highlighting the important steps forward that have been made with RIC.
The First Steps: Myeloablative Conditioning (MAC) Regimens for Allogeneic HCT
The first allogeneic HCT for HLH was described in 1986 using a matched sibling donor (Fischer, et al 1986), followed by several case reports and case series over the next 10 years confirming that allogeneic HCT was curative for HLH. In 1996, the outcomes of 122 patients included in an international registry were reported by (Arico, et al 1996). The estimated 5-year survival was 66% for patients undergoing HCT, as opposed to the estimated 5-year survival of 10% for patients not undergoing HCT. This report proved the need for HCT for long-term survival, but also revealed a high mortality rate in patients even with HCT, as it was performed in that era.
Following these registry findings, 4 additional small series were reported. The 3 largest series of patients during this time frame included 14 (Jabado, et al 1997), 20 (Baker, et al 1997) and 17 patients(Imashuku, et al 1999). The majority of patients received conditioning regimens consisting of busulfan and cyclophosphamide, with or without etoposide and ATG. In the first study (Jabado et al 1997) 64% of patients were surviving. Deaths in that series were related to progressive disease, hepatic veno-occlusive disease (VOD), fungal infection, and aplasia. The 3-year probability of survival in the largest series (Baker et al 1997) was also poor, at 45%, with deaths attributed to relapse of HLH, infection, pneumonia, adult respiratory distress syndrome, and multi-organ failure. Notably, only 17% of patients with active disease at the time of transplant survived in this series, and no patients with active central nervous system disease (CNS) disease at the time of transplant survived to 3 years. Two-year probability of survival was 54% for patients included in the series reported by Imashuku et al (1999), with 3/5 deaths related to disease relapse following HCT. In a single, contrasting, single-centre series of patients(Durken, et al 1999), a remarkable 100% survival was reported. However, it is notable that 5/12 patients developed hepatic VOD and that 3/12 patients developed pneumonia or sepsis requiring mechanical ventilation, though all patients recovered. Four of 12 patients experienced psychomotor retardation following HCT; 3 had CNS HLH prior to HCT. The collectively disappointing outcomes revealed in these studies were generally speculated to be related to a degree of active HLH at the time of transplantation and pre-existing organ damage (especially liver) in the setting of busulfan-based preparative regimens.
In 2002, the Histiocyte Society reported their results of the HLH-1994 study, the first large, collaborative effort to standardize the pre-HCT treatment of patients with HLH and prospectively monitor progress through HCT (Henter, et al 2002). The suggested HCT conditioning regimen consisted of busulfan, cyclophosphamide, and etoposide, with or without ATG, depending on donor relationship. Sixty-five patients underwent HCT, with a 62% 3-year probability of survival. Four of 25 deaths post-transplantation were related to reactivation of HLH, with the majority of remaining deaths related to unspecified HCT complications prior to day +100 (Table I).
Table I.
Outcomes of MAC HCT for HLH since 2002.
| Refernce | N | Predominant MAC Regimen | MRD/MUD | MMRD/MMUD | Haplo | Engrafted (%) | Mixed Chimerism (%) | Graft-Related Acute GVHD (I-IV or II-IV) | Recurrent Disease post-HCT (%) | VOD (%) | Death Prior to Day +100 | Predominant Causes of Death | Survival (years) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Henter et al (2002) | 65 | Bu, Cy, Etop, +/- ATG | 62% | 6% | 22% | 89% | 4% | Not Specified | 5% | Not Specified | 31% | BMT Complications, Not Otherwise Specified, HLH | 62% (3-year POS) |
| Horne et al (2005) | 86 | Bu, Cy, +/- Etop, +/- ATG | 66% | 15% | 19% | 90% | 19% | 32% | 8% | Not Specified | 27% | VOD, Pneumonia, BOOP, Respiratory Failure, CMV, GVHD, Septicemia, Interstitial Pneumonia with Adenovirus and CMV, HLH | 64% (3-year POS) |
| Ouachee-Chardin et al (2006) | 48 | Bu, Cy, +/- Etop, +/- ATG | 38% | 2% | 60% | 78% | 50% | 17% | Not Specified, at least 21% | 29% | Not Specified | HLH, Infections, Not Otherwise Specified | 59% (10-year POS) |
