

Abstract
Metabolomics involves the unbiased quantitative and qualitative analysis of the complete set of metabolites present in cells, body fluids and tissues (the metabolome). By analyzing differences between metabolomes using biostatistics (multivariate data analysis; pattern recognition), metabolites relevant to a specific phenotypic characteristic can be identified. However, the reliability of the analytical data is a prerequisite for correct biological interpretation in metabolomics analysis. In this review the challenges in quantitative metabolomics analysis with regards to analytical as well as data preprocessing steps are discussed. Recommendations are given on how to optimize and validate comprehensive silylation-based methods from sample extraction and derivatization up to data preprocessing and how to perform quality control during metabolomics studies. The current state of method validation and data preprocessing methods used in published literature are discussed and a perspective on the future research necessary to obtain accurate quantitative data from comprehensive GC-MS data is provided.
Keywords: Quantitative metabolomics, Method validation, Data preprocessing, Quality control, Gas chromatography mass spectrometry
Introduction
Functional genomics technologies (transcriptomics, preoteomics, metabolomics) are increasingly important in the fields of microbiology, plant and medical sciences, and are increasingly used in a systems biology approach. Metabolomics evolved from conventional profiling techniques and the view to study organisms or biological systems as integrated and interacting systems of genes, proteins, metabolites, cellular and pathway events, the so-called systems biology approach (van Greef et al. 2004a). Metabolomics involves the unbiased quantitative and qualitative analysis of the complete set of metabolites present in cells, body fluids and tissues (the metabolome). Biostatistics (multivariate data analysis; pattern recognition) plays an essential role in analyzing differences between metabolomes, enabling the identification of metabolites relevant to a specific phenotypic characteristic.
In analogy with other functional genomics techniques, a comprehensive, generally non-targeted approach is used to gain new insights and a better understanding of the biological functioning of a cell or organism. To answer biological questions, it is crucial that all steps from the clear definition of the biological questions, the choice of a suitable experimental design, the proper sampling procedure, sample preparation, data acquisition and data processing are addressed to obtain quantitative data, which can be then used for data analysis and final biological interpretation (Fig. 1). Obviously, optimization, validation and proper quality control of analytical methods is of key importance.
Fig. 1.
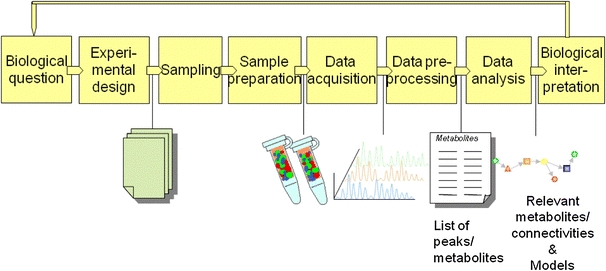
Schematic of a typical workflow in metabolomics
Strategies in metabolomics related research
In present-day research several different analytical strategies are applied for the analysis of a wide range of metabolites, i.e. metabolic target analysis, metabolic profiling, metabolic fingerprinting, metabonomics and metabolomics (Table 1). Depending on the biological question, different analytical approaches are required and different demands are posed on analytical performance (detection limits, precision, accuracy, etc.).
Table 1.
Analytical strategies for metabolic research
| Metabolite target analysis | Quantitative (absolute or relative) analysis of one or few target metabolites. Typical strategy: selective sample pretreatment followed by separation (GC, LC, CE) coupled to sensitive selective detection |
| Metabolic profiling | Quantitative (absolute or relative) and qualitative (identification) multi-component analyses that define or describe metabolic patterns for a group of metabolically or analytically related metabolites (Horning and Horning 1971). Typical strategy: sample pretreatment selective for compound class or compounds from certain pathway followed by separation coupled to MS detection |
| Metabolic fingerprinting | High throughput screening of samples to provide sample classification. Generally no quantification (or only relative quantification) and no identification of individual metabolites (Dunn and Ellis 2005; Fiehn 2002) Typical strategy: Simple sample pretreatment followed by NMR, FTIR, or direct infusion mass spectrometry (DIMS). |
| Metabolomics (Metabonomics) | Quantitative (mostly relative quantification) and qualitative analysis of the complete set of metabolites present in a biological system (cells, body fluids, tissues). Typical strategy: generic sample pretreatment followed by separation coupled to MS detection |
The terminology in metabolic research is still not standardized and different definitions of the terms are proposed in different papers. Metabolic target analysis and metabolic profiling are commonly used strategies in classically hypothesis-driven metabolic research, where the interest is focused on a limited number of metabolites, or a certain compound class or metabolic pathway. Due to the selective sample pretreatment and/or sample cleanup used in this approach, low detection limits and high precision and accuracy can be achieved. For rapid screening and classification of samples identification and quantification of each individual metabolite is not always necessary and metabolic fingerprinting approaches are commonly applied. Metabolomics is the comprehensive non-target analysis of all (or at least as many as possible) metabolites in a biological system. Ultimately, metabolomics analysis would provide information on the absolute concentrations of all extractable metabolites in a sample (absolute quantification), as this would help to make data comparable. However, due to practical limitations, e.g. the absence of standard reference materials for metabolomics analysis, metabolites are mostly quantified using relative quantification, i.e. determining the response ratio between the metabolite and an internal standard or other metabolite. In addition, unidentified metabolites present in a sample can also be quantified using relative quantification. Metabonomics is sometime distinguished as a separate approach in metabolomic research. Metabonomics is defined as the quantitative measurement of the dynamic multiparametric metabolic response of living systems to pathophysiological stimuli or genetic modification (Nicholson et al. 1999; Nicholson and Lindon 2008) In practice, the terms metabolomics and metabonomics are often used interchangeably, and the analytical and modeling procedures are the same. Throughout this review the terminology and definitions as described in Table 1 are used.
Analytical techniques in metabolomics research
Development of generic methodologies to analyze the complete metabolome, or at least as many metabolites as possible, is very challenging considering the complexity of the metabolome. The extent of the full metabolome is dependent on the organism studied, varying from a few hundred endogenous metabolites for microorganisms (Forster et al. 2003; Hall et al. 2002) to a few thousands endogenous human metabolites (without taking lipids into account). For lipids tens of thousands different metabolites might be expected, but a definite estimation is not possible at the moment. In addition, more than 100,000 small molecules can be expected to be present in humans due to the consumption of food, drugs, etc. (Wishart et al. 2007). Moreover, metabolites consist of a wide variety of compound classes with different physical and chemical properties and are present in a large range of concentrations. For example, in human-blood-plasma samples, normal glucose concentrations are as high as 5000 μM (925 mg/l), while estradiol has a concentration of approximately 0.00009 μM (24 ng/l) (Wishart et al. 2009), covering a range of more than seven decades.
Currently, the main analytical techniques used for the analysis of the metabolome are nuclear magnetic resonance spectroscopy (NMR) and hyphenated techniques such as gas chromatography (GC) and liquid chromatography (LC) coupled to mass spectrometry (MS). In addition, other combinations are possible, e.g. capillary electrophoresis (CE) coupled to MS or LC coupled to electrochemical detection. Alternatively, Fourier transform infrared spectroscopy and direct infusion mass spectrometry (DIMS) have been applied (Dunn and Ellis 2005; van Greef et al. 2004a; Greef and Smilde 2005; Lindon et al. 2007) without any prior separation, except for eventual sample preparation. NMR, FTIR and DIMS are high throughput methods and require minimal sample preparation and may be preferred techniques for metabolic fingerprinting. However, the obtained spectra are composed of the signals of very many metabolites and elucidation of these complex spectra can be very complicated. In addition, detection limits for NMR and FTIR are much higher than for MS-based techniques, limiting the application range to metabolites present in higher concentrations. Therefore hyphenated techniques, e.g. GC-MS, LC-MS and CE-MS, are generally preferred in metabolomics to allow quantification and identification of as many as possible (individual) metabolites. However none of the individual methods will cover the full metabolome and a combination of techniques is necessary to ultimately measure the full metabolome.