| Eapen et al (2007) | 35 (Chediak-Higashi) | Bu, Cy, +/- Etop, +/- ATG | 60% | 29% | 11% | 94% | 23% | 52% | 23% | Not Specified, at least 3% | 26% | HLH, ARDS, VOD, Infection, Pneumonitis, GVHD | 62% (5-year POS) |
| Baker et al (2008) | 91 | Bu, Cy, Etop, +/- ATG | 59% | 39% | 0% | 91% | 10% | 41% | Not Specified, at least 8% | 18% | 35% | Infections, Interstitial Pneumonitis, VOD, Organ Failure, GVHD, Haemorrhage, HLH | 45% (5-year POS) |
| Cesaro et al (2008) | 61 | Bu, Cy, +/- Etop or Mel, +/- ATG | Not Specified | Not Specified | Not Specified | 95% | 21% | 31% | Not Specified, at least 10% | Not Specified, at least 11% | 18% | VOD, Infection, HLH | 59% (8-year POS) |
| Pachlopnik Schmid et al (2009) | 10 (Griscelli type 2) | Bu, Cy, ATG | 50% | 30% | 20% | 90% | Not Specified, at least 10% | 70% | 10% | 50% | 30% (Prior to Day 110) | VOD, Acute Respiratory Distress Secondary to Capillary Leak Syndrome or Alveolar Haemorrhage, HLH | 70% (Various) |
| Al-Ahmari et al (2010) | 11 (Griscelli type 2) | Bu, Cy, +/- Etop, +/- ATG | 73% | 27% | 0% | 100% | 33% | 9% (Grades II-IV) | 0% | 9% | 0% | Sepsis | 91% (Various) |
| Yoon et al (2010) | 19 | Bu, Cy, Etop, +/- ATG | 74% | 26% | 0% | 84% | Not Specified | 26% | 5% | 11% | 26% | Infection, Pneumonitis, Pulmonary Haemorrhage, HLH | 73% (5-year POS) |
| Marsh et al (2010b) | 14 | Bu, Cy, +/- Etop, ATG | 64% | 36% | 0% | 100% | 18% | 14% | 7% | 0% | 29% | Pulmonary Haemorrhage, ARDS, Infection, Sepsis, Multi-Organ Failure, GVHD, Organ Failure | 43% (3-year POS) |
| Ohga et al (2010) | 43 (FHLH; 1 autologous) | Bu, Cy, Etop, +/- ATG | 65% | 28% | 5% | 83% | 19% | Not Specified | 5% | Not Specified | 17% | Transplant Related Mortality, Not Otherwise Specified, HLH | 65% (10-year POS) |
MAC Myeloablative conditioning
MRD Matched related donor
MUD Matched unrelated donor
MMRD Mismatched related donor
MMUD Mismatched unrelated donor
Haplo Haploidentical
GVHD Graft-versus-host disease
HLH Haemophagocytic lymphohistiocytosis
HCT Haematopoietic cell transplantation
VOD (hepatic) Veno-occlusive disease
BOOP Bronchiolitis obliterans organizing pneumonia
Bu Busulfan
Cy Cyclophosphamide
ATG Anti-thymocyte globulin
Etop Etoposide
FHLH Familial Haemophagocytic lymphohistiocytosis
ARDS Adult respiratory distress syndrome
POS Probability of survival
Following this, three moderate-sized studies in the late 2000s of similar conditioning regimens essentially supported the observation of an approximate 60% survival of patients with HLH undergoing allogeneic HCT. These studies (Horne, et al 2005 [n=86]; ,Ouachee-Chardin, et al 2006 [n=48]; Baker, et al 2008 [n=91]) observed 3-5 year probabilities of survival ranging from 49%-64% (Table I). Recent observations of predominantly Italian, Japanese, or Korean patients are similar (Cesaro, et al 2008, Ohga, et al 2010, Yoon, et al 2010), and experiences with Chediak-Higashi syndrome (Eapen, et al 2007) and Griscelli syndrome (Al-Ahmari, et al 2010, Pachlopnik Schmid, et al 2009) have also been reported (Table I). Deaths in most series were predominantly before day +100, and related to early transplant-related complications including non-engraftment, hepatic VOD, pulmonary toxicity, pneumonia, interstitial pneumonitis, bronchiolitis obilterans, idiopathic pneumonia syndrome, infections, organ failure, haemorrhage, and graft-versus-host disease (GVHD), or to HLH. Importantly, many studies noted an adverse effect of active disease at the time of HCT upon outcomes, supporting the initial presumptions that active HLH at HCT portends a worse prognosis.
When taken together, these studies reveal that myeloablative protocols consisting predominantly of busulfan, cyclophosphamide, with or without etoposide and/or ATG prevents HLH from recurring in 80-90% of patients (Table I). Mixed chimerism occurs in approximately 20% of patients, with one study that included a large number of haplo-identical donors observing a higher rate of 50% (Table I). Insufficient donor chimerism or graft rejection often precedes the development of HLH following HCT, although disease recurrence may take many years to manifest (Ardeshna, et al 2001). Overall, approximately one-quarter of patients with HLH died within 100 days following HCT with MAC, with an approximate 60% overall long-term probability of survival (Table I).
An Important Step Forward: RIC Regimens
Two patients with XLP were reported to undergo RIC HCT in 2000 and 2005, using fludarabine and melphalan with either alemtuzumab or ATG. Both patients developed mixed donor and recipient chimerism. Following HCT, one patient developed EBV viraemia, which cleared after receipt of a stem cell boost (Slatter, et al 2005). The second patient developed HLH and subsequently died of pseudomonas sepsis after starting treatment for HLH (Amrolia, et al 2000).
Shenoy at al (2005) published their results using a RIC regimen consisting of alemtuzumab, fludarabine, and melphalan for a variety of patients with non-malignant diseases, including 2 patients with HLH. One patient with HLH died at day +14 of drug-resistant pseudomonas sepsis.
A year later, another group described the outcomes of 12 patients with HLH who underwent HCT with RIC regimens (Cooper et al 2006; Table II). The predominant regimen used was an alemtuzumab, fludarabine, and melphalan regimen that was similar to that reported earlier (Shenoy et al 2005), with the exception of the timing and dose of alemtuzumab (Cooper, et al 2006). Patients with haploidentical donors received ATG instead of alemtuzumab, and additionally received busulfan. They observed a notable lack of VOD, and 75% of patients survived. Of the 9 survivors, 3 patients developed mixed donor/recipient chimerism, but maintained disease remission. Notably, one of these patients possessed donor chimerism in only the T-cell compartment. A follow-up report in 2008 of 25 patients with HLH or Langerhan’s Cell Histiocytosis (including the 12 patients reported earlier) by the same group described an 84% survival rate (Table II).(Cooper, et al 2008)
Table II.
Outcomes of RIC HCT for HLH.
| Reference | N | Predominant RIC Regimen | MRD/MUD | MMRD/MMUD | Haplo | Engrafted (%) | Mixed Chimerism (%) | Graft-Related Acute GVHD (I-IV or II-IV) | HLH post-HCT (%) | VOD (%) | Death Prior to Day +100 | Predominant Causes of Death | Survival (years) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cooper et al (2006) | 12 | Alem, Flu, Mel *Haplos received ATG instead of alem, and also busulfan | 50% | 25% | 25% | 100% | 33% | 33% | Not Specified | 0% | Not Specified | Pneumonitis | 75% (30-month Median Follow-Up) |
| Cooper et al (2008) | 25 | Alem, Flu, Mel *Haplos received ATG instead of alem, and also busulfan | 40% | 44% | 16% | 100% | 29% | Not Specified | 0% | 0% | Not Specified | Pneumonitis | 84% (3- year Median Follow-Up) |
| Marsh et al (2010b) | 26 | Alem, Flu, Mel | 73% | 27% | 0% | 100% | 65% | 8% | 4% | 0% | 0% | Infection, GVHD | 92% (3-year POS) |
RIC Reduced intensity conditioning
MRD Matched related donor
MUD Matched unrelated donor
MMRD Mismatched related donor
MMUD Mismatched unrelated donor
Haplo Haploidentical
GVHD Graft-versus-host disease
HLH Haemophagocytic lymphohistiocytosis
HCT Haematopoietic cell transplantation
VOD (hepatic) Veno-occlusive disease
Alem Alemtuzumab
Flu Fludarabine
Mel Melphalan
POS Probability of survival
While these results were promising, a direct comparison of RIC to MAC regimens was lacking. Ohga et al (2010) reviewed their experience with FHLH and also EBV-Associated HLH in Japan. No difference was noted between patients receiving MAC versus RIC, though RIC patients received fludarabine and melphalan-based regimens that included low-dose total body irradiation, and the total number of patients receiving RIC was small (N=11). A recent survey-based study of XLP compared 23 myeloablative transplants to 23 RIC transplants, and observed no difference in survival (83% versus 79%, respectively).(Booth, et al 2011)
Also in 2010, we reported our experience at Cincinnati Children’s Hospital with 26 HLH patients who underwent allogeneic HCT using alemtuzumab, fludarabine, and melphalan (Table II) (Marsh, et al 2010b). We noted a remarkable improvement in 3-year probability of survival of patients receiving RIC, 92%, as compared to MAC patients treated at our centre, 43%. The current long-term survival of patients is similar (Figure 1). There were no cases of VOD, and there were no deaths prior to day +100 in the RIC group. Furthermore, the 2 deaths in this group were not related to toxicity from the conditioning protocol.