Comprehensive analysis with GC-MS
GC-MS is a very suitable technique for comprehensive analysis, as it combines a high separation efficiency with versatile, selective and sensitive mass detection. In Table 2 an overview of GC-based applications in metabolomics research is presented from different fields of research, such as microbiology, plant- and medical science (pharmacology, clinical research). Only papers targeting more than three different metabolite classes were included in the table. Nearly all GC-based metabolomics applications combine GC with MS detection using electron ionization (EI). As the full scan response in EI mode is approximately proportional to the amount of compound injected, i.e. more or less independently of the compound, all compounds suitable for GC analysis are detected non-discriminatively. Furthermore, problems with ion suppression of co-eluting compounds as observed in LC-MS are virtually absent in GC-EI-MS. Also, the assignment of the identity of peaks via a database of mass spectra is straightforward, due to the extensive and reproducible fragmentation patterns obtained in full-scan mode. In addition, the fragmentation pattern can be used to identify or classify unknown metabolites.
Table 2.
Overview of GC(-MS) based metabolomics papers
| Authors | Technique | Focusb | Matrix | Validation parametersc |
|---|---|---|---|---|
| Aura et al. (2008) | S-GC-MS & S-GC×GC-MS | 7 | feaces | – |
| Birkemeyer et al. (2005) | GC-MS | 4 | microbial | – |
| Chang et al. (2006) | OS-GC-MS | 7 | plant | – |
| Coucheney et al. (2008) | OS-GC-MS | 3, 6, 7 | microbial | 4 |
| De Souza et al. (2006) | OS-GC-MS | 6 | microbial | – |
| Fan et al. (1993) | S-GC-MS | 7 | plant | 3, 4 |
| Fan et al. (2001) | GC-MS | 7 | plant | – |
| Fiehn et al. (2000b) | OS-GC-MS | 5 | plant | – |
| Fiehn et al. (2000a) | OS-GC-MS | 1, 2, 3, 4, 6 | plant | 2, 4 |
| Fiehn (2003) | OS-GC-MS | 6 | plant | – |
| Fiehn et al. (2008) | OS-GC-MS | 3, 4, 6, 7 | plant | – |
| Gullberg et al. (2004) | OS-GC-MS | 1 | plant | 4 |
| Guo and Lidstrom (2008) | OS-GC × GC-MS | 5, 6 | microbial | 2 |
| Hiller et al. (2009) | OS-GC-MS | 6 | microbial | 2a |
| Hope et al. (2005a) | S-GC × GC-MS | 7 | plant | – |
| Huang and Regnier (2008) | OS-GC × GC-MS | 4 | serum | – |
| Humston et al. (2010) | SPME(HS)- GC × GC-MS | 6, 7 | plant | – |
| Humston et al. (2008) | OS-GC × GC-MS | 6, 7 | microbial | – |
| Jeong et al. (2004) | OS-GC-MS | 7 | plant | – |
| Jiye et al. (2008) | OS-GC-MS | 6, 7 | urine | – |
| Jonsson et al. (2004) | OS-GC-MS | 6 | plant | – |
| Jonsson et al. (2005) | OS-GC-MS | 6 | plant | – |
| Jonsson et al. (2006) | OS-GC-MS | 6 | urine | – |
| Koek et al. (2006) | OS-GC-MS | 1, 2, 3, 4 | microbial | 2, 3, 4, 5, 6, 7 |
| Koek et al. (2008) | OS-GC × GC-MS | 1, 2, 3, 4 | serum/plasma | 1, 2, 3, 4, 6 |
| Koek et al. (2010a) | S-GC-MS | 2, 3, 7 | mouse CSF | 2, 3, 4, 5 |
| Koek et al. (2010b) | OS-GC × GC-MS | 6, 7 | liver | 4 |
| Kuhara (2001) | OS-GC-MS | 7 | urine | – |
| Kusano et al. (2007) | OS-GC × GC-MS | 6 | plant | 4 |
| Lee and Fiehn (2008) | OS-GC-MS | 1, 7 | microbial | – |
| Li et al. (2009) | S-GC × GC-MS | 6 | plasma | 4 |
| Lu et al. (2008) | OS-GC-MS | 6, 7 | plasma | 4 |
| Ma et al. (2008) | S-GC-MS | 2, 6 | plant | – |
| Martins et al. (2004) | OS-GC-MS | 2, 3 | microbial | 4 |
| Matsumoto and Kuhara (1996) | S-GC-MS | 7 | urine | – |
| Mills and Walker (2001) | SPME(HS)-GC-MS | 7 | urine | – |
| Mohler et al. (2007) | OS- GC × GC-MS | 6, 7 | yeast | – |
| Mohler et al. (2006) | OS-GC × GC-MS | 1, 2, 3, 6 | yeast | 4 |
| Mohler et al. (2008) | OS-GC × GC-MS | 6 | microbial | – |
| Morgenthal et al. (2005) | OS-GC-MS | 1, 2, 3, 6 | plant | 4 |
| O’Hagan et al. (2005) | OS-GC-MS | 3, 4 | serum/yeast | 4 |
| O’Hagan et al. (2007) | OS-GC × GC-MS | 3 | serum | – |
| Oh et al. (2008) | S-GC × GC-MS | 6 | serum | – |
| Ong et al. (2009) | S-GC-MS | 7 | liver | 4 |
| Pan et al. (2010) | OS-GC-MS | 1 | liver | 2, 3, 4, 6, 7 |
| Pasikanti et al. (2008) | S-GC-MS | 4, 6 | urine | 2a, 4, 5, 7 |
| Pauling et al. (1971) | HS-GC-FID | 3, 4 | urine, breath | 4 |
| Pierce et al. (2006b) | S-GC × GC-MS | 6 | plant | – |
| Pierce et al. (2006a) | S-GC × GC-MS | 6 | urine | – |
| Qiu et al. (2007) | ECF-GC-MS | 1, 2, 3, 4 | urine | 2, 3, 4, 6 |
| Ralston-Hooper et al. (2008) | OS-GC × GC-MS | 7 | invertebrates | – |
| Roessner et al. (2000) | OS-GC-MS | 1, 2, 3, 4 | plant | 2, 3, 4 |
| Roessner et al. (2001a) | OS-GC-MS | 6 | plant | – |
| Roessner et al. (2001b) | OS-GC-MS | 6 | plant | 4 |
| Schauer et al. (2005) | OS-GC-MS | 5 | all | – |
| Sangster et al. (2006) | OS-GC-MS | 6 | plasma | – |
| Schmarr and Bernhardt (2010) | SPME(HS)- GC × GC-MS | 6 | plant | – |
| Shellie et al. (2005) | OS-GC × GC-MS | 6 | mouse spleen | – |
| Sinha et al. (2004b) | OS-GC × GC-MS | 6 | urine | – |
| Strelkov et al. (2004) | OS-GC-MS (polar) + S-GC-MS (apolar metabolites) | 1, 2, 3, 4 | microbial | 4 |
| Styczynski et al. (2007) | MCF-GC-MS | 6 | microbial | – |
| Tian et al. (2008) | S-GC-FID/MS | 3, 6 | microbial | 2a, 3a, 4a, 6a |
| Tianniam et al. (2008) | OS-GC-MS | 7 | plant | – |
| Vikram et al. (2004) | HS-GC-MS | 7 | apples | – |
| Villas-Bôas et al. (2003) | MCF-GC-MS | 2, 3 | microbial | 2, 4, 6 |
| Villas-Bôas et al. (2005) | OS-GC-MS/MCF-GC-MS | 1 | yeast | 3 |
| Wagner et al. (2003) | OS-GC-MS | 5, 6 | plant | – |
| Weckwerth et al. (2004a) | OS-GC-MS | 4, 6 | plant | – |
| Weckwerth et al. (2004b) | OS-GC-MS | 1, 2, 3, 4, 6 | plant | 3, 4 |
| Welthagen et al. (2005) | OS-GC × GC-MS | 3, 6 | mouse spleen | 4 |
| Wishart et al. (2008) | OS-GC-MS | 7 | CSF | – |
| Zhang et al. (2007) | OS-GC-MS | 3, 4 | urine | 2, 4, 5, 6, 7 |
O oximation, S silylation, CF chloroformate derivatization, HS headspace sampling, SPME solid phase micro extraction
aValidation parameter only assessed in academic standard, i.e. standard without matrix
bFocus: 1 = extraction, 2 = derivatization, 3 = analysis, 4 = detection/quantification, 5 = identification, 6 = data preprocessing and analysis, 7 = application
cAnalytical validation parameters: 1 = selectivity (peak capacity), 2 = calibration model, 3 = accuracy (recovery), 4 = repeatability, 5 = intermediate precision, 6 = LLOQ/LLOD, 7 = stability
Volatile, low-molecular-weight metabolites can be sampled and analyzed directly, e.g. in breath analysis often a direct approach without derivatization is used (Pauling et al. 1971). However, many metabolites contain polar functional groups and are thermally labile at the temperatures required for their separation or are not volatile at all. Therefore, derivatization prior to GC analysis is needed to extend the application range of GC based methods. The majority of GC methods reviewed (Table 2) rely on derivatization with an oximation reagent followed by silylation, or solely silylation. As silylation reagents are the most versatile and universally applicable derivatization reagents, these are most suitable for comprehensive GC(-MS) analysis. Only few authors used an alternative derivatization, e.g. chloroformates (Qiu et al. 2007) or no derivatization at all. Therefore this review focuses on GC-MS methods using oximation and subsequently silylation or solely silylation prior to analysis.
Obtaining quantitative data
Ultimately the goal in metabolomics analysis is to identify and quantify all metabolites in order to find answers to biological questions. This review focuses on how quantitative data can be obtained from silylation-based GC-MS methods, thereby covering sample preparation, data acquisition and data processing. The challenges in comprehensive GC-MS based metabolomics analysis are discussed and recommendations on method development, data processing, method validation and quality control during studies are given. Validation and data-processing strategies applied in published comprehensive GC-based metabolomics methods are evaluated. Moreover, a perspective on the future research necessary to obtain accurate quantitative data from comprehensive GC-MS data is provided.
Recommendations on method development, data processing, validation and quality control
The reliability and suitability of sample preparation, data acquisition, data preprocessing and data analysis are prerequisites for correct biological interpretation in metabolomics studies. The significance of differences between samples can only be determined when the performance characteristics of the entire method (from sample preparation to data preprocessing) are known. Therefore it is important to perform method validation to assess the performance and the fitness-for-purpose of a method or analytical system for metabolomic research, including ultimately error models per metabolite.
In the following sections the challenges and recommendations for method development and data processing and some commonly used data analysis tools are discussed. Furthermore, strategies for method validation and quality control are provided.
Analytical method development and analysis
The development of silylation based GC-MS methods poses serious challenges for analytical chemists considering the large range of compound classes and the large differences in concentrations within and between biological samples.
Inconsistencies in quantification of metabolites can arise from many sources during sampling, sample storage, sample extraction, derivatization, analysis and/or detection. For example, during sampling, sample storage and extraction of the metabolites, undesired changes in metabolite composition may occur due to for example enzyme activity, high reactivity and/or breakdown of metabolites. One way to avoid this is to use ‘snapshot’ sampling, i.e. fast cooling of the sample to low temperatures, maintain low storage temperatures (−80°C) and/or use low temperatures and appropriate additives to inhibit enzyme activity during extraction. Furthermore, irreproducible extraction and/or derivatization as well as degradation of derivatized metabolites in the analytical system are common problems that can introduce errors in the quantification. To detect the occurrence of these problems, an extensive set of test metabolites with different functional groups, polarity, molecular mass, etc. is required to optimize the method performance along the entire trajectory from sampling up to detection.
In a previous paper we introduced three performance classes based on the differences in reactivity towards silylation and the stability of derivatized metabolites (Koek et al. 2006) Performance class-1 metabolites are metabolites containing hydroxylic and carboxylic functional groups, such as sugars, fatty acids and organic acids. The analytical performance for these metabolites is generally very satisfactory with performance characteristics that fit the FDA requirements for target analysis in bioanalysis. Performance class-2 type metabolites, metabolites containing amine or phosphoric functional groups, can also be measured with satisfactory derivatization efficiencies, repeatability and intermediate precision. However the analysis of these metabolites is more critical compared to class-1 metabolites, when the method is not carried out under ‘optimal’ conditions. Metabolites with amide, thiol or sulfonic functional groups, so-called performance class-3 compounds, are more difficult to derivatize and analyze. We recommend using representative metabolites from all three performance classes, preferably isotopically labeled and with different volatilities and molecular mass for method optimization and validation. It should be noted that the mass difference between the labeled and naturally occurring metabolite must be sufficient to avoid isotopic interference from the naturally occurring metabolite in the quantification of the reference compound. The amount of mass difference needed depends on the amount of silyl groups after derivatization, the ratio between the naturally occurring metabolite and the labeled metabolite and the mass fragmentation of the metabolites. For example, phenylalanine can be distinguished from phenylalanine-d5 in most extracts by using mass fragments m/z 192 (endogenous) and m/z 197 (labeled) for quantification, as these fragments contain five deuterium and only one silicon atom. However, due to the large amounts of endogenous glucose in matrices such as blood plasma, the accurate quantification of glucose-d7 is (almost) impossible due to isotopic interference, using m/z 319 and m/z 323 (highest significant mass fragments in EI spectrum) as quantification masses. By adding labeled metabolites at different stages during from sample workup till injection, extraction and derivatization can be optimized to maximize the coverage of the entire analytical method (and minimize errors due to insufficient and/or irreproducible extraction and/or derivatization and artifact formation). Silylation has the advantage of a wide application range (Blau and Halket 1993; Knapp 1979). In metabolomics, trimethylsilylation (TMS) reagents, such as N-methyltrimethylsilyltrifluoroacetamide (MSTFA) and N,O-bis(trimethylsilyl)acetamide (BSA), are most commonly used. Both MSTFA and BSA are general purpose reagents with a wide application range and comparable silylation strength. In some cases trimethylsilylchlorosilane is added as a possible catalyst. Furthermore, several other reagents or mixes of reagents are available with more selective reagents, e.g. trimethylsilylimidazole (TMSI) or a mix of hexamethyldisilazane (HMDS) with TMCS and a mix of TMSI/BSA/TMCS, all developed to derivatize (sterically hindered) hydroxylgroups.
Early on in the development of our derivatization method as described in Koek et al. (2006), several derivatization reagents were compared, e.g. MSTFA, MSTFA with 1% TMCS, BSA, TMSI and TMSI/BSA/TMCS 3:3:2 (unpublished data). The byproduct of TMSI, i.e. imidazole, eluted as a (large) tailing peak in the chromatogram, and TMSI did not improve the recovery or performance (RSD) for typical targets such as monosaccharides compared to MSTFA. In our experiments the best results (highest recoveries and smallest RSDs) were obtained with MSTFA, the addition of TMCS did not improve the performance. The results with BSA were for the most part, comparable with the result for MSTFA, however, the RSDs for sugars were slightly higher. In addition, the byproduct of MSTFA is more volatile than the byproduct of BSA and therefore allows for the quantification of more volatile metabolites compared to BSA. A drawback of silylation reagents is their susceptibility towards hydrolysis. Therefore extracts should be as dry as possible before derivatization. As an example, only 1 μl of water in an extract will use up approximately 20 μl of MSTFA. In addition, derivatized extracts should be kept free of water after derivatization to avoid hydrolysis of the derivatized metabolites. By using a bulkier silylgroup, the hydrolytic stability of derivatized metabolites can be improved. However, in experiments with N-methyl-N-(tert-butyldimethylsilyl)trifluoroacetamide (MTBSTFA), sugars and some amino acids could not be derivatized completely (not even under extreme conditions) resulting in several derivates for one metabolite (unpublished data). In addition, the elution temperature of derivatized metabolites is increased compared to TMS derivates, limiting the application range for large molecules. Still, the use of MTBSTFA can be useful for identification purposes. Due to a more favorable fragmentation behavior EI mass spectra of TBS derivates contain higher characteristic M-57 (loss of tert-butyl) peak compared to the M-15 (loss of methyl) found with TMS derivates.