Figure 1.
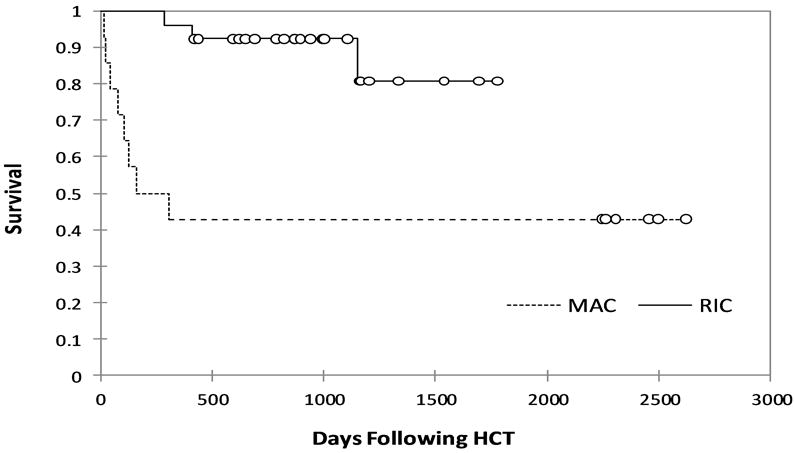
Updated long-term probability of survival (Kaplan-Meier Survival Curves generated with XLSTAT software) for 40 patients who underwent haematopoietic cell transplantation with a reduced-intensity preparative regimen (RIC) or a myeloablative preparative regimen (MAC) at Cincinnati Children’s Hospital between 2003-2009.(Marsh, et al 2010b)
Notably, there was a high incidence of mixed donor and recipient chimerism in the RIC group, occurring in 65% of patients, despite that the grafts were from fully matched or single allele mismatched donors. This may be due to the inclusion of alemtuzumab in the preparative regimen, as a significant effect of timing of alemtuzumab was noted upon the incidence of mixed chimerism. Many patients were treated with donor lymphocyte infusion(s) (DLI) and/or stem cell boost, to stabilize or increase the donor contribution to haematopoiesis. In this cohort, 13 patients received a first or only DLI at a median of 121 days following HCT (range 54-230). The median whole blood donor chimerism level at the time of DLI was 42% (range 9-69%). Nine patients received further DLI(s). Two patients additionally received a stem cell boost. These interventions resulted in stabilization or increase in the donor contribution to haematopoiesis in all but 1 patient who required a second HCT.
Despite the incidence of mixed chimerism and need for intervention in this group of patients, RIC HCT was successful in 96% of patients. One patient developed HLH as donor chimerism fell to less than 10% of whole blood samples. This patient underwent a second HCT, which was successful. Though no direct analysis was performed regarding the influence of active HLH at the time of transplantation, it is notable that, in the only RIC patient to require treatment of systemic HLH following transplantation and subsequent second HCT, HLH was very difficult to treat, with three reactivations prior to transplantation.
The Steps Ahead
Work thus far reveals that RIC HCT using alemtuzumab, fludarabine, and melphalan has dramatically improved patient survival at 2 centres in London and Cincinnati. However, clinical experience following RIC HCT remains relatively short, and further time is needed to confirm that patients will maintain sufficient donor contribution to haematopoiesis to sustain disease remission. Additionally, multi-centre trials are needed to confirm that the improved results observed with RIC HCT can be reproduced at other centres.
RIC HCT can be associated with a high incidence of mixed donor and recipient chimerism. This often complicates and lengthens the treatment of patients following HCT due to the need for aggressive monitoring of donor/recipient chimerism and interventions such as DLI or stem cell boost. RIC regimens could possibly be improved by optimizing the timing and dosing of alemtuzumab, such that an adequate amount is given to limit the incidence of acute GVHD, but not offer such profound graft T lymphocyte depletion as to contribute unnecessarily to the development of mixed chimerism. However, until RIC regimens are improved, DLI and stem cell boost may continue to be necessary for many patients.