The derivatization efficiency is an important factor, that should be addressed during the method optimization, as metabolites can be only be analyzed reproducibly if the derivatization efficiency is sufficiently large. Due to the absence of commercially available reference standards that are silylized, the recovery cannot be determined by comparing the response of a metabolite spiked to a sample prior to derivatization and a standard solution of a reference standard. However, the derivatization efficiency can be estimated by using the assumption that the full scan response of a metabolite is proportional to the amount of mass injected. By comparing the response for the derivatized metabolites with the response of n-alkanes as reference compounds, the derivatization efficiency can be estimated (Koek et al. 2006).
Due to differences in stability of derivatized metabolites, some metabolites, especially derivatized class 3 metabolites, are more prone to degradation during storage or decomposition in the analytical system. Also, the degree of adsorption and/or degradation can vary between different samples with different biomass concentrations and different matrix compositions. For example, the presence of large amounts of extraction buffer components, such as HEPES or sulfate can significantly decrease the response of metabolites, while large amounts of other compounds, such as glucose or urea, can increase the response (unpublished results). In addition the extend of these effects can vary depending on the concentration of the matrix compound and the class and concentration of the metabolite. In Fig. 2 the matrix enhancement effect of glucose on different metabolites measured on GC × GC-MS is illustrated. In the ‘conventional’ setup, using a narrow bore thin film column, the response of the same amount of lysine and citric acids in extracts with low levels of glucose are lower compared to extracts with high levels of glucose, due to reduced adsorption of metabolites on active sites in the analytical system. In the high capacity setup using a more inert thicker film column in the second dimension virtually no adsorption of these metabolites occurs. Consequently, such matrix effects should be evaluated. This also illustrates the importance of an inert analytical system (sample storage vials, injection liners, analytical columns, etc.) to minimize adsorption and degradation of especially relatively unstable derivatized metabolites.
Fig. 2.
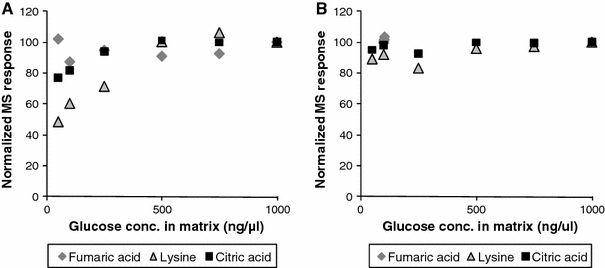
Illustration of the matrix enhancement effect of glucose on different metabolites measured with two different GC × GC-MS configurations. a ‘conventional’ setup with 30 m × 0.25 mm × 0.25 μm HP5-MS in the first and 1 m × 0.1 mm × 0.1 μm BPX-50 in the second dimension. b ‘high capacity’ setup with 30 m × 0.25 mm × 0.25 μm HP5-MS in the first and 2 m × 0.32 mm × 0.25 μm BPX-50 in the second dimension. In the ‘conventional’ setup in extracts with smaller amounts of glucose, the class-2 metabolite lysine and, to a lesser extent, citric acid adsorb and/or degrade on active sites present in the analytical system. In the extracts with high levels of glucose, the response for these metabolites increases, most probably because active sites are blocked. In the ‘high capacity’ setup using the more inert thicker film second dimension column the absorption is not present even at low levels of glucose in the matrix (Koek et al. 2008)
Data processing
Prior to statistical analysis the acquired analytical data needs to be processed such that equal identity is assigned to the same variable in each sample. For this purpose, essentially three types of methods are available: target analysis, peak picking and deconvolution. Each method requires its own tactics to tackle problems such as peak shift and peak overlap. The main challenges for data processing are (i) the amount of data (hundreds up to thousands of peaks in one sample), (ii) unbiased data processing, (iii) alignment of peaks shifted along the retention time axis and (iv) obtaining only one entry for each metabolite.
For target analysis, a list is prepared that contains a specific m/z value and a small retention time window within which a certain metabolite is expected to appear in all data files. Software provided by the instrument vendor is then able to determine the peak area of each metabolite based on the so called target list. This results in a peak area per metabolite and per sample. The advantages of this method are good precision, identities can be assigned beforehand and only one entry is obtained per peak. Disadvantages are that building the target table is time consuming and small peaks overlapping with larger peaks are easily overlooked.
A more comprehensive method that ensures the inclusion of most peaks, if not all, is peak picking. For peak picking methods such as the second derivative per m/z channel are used to detect the location of peaks in a chromatogram. Often, the peak height is then used as an estimate of the peak area. Methods for peak picking are automated and therefore much faster than for instance target analysis if the target list has to be prepared. There are however many drawbacks: (i) precision is lower, (ii) multiple entries per metabolite are usually obtained because peaks found for all m/z value are reported and (iii) the quality of the final results are difficult to check because the peak identities are not known. Furthermore, the peaks require alignment after peak picking due to retention time shifts. A summary of commonly used alignment techniques and algorithms is given by Jellema 2009).
The third generic class of data processing methods is deconvolution, a mathematical method that enhances the analytical resolution even further. Deconvolution makes use of the differences in mass-spectral information between different metabolites to separate overlapping peaks (Fig. 3). Furthermore, the method reports mass spectra rather than individual mass signals which offers a great advantage over peak picking where 20–30 peak areas (corresponding to the number of m/z values) per metabolite are common. Generally, in metabolomics research, deconvolution resolves unresolved peaks and transforms the raw data into peak tables with integrated peak areas per metabolite and per sample plus a list of mass spectra. Deconvolution can also be automated and is therefore faster than target analysis. Another advantage is that complete mass spectra are reported that can be used for annotation of peak identities to each reported peak. In comparison to peak picking the alignment step can be skipped because deconvolution can be performed on a complete dataset simultaneously rather than on individual chromatograms. However, the lack of a perfect computer program can result in poor spectra, multiple entries per metabolite and poor precision. For example, in automated data processing in GC × GC-MS, which requires the merging of peaks from different modulations originating from one peak after deconvolution, lower precision was observed using currently available methods compared to a targeted approach (Koek et al. 2010b). Actually, automated deconvolution, peak integration and peak merging is currently the only possibility to get from raw GC × GC-MS data to a peak list with corresponding areas.
Fig. 3.
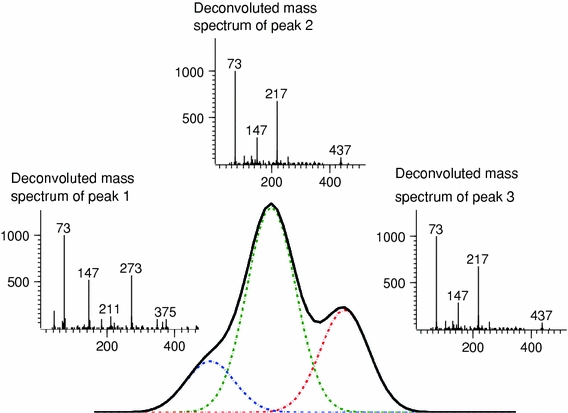
Example of deconvolution: three overlapping peaks were separated, making use of the mass spectral information. This results in a peak table with the response for all three individual metabolites and their corresponding mass spectrum
In terms of quality the target analysis results are up till now the best that can be obtained for any given GC-MS dataset if a proper target table is prepared. However, it can easily take more than a full week for an experienced analyst to produce targeted results for approximately 20–40 samples, because of the large amount of different peaks (components) present in the data files. However, the drawback of missing minor peaks in a targeted approach is probably of much more importance than a reduced precision which is currently still the case in deconvolution based methods (in GC-MS and GC × GC-MS).
Deconvolution is the most promising method for processing of gas chromatography mass spectrometry based metabolomics data as it fits all requirements: (i) handling huge datasets, (ii) automated processing, (iii) automatic peak alignment and (iv) just one quantitative value per metabolite per sample. Major issues in the development of deconvolution procedures are still the estimation of the number of metabolites present in a cluster of peaks and the variability of the mass spectral information which needs to be assumed equal for a single metabolite measured in multiple samples. However, this assumption cannot be met in some cases, for example, when large differences exist between the concentrations of a metabolite in different samples, some masses of a mass spectrum are outside the linear range, or when peaks with higher concentration are disturbing the measurements of nearby low concentration metabolites. These issues need to be resolved to come to an optimal deconvolution algorithm. Still, it is the authors’ opinion that a deconvolution approach, in which the chromatograms of all samples are automatically processed resulting in peak tables and metabolite spectra, is the most optimal solution.
Data analysis
Data analysis or statistical analysis is used to extract relevant biological information from the analytical data obtained. The quantitative aspects of analytical data are not influenced by data analysis and therefore these were considered beyond the scope of this paper. However, the applicability of data analysis tools is largely dependent on the quality of the analytical data. Therefore, we want to shortly reflect on some commonly used statistical methods for data analysis and their application in metabolomics data analysis. The proper way of statistical analysis depends highly on characteristics of the data set such as: design of the study, the data preprocessing method that was used, aim of the study and availability of prior knowledge such as metabolic pathway information. Therefore, the ideal strategy to perform statistics on metabolomics data is not limited to one single method. However, all statistics should include some means to validate the model in order to prevent optimistic models that don’t hold when applied in practice. In the third paragraph of the next section an overview of statistical tools and validation strategies applied in metabolomics research is provided.
Validation strategy
Due to the complexity of the metabolome (hundreds up to thousands of different metabolites), the comprehensiveness of silylation based GC-MS methods, the elaborate sample workup and difficulties in data processing, an extensive method validation is needed to assess the overall performance of the method from sample pretreatment through data preprocessing. The Metabolomics Standardization Initiative (MSI) provides guidelines on reporting of studies and methods (Fiehn et al. 2006), enabling the exchange of metabolomics methods and data. However, no guidelines on how to validate analytical metabolomics methods and data preprocessing tools have been provided so far.
In several guidelines the requirements for method validation of usually a limited and defined number of analytes have been described. In quantitative procedures at least the following validation parameters should be considered: selectivity, calibration model (linearity and range), accuracy, precision (repeatability and intermediate precision) and limit of quantification (LLOQ) (Table 3) (ICH 2005; Peters and Maurer 2002; Thompson et al. 2002; U.S. Department of Health and Human Services et al. 2001). Additional parameters that are generally recommended to be evaluated are: limit of detection, recovery, reproducibility and robustness.
Table 3.
Definitions of validation parameters
| Selectivity | The ability of an analytical method to differentiate and quantify an analyte in the presence of other components in the sample. One way to establish method selectivity is to prove the lack of response in blank matrix, an approach not suitable for metabolomics analysis. The second approach is based on the assumption that small interferences can be accepted as long as precision and bias (at LLOQ level) remain within certain acceptance limits |
| Calibration model | The relationship between the concentration of an analyte in the sample and the corresponding detector response. There is general agreement that calibration samples should be prepared in blank matrix and that their concentrations must cover the whole calibration range. Recommendations on how many concentration levels should be studied with how many replicates per concentration level differ significantly. To establish a calibration model, we suggest measuring at least six different calibration levels, evenly spread over the whole calibration range, in duplicate (Table 3) |
| Accuracy | The closeness of mean test results obtained by the method to the true value (concentration) of the analyte. Accuracy is determined by replicate analysis of samples containing known amounts of the analyte. Ideally, the accuracy or trueness of an analytical method is assessed by comparing the value found with a certified reference value or ‘true’ value (Hartmann et al. 1998; International conference on harmonisation. Q2(R1). Validation of analytical procedures: text and methodology, 2005; Peters and Maurer 2002; Thompson et al. 2002). However, in the absence of reference materials, as is the case in metabolomics analysis, the accuracy of an analytical method can be investigated by recovery experiments of (isotopically labeled) metabolites spiked to samples |
| Precision | The closeness of individual measures of an analyte when the procedure is applied repeatedly to multiple aliquots of a single homogeneous volume of biological matrix. Three different levels of precision can be determined, i.e. repeatability, intermediate precision and reproducibility. The repeatability or intra-batch precision is the precision over a short period of time using the same operating condition and is determined by repeated injection of individually prepared samples of the same test material. Intermediate precision or inter-batch precision expresses the within-laboratories variations, e.g. different days, different analyst, different equipment, etc. Reproducibility describes the precision between different laboratories and only has to be studied when the method is to be used in different laboratories |
| Limit of quantification | The lowest amount of metabolite that can be quantified with suitable precision and accuracy (Hartmann et al. 1998; International conference on harmonisation.Q2(R1).Validation of analytical procedures: text and methodology, 2005; Peters and Maurer 2002; Thompson et al. 2002). The LLOQ can be based on precision and accuracy data (lowest concentration with a precision and accuracy better than 20%), signal-to-noise or calculated from the standard deviation (SD) of in a blank sample or preferably the lowest point of the calibration line (LLOQ = k × SD/slope). For LLOQ a S/N ratio or k-factor equal to or greater than ten is usually chosen |
In principle the same validation parameters as mentioned above should be considered in quantitative comprehensive analysis. The question remains: “How to assess the validation parameters for a comprehensive analytical method for metabolomics analysis”? Ideally, the method performance for every individual metabolite should be assessed by spiking isotopically labeled metabolites to the matrix of interest. However, the availability of isotopically labeled standards is limited and such an approach would be very time consuming and expensive, especially since method performance can vary depending on the composition of the sample matrix studied and validation needs to be performed in all matrices of interest. An alternative could be to use different dilutions of a pooled sample of the samples to be analyzed to establish the calibration model (Koek et al. 2010b). However, only relative quantification of metabolites is possible using this strategy as metabolite concentrations are unknown and only linearity and precision can be determined. In addition, method performance can differ significantly with changing matrix composition. In general, the recovery of critical (class-3) metabolites is lower when the amount of total sample matrix injected is lower, and the calibration results obtained by this strategy can deviate from the linearity obtained when similar amounts of total biomass are injected. Another approach could be to use standard addition of metabolites to the matrix. However, if the metabolite of interest is present in the matrix the LOD cannot be determined. A more feasible and straightforward approach is the use of an extensive set of representative isotopically labeled metabolites from different performance classes (Sect. 2.1) with different functional groups, polarities and molecular mass. By performing the validation for these representative metabolites a good insight into method performance and reliability of the analytical data for different compound classes of the method can be obtained. Furthermore the use of representative quality control samples (Sect. 2.5) measured multiple times during a study can be used to assess the precision (inter- en intra-batch) of all metabolites present in the pooled sample.
For metabolomics studies we propose a minimum validation scheme as shown in Table 4. In this validation scheme the calibration model, repeatability, intermediate precision, LLOQ, recovery and matrix effect are addressed. The guidelines proposed were derived from the FDA validation guidelines for bio analysis (U.S. Department of Health and Human Services et al. 2001) and from experience in daily practice. For initial validation of a method a minimum number of 80 sample injections is proposed. For studies with a limited number of samples, measured within a few days, or when evaluating a new sample matrix a validation with a minimum of 35 sample injections is recommended. Obviously, when (larger) sample sets are measured over larger periods of time or more information on selectivity is needed, validation should be extended accordingly, and, for example intermediate precision over a larger period of time, stability of samples and selectivity should be investigated.
Table 4.