Unfortunately, there is currently limited data upon which to base decisions regarding instigation of DLI or stem cell boost. Formal studies are needed to accurately define the risk of HLH with low-level donor chimerism and to determine which specific donor lymphocyte populations are required for permanent remission. HLH has been observed at whole blood donor chimerism levels of less than 20%,(Marsh, et al 2010b, Ouachee-Chardin, et al 2006) but it is notable that long-term remission has been observed in a patient with donor cells found only in the T cell compartment.(Cooper, et al 2006)
Decisions regarding DLI should currently be made on a case-by-case basis, based on patient chimerism trends, availability of DLI product, active infections, history of GVHD, and other factors. At our centre, DLIs are often pursued when donor contribution to overall haematopoiesis and/or T- and/or NK-cell chimerism is rapidly or persistently declining during the early post-transplant period towards thresholds that cause concern for eventual HLH relapse. In practical terms, patients with trends of donor contribution to haematopoiesis that are decreasing to levels lower than 40-60% within the first 6 months post-HCT are often considered for DLI. Consideration is at this point so as to avoid further decreases to levels below 20%. The onset and rate of decline of donor chimerism are also useful indices. Patients with earlier onset or more rapid decline of donor chimerism appear to be at higher risk of graft loss or HLH recurrence.
Despite the association of RIC HCT with mixed chimerism, it is the authors’ opinion that RIC HCT using alemtuzumab, fludarabine, and melphalan should be considered for all patients with HLH when an appropriate related or unrelated donor bone marrow or peripheral blood stem cell graft is available. Caution should be used when considering RIC for cord blood transplantation. Cord blood offers no source of additional donor stem cells or T cells after transplant, which prevents the possibility of stem cell boost or DLI should they be needed. The risks and benefits of RIC HCT versus traditional myeloablative HCT should be contemplated on a case-by-case basis for patients with cord blood as their only option for HCT.
In addition to the future improvements in the transplantation process for patients with HLH, ongoing efforts to improve the pre-transplant treatment of HLH will improve the outcomes of HCT. Patients with active HLH at the time of transplantation generally have worse outcomes compared to patients with inactive disease, (Baker, et al 2008, Horne, et al 2005, Ouachee-Chardin, et al 2006) and complete responses to conventional HLH therapies are only observed in 50%-75% of patients.(Henter, et al 2002, Mahlaoui, et al 2007) Formal trials of salvage therapies and standardization of second-line treatment schemas are needed, and will improve HCT outcomes. Additionally, further experience with post-HCT CNS HLH will improve transplant outcomes. In our experience, early post-transplant CNS disease is not uncommon. Children with low donor chimerism also appear to be at increased risk for the development of CNS disease. We have found that cerebrospinal fluid analysis and brain magnetic resonance imaging (MRI) can be helpful in the early post-transplant period, and that systemic dexamethasone and intrathecal treatment can be beneficial in affected patients. However, there is very little data regarding the incidence, course, treatment, or outcome of CNS HLH that occurs in the post-transplant period. Formal studies are needed.
In summary, we have reviewed the current experience with allogeneic HCT for patients with HLH. Although important steps forward have been made with regard to patient survival using RIC-HCT, many steps remain to optimize this approach and further improve patient outcomes.
Acknowledgments
The authors wish to thank the patients, physicians, nurses, and staff who have received and provided care at Cincinnati Children’s Hospital. We also wish to acknowledge all researchers and care providers whose efforts have contributed to the improved outcomes of patients with HLH. R.M. is supported by NIH R0AI079797 and by a grant from the Clinical Immunology Society. M.J. is supported by NIH R01HL091769.
Footnotes
Contributions