Proposed minimum validation of analytical metabolomics methodsa
| Sample characteristics used for validation experiments | Validation parameters investigated | |||||||
|---|---|---|---|---|---|---|---|---|
| Concentration | Biol. sample | Standard solution | Added prior to sample preparation | Calibration curve + repeatability | Intermediate precision | Recovery and matrix effecte | LLOQ | Total number |
| Number of samples on days 1–14 | ||||||||
| Day 1 | Day 2 & 3, (&7, 10 and 14)d | Day 1 | ||||||
| C0 | x | No | 2 | 2 | ||||
| C1 | x | Very low | 3b | (5 × 3) | (3 after sample preparation) | f | 3 (21) | |
| C2 | x | Low | 2 | 2 | ||||
| C3 | x | x | Intermediate | 3b | 2 × 3(+3 × 3) | 3 std + 3 after sample prep. prior to derivatization | 15 (24) | |
| C3 | x | Intermediate | 6 × 1c | 6 | ||||
| C4 | x | Higher | 2 | 2 | ||||
| C5 | x | High | 3b | (5 × 3) | (3 after sample preparation) | 3 (21) | ||
| C6 | x | Highest | 2 | 2 | ||||
| Total | 35 (80) | |||||||
aMinimum validation for initial validation of a method all samples (also the samples between brackets) should be measured. For studies with a limited number of samples, analyzed within a few days, the samples between brackets could be discarded
bIt is recommended to analyze 3 samples so that data for a calibration line can also be used for determining intermediate precision (C1, C3, C5), recovery (C1, C3, C5) and LLOQ (C1)
cDetermination of analytical repeatability, one sample injected six times
dDetermination of intermediate precision over 3 days or 14 days (between brackets), analysis of three samples per day including sample preparation
eThe recovery of the extraction (excluding derivatization) can be calculated by determining the ratio between the response of the metabolites spiked before and after extraction. The matrix effect is determined by determining the ratio of the response of the metabolites spiked after extraction and the metabolites in a standard solution. The matrix effect calculation covers matrix effects during derivatization (generally decreasing response) and matrix effects during analysis (increase (matrix enhancement) or decrease in response due to matrix present)
fCalculated from RSD of lowest concentration point of calibration line (LOQ = 10 × SD/slope)
In view of the unbiased non-targeted analysis used in metabolomics research, some validation parameters such as accuracy, require a different approach compared to targeted analysis. In general, no standard reference materials (SRM) are available for determining the accuracy of metabolomics methods. NIST is developing a SRM for metabolites in human blood plasma (NIST 2010), however this is still not commercially available. In the absence of reference materials with known metabolite concentrations, we propose to investigate the accuracy of the analytical method by determination of the recovery of metabolites spiked to samples. The recovery of the method (excluding the derivatization) is determined by comparing the response of (labeled) metabolites spiked to a biological sample prior to the sample workup with the response of the same metabolites spiked after extraction prior to derivatization (Table 4). The derivatization recovery is not determined, due to the absence of commercially available reference standards of silylized metabolites. Still, when the method performance is reproducible, quantitative results can be obtained without knowing the actual derivatization efficiency.
As mentioned earlier, matrix effects, such as degradation or adsorption in the analytical system can differ depending on the matrix composition. Therefore it is important to investigate whether the same concentration of a metabolite gives similar response in different matrices, to justify the comparison of relative metabolite concentrations between samples. The matrix effect is determined by determining the ratio of the response of the metabolites spiked after extraction and the metabolites in a standard solution. The matrix effect calculation covers matrix effects during derivatization (generally decreasing response) and matrix effects during analysis (increase (matrix enhancement; Anastassiades et al. 2003; Hajslova and Zrostlikova 2003; Koek et al. 2006, 2008) or decrease in response due to matrix present) (Table 4).
The selectivity is the ability of a method to differentiate and quantify an analyte in the presence of other components in a sample. Metabolomics samples contain large numbers of different metabolites that are all of interest. Therefore, the conventional ways of determining the selectivity, i.e. proving the absence in blank samples or to determine the precision and accuracy at the LLOQ level for every metabolite, are not feasible. A compromise could be to assess the selectivity (accuracy and precision) in specific ‘worst case’ scenarios, for example when analyzing monosaccharides (e.g. hexoses) with similar molecular weight, retention behavior and very similar mass spectra, or in case of coelution of low-abundant metabolites with very-high-abundant metabolites.
The evaluation of the fitness-for-purpose of a method is the most important goal in method validation. In metabolomics this means that one has to assess whether the method is suitable to answer the underlying biological question. This is a difficult question to answer, because it is often not known in advance which metabolites are most interesting (high correlation with a biological characteristic), at what levels of concentration these metabolites will be present and how small the differences in concentrations will be. In addition, due to the large differences in physicochemical properties of the metabolites targeted in the GC based methods in metabolomics research, method performance can differ significantly for different metabolites. Therefore, the formulation of general acceptance criteria for the different method-performance characteristics is complicated. One way to overcome this constraint is to classify metabolites in view of their analytical performance and formulate acceptance criteria per group of metabolites (Koek et al. 2006). Data obtained during optimization of a metabolomics method can be used to formulate realistic and manageable acceptance criteria. In addition, the performances and results from validations of GC-based metabolomics methods described in literature (Sect. 3.3) can be useful for that purpose.
Quality control
When a validated analytical method is implemented, quality control is essential to ensure the quality and reliability of the analytical data obtained. Quality control is needed to monitor and/or correct for deviations that occur during sample workup or analysis, as discussed in Sect. 2.1. Other known sources of variation in metabolomics analysis are, for instance, differences between instruments, operators, changes in instrumental sensitivity, fouling of mass spectrometers etc. As all endogenous metabolites are of interest and the identities of many metabolites are unknown a priori, quality control is complex. Several strategies can be followed to monitor the quality and correct for deviations in metabolite response, such as the use of external standards, internal standards or a combination of both internal and external standards (Table 5). It should be noted that quality standards should either be used for the detection of deviations or the correction of deviations. Only in this way quality standards for control (detection) can be used to check the quality of the data after eventual corrections.
Table 5.
Different quality control standards and their function
| External standards | Internal standards | ||||
|---|---|---|---|---|---|
| Academic standard (no matrix) | Pooled QC | Exogenous standardd | Spike isotopically labeled metabolites | Labeled standard for every metabolite | |
| Control/detect | |||||
| • Storage | − | − | − | + | + |
| • Extraction | − | − | − | + | + |
| • Derivatization | − | − | − | + | + |
| • Injection vol. | − | − | + | − | − |
| • Detector sensitivitya | − | − | + | − | − |
| • Detector driftb | − | + | − | − | − |
| • Inertness analytical system | + | + | − | ±c | ±c |
| Correction | |||||
| • Detector responsea | − | − | + | − | − |
| • Detector driftb | − | + | − | − | − |
| • Batch correction | − | + | + | − | − |
| • Recovery metabolites | − | ± | − | ± | + |
aOverall sensitivity of the detector
bDetector drift, i.e. the change in detector response with mass-to-charge ratio (m/z) can vary with different masses (e.g. due to fouling) and should be addressed separately from the overall sensitivity
cThe ratio of different labeled metabolites, e.g. class 3/class 1 (critical/good performing metabolite; §2.1), can be used as an indicator for the inertness of the analytical system. However, deviations in the ratio can also be caused by other deviations, e.g. during sample workup
dStable compound that is not derivatized and not present in biological samples
External standards are especially suitable to detect and/or correct for detector drift and to control the inertness of the analytical system. For example, academic standards, i.e. standard solutions without matrix, can be used as early markers for the decline of the performance of the analytical system, as metabolites are more prone to adsorb or degrade on the surface of the analytical column in the absence of sample matrix (Anastassiades et al. 2003; Hajslova and Zrostlikova 2003; Koek et al. 2006; Koek et al. 2008). Another very useful external standard is a pooled sample of all individual samples (pooled QC) measured during a study (Sangster et al. 2006). A pooled QC can be used to calculate the repeatability and intermediate precision of all detectable metabolites present in the samples and to correct for detector drift and/or variations in MS response between batches. In addition, a pooled QC representative of the samples measured, can be used to correct MS responses of metabolites in individual samples, as proposed by Greef et al. (2007) and Kloet et al. (2009). However, this correction will only work when the matrix effects are not varying between samples, e.g. when the variation of the sample composition is limited.