RAM performed the literature review and wrote the manuscript. MBJ and AHF contributed invaluable expertise and reviewed and edited the manuscript.
Conflict of Interest The authors declare that there are no conflicts of interest.
References
- Al-Ahmari A, Al-Ghonaium A, Al-Mansoori M, Hawwari A, Eldali A, Ayas M, Al-Mousa H, Al-Jefri A, Al-Saud B, Al-Seraihy A, Al-Muhsen S, Al-Mahr M, Al-Dhekri H, El-Solh H. Hematopoietic SCT in children with Griscelli syndrome: a single-center experience. Bone Marrow Transplant. 2010;45:1294–1299. doi: 10.1038/bmt.2009.358. [DOI] [PubMed] [Google Scholar]
- Amrolia P, Gaspar HB, Hassan A, Webb D, Jones A, Sturt N, Mieli-Vergani G, Pagliuca A, Mufti G, Hadzic N, Davies G, Veys P. Nonmyeloablative stem cell transplantation for congenital immunodeficiencies. Blood. 2000;96:1239–1246. [PubMed] [Google Scholar]
- Ardeshna KM, Hollifield J, Chessells JM, Veys P, Webb DK. Outcome for children after failed transplant for primary haemophagocytic lymphohistiocytosis. Br J Haematol. 2001;115:949–952. doi: 10.1046/j.1365-2141.2001.03177.x. [DOI] [PubMed] [Google Scholar]
- Arico M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, Martinetti M, Rusca MP. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996;10:197–203. [PubMed] [Google Scholar]
- Baker KS, DeLaat CA, Steinbuch M, Gross TG, Shapiro RS, Loechelt B, Harris R, Filipovich AH. Successful correction of hemophagocytic lymphohistiocytosis with related or unrelated bone marrow transplantation. Blood. 1997;89:3857–3863. [PubMed] [Google Scholar]
- Baker KS, Filipovich AH, Gross TG, Grossman WJ, Hale GA, Hayashi RJ, Kamani NR, Kurian S, Kapoor N, Ringden O, Eapen M. Unrelated donor hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis. Bone Marrow Transplant. 2008;42:175–180. doi: 10.1038/bmt.2008.133. [DOI] [PubMed] [Google Scholar]
- Barbosa MD, Nguyen QA, Tchernev VT, Ashley JA, Detter JC, Blaydes SM, Brandt SJ, Chotai D, Hodgman C, Solari RC, Lovett M, Kingsmore SF. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature. 1996;382:262–265. doi: 10.1038/382262a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth C, Gilmour KC, Veys P, Gennery AR, Slatter MA, Chapel H, Heath PT, Steward CG, Smith O, O’Meara A, Kerrigan H, Mahlaoui N, Cavazzana-Calvo M, Fischer A, Moshous D, Blanche S, Pachlopnick-Schmid J, Latour S, de Saint-Basile G, Albert M, Notheis G, Rieber N, Strahm B, Ritterbusch H, Lankester A, Hartwig NG, Meyts I, Plebani A, Soresina A, Finocchi A, Pignata C, Cirillo E, Bonanomi S, Peters C, Kalwak K, Pasic S, Sedlacek P, Jazbec J, Kanegane H, Nichols KE, Hanson IC, Kapoor N, Haddad E, Cowan M, Choo S, Smart J, Arkwright PD, Gaspar HB. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood. 2011;117:53–62. doi: 10.1182/blood-2010-06-284935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesaro S, Locatelli F, Lanino E, Porta F, Di Maio L, Messina C, Prete A, Ripaldi M, Maximova N, Giorgiani G, Rondelli R, Arico M, Fagioli F. Hematopoietic stem cell transplantation for hemophagocytic lymphohistiocytosis: a retrospective analysis of data from the Italian Association of Pediatric Hematology Oncology (AIEOP) Haematologica. 2008;93:1694–1701. doi: 10.3324/haematol.13142. [DOI] [PubMed] [Google Scholar]
- Coffey AJ, Brooksbank RA, Brandau O, Oohashi T, Howell GR, Bye JM, Cahn AP, Durham J, Heath P, Wray P, Pavitt R, Wilkinson J, Leversha M, Huckle E, Shaw-Smith CJ, Dunham A, Rhodes S, Schuster V, Porta G, Yin L, Serafini P, Sylla B, Zollo M, Franco B, Bolino A, Seri M, Lanyi A, Davis JR, Webster D, Harris A, Lenoir G, de St Basile G, Jones A, Behloradsky BH, Achatz H, Murken J, Fassler R, Sumegi J, Romeo G, Vaudin M, Ross MT, Meindl A, Bentley DR. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet. 1998;20:129–135. doi: 10.1038/2424. [DOI] [PubMed] [Google Scholar]
- Cooper N, Rao K, Gilmour K, Hadad L, Adams S, Cale C, Davies G, Webb D, Veys P, Amrolia P. Stem cell transplantation with reduced-intensity conditioning for hemophagocytic lymphohistiocytosis. Blood. 2006;107:1233–1236. doi: 10.1182/blood-2005-05-1819. [DOI] [PubMed] [Google Scholar]
- Cooper N, Rao K, Goulden N, Webb D, Amrolia P, Veys P. The use of reduced-intensity stem cell transplantation in haemophagocytic lymphohistiocytosis and Langerhans cell histiocytosis. Bone Marrow Transplant. 2008;42(Suppl 2):S47–50. doi: 10.1038/bmt.2008.283. [DOI] [PubMed] [Google Scholar]