With isotopically labeled metabolites or non-endogenous as internal standards, disturbances can be detected or corrected, for every single metabolite in every individual sample. By adding labeled metabolites (e.g. prior to extraction, derivatization or analysis) the different steps of the sample work-up can be controlled. A endogenous metabolite can be corrected by the addition of its isotopologues (same molecule with different isotopic composition), or an isotopically labeled or non-endogenous metabolite with different composition but similar in performance characteristics (e.g. of the same class). Despite the fact that isotopically labeled metabolites are relatively expensive and their availability is limited, the addition of labeled metabolites is essential to monitor and eventually correct metabolite responses in metabolomics studies. Another approach is to use in vivo isotopically labeled microorganisms as internal standards. In this setup microorganisms are grown on isotopically labeled growth media to label all intracellular metabolites. Extracts of this microorganism are then mixed with non-labeled microbial extracts, resulting in an extract containing isotopically labeled metabolites as internal standards for every metabolite (Birkemeyer et al. 2005). However, these labeled reference materials are not available for most matrices (e.g. mammalian metabolomics), and the labeling efficiency has to be high. In addition, the retention behavior of labeled internal standards is very similar to the endogenous metabolite and when silylation is used their mass spectra can contain many similar mass fragments. Therefore, labeled internal standards can complicate the data preprocessing and quantification (e.g. deconvolution, peak picking and integration).
In this section we propose a quality control scheme using a combination of isotopically labeled internal standards and external quality standards (Fig. 4). This scheme is suitable for the most commonly used GC-MS methods using an oximation and subsequent silylation as derivatization prior to analysis, although it can also be used when applying different derivatization methods or no derivatization at all.
Fig. 4.
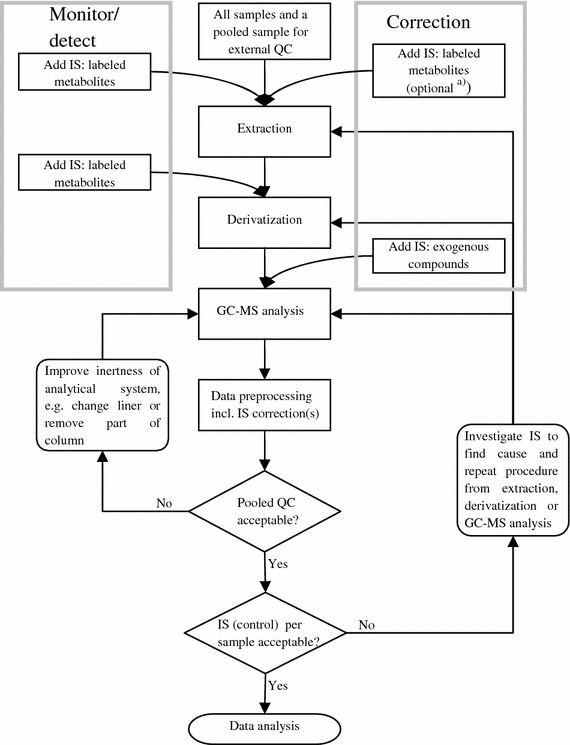
Suggested quality-control scheme for GC-MS metabolomics studies; IS internal standard(s), QC quality-control sample. a)Depending on the matrix analyzed, one can choose to add IS for correction or leave these standards out (see Sect. 2.5)
The amount of internal standards needed and how to correct the MS response for metabolites in individual samples depends on the variability of the sample composition. When differences between sample compositions are small (e.g. plasma or serum) the differences in matrix effects between different samples can be expected to be small as well. In that case the correction of individual metabolites can be performed by using an external standard. In these studies we suggest using a set of at least six labeled metabolites as internal standards for quality control. Three standards should be added before extraction (one for every performance class; cf. Sect. 2.1, i.e. favorable as well as unfavorable metabolites), and three (one for every performance class) added before derivatization. In addition, at least one exogenous standard, i.e. a stable compound that is not derivatized, should be added to every sample before injection to correct for injection volume and MS response; this is the only internal standard used for correction purposes. To monitor and eventually correct for differences in the MS response within or between batches for all individual metabolites a pooled QC should be analyzed repeatedly, for example at the beginning and end of a batch of samples and between every set of five samples. The pooled QC is used to calculate the repeatability and precision of response for each metabolite. In common practice, the correction for small variations in injection volume and MS response with the internal standard described above is always performed. If needed, for example in large studies when differences between batches are significant, each metabolite can be corrected by using the QC samples (Kloet et al. 2009). In Fig. 5 the effects of IS and QC correction are illustrated in a real-life study. During the analysis of this study, consisting of 5 batches of urine samples (total of approximately 200 samples), the MS-ion source was replaced between batch 3 and 4, causing an offset in the peak areas between batch 3 and 4. As an example, the MS response of phenylalanine could be corrected properly by correction on only the internal standard (dicyclohexylphthalate), however the peak area of glycolic acid was only properly corrected for after IS and QC correction.
Fig. 5.
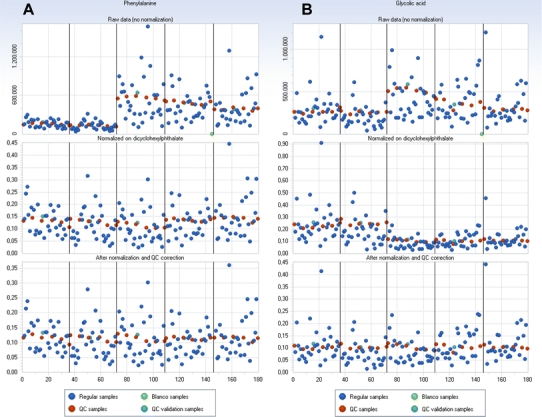
Example of the effects of correction of peak areas of phenylalanine (a) and glycolic acid (b) measured in 5 consecutive batches (approximately 35 samples per batch, total of 180 samples); on the x-axis: sample number, and on the y-axis: (normalized) peak areas. Upper: absolute peak areas of uncorrected data, middle: normalized peak areas after IS correction, lower: normalized peak areas after IS and QC correction. In blue: regular samples, in red: QC samples, in green: blank samples and in turquoise: QC validation samples (the same as QC samples, but not used for correctional purposes) (Color figure online)
When the differences in matrix composition are larger, for example microbial samples, the pooled QC generally cannot be used to correct for variations in MS response for individual samples. In these studies the matrix effects can differ between samples and a correction with an external standard could even decrease the reliability of the data. In these cases, the set of internal standards added before extraction should be extended. Especially, labeled metabolites from compound classes that are more prone to degradation or adsorbtion on the surface of the analytical column (performance class 3, e.g. thiols, amides and amines, Koek et al. 2006), should be added to be able to control or correct for matrix-dependent variations in metabolite responses for individual samples.
Still the pooled QC is useful to monitor detector drift, monitor the inertness of the analytical column and to calculate the repeatability and precision of response for all metabolites. In addition, the pooled QC samples can be used to determine the most suitable internal standard to correct for deviations from the extended set of corrective internal standards for every individual metabolite.
Besides the use of internal standards and pooled QC, the quality of the sample work-up and/or analysis can be further controlled by repeated sample workup and/or injection of samples. In this way the repeatability of duplicates can be evaluated.
Based on daily practice we find that RSDs (without QC correction) of internal quality control standards (from compound classes: organic acids, sugars, amino acids) within one batch are generally less than 5% (repeatability) and 10–15% between batches within one study (intermediate precision). However, the method performance depends on both the physicochemical properties of the metabolite measured and the matrix (plasma, urine, microbial, tissue) and may therefore deviate from the values mentioned above (Koek et al. 2006). Therefore, as already mentioned in Sect. 2.4, different acceptance criteria are set depending on the compound class and matrix.