- Cote M, Menager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, Al-Manjomi F, Al-Harbi M, Alangari A, Le Deist F, Gennery AR, Prince N, Cariou A, Nitschke P, Blank U, El-Ghazali G, Menasche G, Latour S, Fischer A, de Saint Basile G. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. 2009;119:3765–3773. doi: 10.1172/JCI40732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durken M, Horstmann M, Bieling P, Erttmann R, Kabisch H, Loliger C, Schneider EM, Hellwege HH, Kruger W, Kroger N, Zander AR, Janka GE. Improved outcome in haemophagocytic lymphohistiocytosis after bone marrow transplantation from related and unrelated donors: a single-centre experience of 12 patients. Br J Haematol. 1999;106:1052–1058. doi: 10.1046/j.1365-2141.1999.01625.x. [DOI] [PubMed] [Google Scholar]
- Eapen M, DeLaat CA, Baker KS, Cairo MS, Cowan MJ, Kurtzberg J, Steward CG, Veys PA, Filipovich AH. Hematopoietic cell transplantation for Chediak-Higashi syndrome. Bone Marrow Transplant. 2007;39:411–415. doi: 10.1038/sj.bmt.1705600. [DOI] [PubMed] [Google Scholar]
- Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, Lambert N, Ouachee-Chardin M, Chedeville G, Tamary H, Minard-Colin V, Vilmer E, Blanche S, Le Deist F, Fischer A, de Saint Basile G. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3) Cell. 2003;115:461–473. doi: 10.1016/s0092-8674(03)00855-9. [DOI] [PubMed] [Google Scholar]
- Fischer A, Cerf-Bensussan N, Blanche S, Le Deist F, Bremard-Oury C, Leverger G, Schaison G, Durandy A, Griscelli C. Allogeneic bone marrow transplantation for erythrophagocytic lymphohistiocytosis. J Pediatr. 1986;108:267–270. doi: 10.1016/s0022-3476(86)81002-2. [DOI] [PubMed] [Google Scholar]
- Henter JI, Samuelsson-Horne A, Arico M, Egeler RM, Elinder G, Filipovich AH, Gadner H, Imashuku S, Komp D, Ladisch S, Webb D, Janka G. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100:2367–2373. doi: 10.1182/blood-2002-01-0172. [DOI] [PubMed] [Google Scholar]
- Horne A, Janka G, Maarten Egeler R, Gadner H, Imashuku S, Ladisch S, Locatelli F, Montgomery SM, Webb D, Winiarski J, Filipovich AH, Henter JI. Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol. 2005;129:622–630. doi: 10.1111/j.1365-2141.2005.05501.x. [DOI] [PubMed] [Google Scholar]
- Imashuku S, Hibi S, Todo S, Sako M, Inoue M, Kawa K, Koike K, Iwai A, Tsuchiya S, Akiyama Y, Kotani T, Kawamura Y, Hirosawa M, Hasegawa D, Kosaka Y, Yamaguchi H, Ishii E, Kato K, Ishii M, Kigasawa H. Allogeneic hematopoietic stem cell transplantation for patients with hemophagocytic syndrome (HPS) in Japan. Bone Marrow Transplant. 1999;23:569–572. doi: 10.1038/sj.bmt.1701620. [DOI] [PubMed] [Google Scholar]
- Jabado N, de Graeff-Meeder ER, Cavazzana-Calvo M, Haddad E, Le Deist F, Benkerrou M, Dufourcq R, Caillat S, Blanche S, Fischer A. Treatment of familial hemophagocytic lymphohistiocytosis with bone marrow transplantation from HLA genetically nonidentical donors. Blood. 1997;90:4743–4748. [PubMed] [Google Scholar]
- Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, Neven B, Picard C, Blanche S, Fischer A. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. 2007;120:e622–628. doi: 10.1542/peds.2006-3164. [DOI] [PubMed] [Google Scholar]
- Marsh RA, Madden L, Kitchen BJ, Mody R, McClimon B, Jordan MB, Bleesing JJ, Zhang K, Filipovich AH. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood. 2010a;116:1079–1082. doi: 10.1182/blood-2010-01-256099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh RA, Vaughn G, Kim MO, Li D, Jodele S, Joshi S, Mehta PA, Davies SM, Jordan MB, Bleesing JJ, Filipovich AH. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood. 2010b;116:5824–5831. doi: 10.1182/blood-2010-04-282392. [DOI] [PubMed] [Google Scholar]
- Menasche G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, Wulffraat N, Bianchi D, Fischer A, Le Deist F, de Saint Basile G. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000;25:173–176. doi: 10.1038/76024. [DOI] [PubMed] [Google Scholar]
- Nagle DL, Karim MA, Woolf EA, Holmgren L, Bork P, Misumi DJ, McGrail SH, Dussault BJ, Jr, Perou CM, Boissy RE, Duyk GM, Spritz RA, Moore KJ. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet. 1996;14:307–311. doi: 10.1038/ng1196-307. [DOI] [PubMed] [Google Scholar]
- Nichols KE, Harkin DP, Levitz S, Krainer M, Kolquist KA, Genovese C, Bernard A, Ferguson M, Zuo L, Snyder E, Buckler AJ, Wise C, Ashley J, Lovett M, Valentine MB, Look AT, Gerald W, Housman DE, Haber DA. Inactivating mutations in an SH2 domain-encoding gene in X-linked lymphoproliferative syndrome. Proc Natl Acad Sci U S A. 1998;95:13765–13770. doi: 10.1073/pnas.95.23.13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohga S, Kudo K, Ishii E, Honjo S, Morimoto A, Osugi Y, Sawada A, Inoue M, Tabuchi K, Suzuki N, Ishida Y, Imashuku S, Kato S, Hara T. Hematopoietic stem cell transplantation for familial hemophagocytic lymphohistiocytosis and Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in Japan. Pediatr Blood Cancer. 2010;54:299–306. doi: 10.1002/pbc.22310. [DOI] [PubMed] [Google Scholar]
- Ouachee-Chardin M, Elie C, de Saint Basile G, Le Deist F, Mahlaoui N, Picard C, Neven B, Casanova JL, Tardieu M, Cavazzana-Calvo M, Blanche S, Fischer A. Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis: a single-center report of 48 patients. Pediatrics. 2006;117:e743–750. doi: 10.1542/peds.2005-1789. [DOI] [PubMed] [Google Scholar]
- Pachlopnik Schmid J, Moshous D, Boddaert N, Neven B, Dal Cortivo L, Tardieu M, Cavazzana-Calvo M, Blanche S, de Saint Basile G, Fischer A. Hematopoietic stem cell transplantation in Griscelli syndrome type 2: a single-center report on 10 patients. Blood. 2009;114:211–218. doi: 10.1182/blood-2009-02-207845. [DOI] [PubMed] [Google Scholar]
- Rigaud S, Fondaneche MC, Lambert N, Pasquier B, Mateo V, Soulas P, Galicier L, Le Deist F, Rieux-Laucat F, Revy P, Fischer A, de Saint Basile G, Latour S. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444:110–114. doi: 10.1038/nature05257. [DOI] [PubMed] [Google Scholar]
- Sayos J, Wu C, Morra M, Wang N, Zhang X, Allen D, van Schaik S, Notarangelo L, Geha R, Roncarolo MG, Oettgen H, De Vries JE, Aversa G, Terhorst C. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature. 1998;395:462–469. doi: 10.1038/26683. [DOI] [PubMed] [Google Scholar]
- Shenoy S, Grossman WJ, DiPersio J, Yu LC, Wilson D, Barnes YJ, Mohanakumar T, Rao A, Hayashi RJ. A novel reduced-intensity stem cell transplant regimen for nonmalignant disorders. Bone Marrow Transplant. 2005;35:345–352. doi: 10.1038/sj.bmt.1704795. [DOI] [PubMed] [Google Scholar]
- Slatter MA, Bhattacharya A, Abinun M, Flood TJ, Cant AJ, Gennery AR. Outcome of boost haemopoietic stem cell transplant for decreased donor chimerism or graft dysfunction in primary immunodeficiency. Bone Marrow Transplant. 2005;35:683–689. doi: 10.1038/sj.bmt.1704872. [DOI] [PubMed] [Google Scholar]
- Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, Henter JI, Bennett M, Fischer A, de Saint Basile G, Kumar V. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957–1959. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]
- Sumegi J, Barnes MG, Nestheide SV, Molleran-Lee S, Villanueva J, Zhang K, Risma KA, Grom AA, Filipovich AH. Gene expression profiling of peripheral blood mononuclear cells from children with active hemophagocytic lymphohistiocytosis. Blood. 2011;117:e151–e160. doi: 10.1182/blood-2010-08-300046. published ahead of print February 16, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HS, Im HJ, Moon HN, Lee JH, Kim HJ, Yoo KH, Sung KW, Koo HH, Kang HJ, Shin HY, Ahn HS, Cho B, Kim HK, Lyu CJ, Lee MJ, Kook H, Hwang TJ, Seo JJ. The outcome of hematopoietic stem cell transplantation in Korean children with hemophagocytic lymphohistiocytosis. Pediatr Transplant. 2010;14:735–740. doi: 10.1111/j.1399-3046.2009.01284.x. [DOI] [PubMed] [Google Scholar]
- zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter JI, Kabisch H, Schneppenheim R, Nurnberg P, Janka G, Hennies HC. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. 2005;14:827–834. doi: 10.1093/hmg/ddi076. [DOI] [PubMed] [Google Scholar]
- zur Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J, Strauss J, Kasper B, Nürnberg G, Becker C, Maul-Pavicic A, Beutel K, Janka G, Griffiths G, Ehl S, Hennies HC. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. American Journal of Human Genetics. 2009;85:482–92. doi: 10.1016/j.ajhg.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]