Data processing, data analysis, method validation and quality control in literature
In Table 2 an overview of GC based applications in metabolomics research is presented. Publications were only included in Table 2 when the number of targeted compound classes was three or higher. Research on metabolic target analysis or metabolic profiling was not included, as a different approach for method development and validation is usually applied for these targeted analyses than for non-target comprehensive analysis. In the next sections the data-processing strategies, data-analysis tools, method validation and quality-control strategies in literature are discussed.
Data preprocessing
As mentioned in Sect. 2.2, there are three ways to preprocess GC-MS data: target analysis, peak picking and deconvolution.
In one-dimensional GC-MS often a targeted approach is followed to obtain a list of metabolites and their corresponding peak areas. For example, Weckwerth et al. (2004a) used a customized reference-spectrum database based on retention indices and mass-spectral similarities to match peaks between chromatograms. Metabolites were quantified using a selective fragment ion for each individual metabolite from their corresponding mass spectrum. Morgenthal et al. (2005) reported a similar approach with the addition of first defining a reference chromatogram with a maximum number of detected peaks that fulfill a predefined signal to noise ratio.
In peak picking, first the m/z traces containing meaningful information are selected, for example with CODA (Windig and Smith 2007), MetAlign (Lommen 2009) or Impress (van Greef et al. 2004b), then the peaks in the selected ion traces are detected using methods such as the second derivative and finally integrated to obtain single intensity measures for complete peak profiles. For GC-MS data this results in a data table, in which one metabolite is represented by many different variables (all masses present in the mass spectrum are separately integrated). Due to this major drawback, peak picking is not frequently used with GC-MS data, and only few examples using this strategy are reported in literature (Lommen 2009; Tikunov et al. 2005).
Deconvolution has been applied for both one-dimensional (1D) and two-dimensional (2D) datasets. For example, Jonsson et al. (2005) and Jellema et al. (2010) demonstrated the advantage of simultaneous deconvolution of all 1D-GC-MS chromatograms at the same time, rather than processing each chromatogram separately and subsequently construct a total data set afterwards from the separate peak tables per chromatogram. In all evaluated GC × GC-MS papers a deconvolution approach was used, when quantitative data on peak areas was extracted from raw chromatograms. Two different software packages were used, i.e. ChromaTOF software (LECO, St. Joseph, MI, USA) (Huang and Regnier 2008; Koek et al. 2008; Kusano et al. 2007; O’Hagan et al. 2007; Oh et al. 2008; Shellie et al. 2005; Welthagen et al. 2005) or parallel factor analysis (PARAFAC; Harshman 1970) (Guo and Lidstrom 2008; Hope et al. 2005b; Humston et al. 2008; Mohler et al. 2006; Mohler et al. 2007; Mohler et al. 2008; Sinha et al. 2004a, b). Although deconvolution was used in all described papers, only few authors used a non-targeted approach in metabolite quantification (Lee and Fiehn 2008; O’Hagan et al. 2007; Oh et al. 2008; all ChromaTOF users). Almost no quantitative data has been reported on the performance of the deconvolution software tools in non-targeted metabolite quantification. Data on the performance that has been published is only for a selected number of target metabolites after deconvolution and peak merging of different modulations (see §3.5). Only Koek et al. (2010a) evaluated the performance of non-target processing in GC × GC-MS using the ChromaTOF software; for approximately 70% of all peaks accurate peak areas could be obtained without manual correction of integration and peak merging. Still, the time required for processing limited the use of the ChromaTOF software for non-target processing (quantification of all metabolites) in large metabolomics studies (>30–50 samples, eventually in duplicate). PARAFAC was used only in a targeted approach, i.e. first a multivariate classification method on segments of aligned raw chromatograms was performed (with the Fisher ratio method (Guo and Lidstrom 2008; Humston et al. 2008; Mohler et al. 2006; Mohler et al. 2007; Mohler et al. 2008) or DotMap algorithm (Hope et al. 2005b; Sinha et al. 2004b)) and subsequently PARAFAC was applied only on time segments of the raw data that discriminate between the different groups of interest. Although PARAFAC could be applied in non-targeted quantification of an entire GC × GC-MS chromatogram (Hoggard and Synovec 2008), the time required to process a single chromatogram (tens of hours, excluding the time-consuming task of ensuring only one entry per metabolite in all samples) is still a major bottleneck to apply PARAFAC for non-target processing.
Oh et al. (2008) developed a GC × GC-MS tool to deal with the difficult task of peak merging and ensuring only one entry per metabolite. Their peak sorting algorithm is based on retention time, correlation of mass spectral information and (optional) peak name which are reported after initial processing (deconvolution and peak integration) by the ChromaTOF software. First the second dimension peaks originating from the same chemical component are merged starting at the first entry of both the first and second dimension run. For both the first- and second-dimension retention time an allowed deviation in retention time is defined. When a peak stays within the allowed retention-time shifts then the underlying mass spectra are compared using the Pearson correlation coefficient (R). The second step in the algorithm then uses a sorting scheme to match equal peaks from different chromatograms. The chromatogram with the most peaks is assigned as the reference sample. Starting with the first peak the sorting algorithm searches for peaks that match as closely as possible the same criteria as were used in the merging step: retention time shifts in the first and second dimension should be less than a preset maximum allowable shift; the Pearson correlation coefficient between the mass spectra should meet a preset minimum correlation and (optional) the peak names, as assigned by the ChromaTOF software, should be the same. All matches are recorded in a new table and the processed peaks are removed from the list, resulting finally in a peak table representing all peaks in all chromatograms. Unfortunately, only qualitative data and no quantitative data on the performance of the software are given.
De Souza et al. (2006) also worked on an algorithm to obtain a single entry per metabolite in all samples, using the peak lists extracted from the raw data by commercial software (ChemStation, Agilent Technologies, Santa Clara, CA, USA). First hierarchical clustering of retention times within replicate measurements was performed resulting in a dendrogram illustrating the distances between the retention times of all detected peaks. The cut-off to determine clusters within the dendrogram was based upon the average number of peaks within the replicate measurements. In a next step the clustered peaks from the replicate measurements are again clustered in a second step to cumulate peaks with the same identity but measured within samples of, for example, different cell states or genotypes. So-called super-clusters are formed which in an ideal situation contain, per cluster, the same peak or metabolite as measured within the complete dataset.
Data analysis
Broadly viewed, data from metabolomics studies are either analyzed using univariate tests from classical statistics, such as the Student t-test (Denkert et al. 2006), or using multivatiate statistics, such as PCA (Denkert et al. 2006) and all sorts of regression and classification methodologies. Key to the success of all statistics is to have both a good statistical validation as well as reliable biological interpretation of the results. Metabolomics data does not fit well to the assumption of normal distribution or the assumption of having more samples ‘n’ than variables ‘m’ per subject or data record. In metabolomics typically the number of variables or metabolites (‘m’) is much larger than the number of samples measured (‘n’). This type of data is also referred to as megavariate data (Rubingh et al. 2009). Given various distributions, the chance of detecting a discriminating variable with a probability of for instance more than 99.9% (P < 0.01) is increased proportional to the number of independent tests one performs. In metabolomics studies often the number of variables is extremely high in comparison to the number of samples and care should be taken not to introduce chance to the scene of marker selection (Broadhurst and Kell 2006).
One way to reduce the chance of finding a coincidental significant effect in univariate data analysis is to take into account the ‘False Discovery Rate’ or FDR (Broadhurst and Kell 2006) using a corrected level of the P-value according to Bonferroni. The P-value for instance in the t-test is thereby reduced such that the chance of obtaining a false discovery due to multiple testing is made proportional to the number of tests being performed. This methodology has been introduced rather recently into metabolomics (Broadhurst and Kell 2006), earlier Fiehn et al. (2000a), Weckwerth et al. (2004b) used a standard P-value to test differential changes. While in the Bonferroni correction methodology the problem of false positives is tackled, it also introduces a problem, because significance levels are rather difficult to reach. For instance, in the case of a P-value of 0.05 and 1000 metabolites, the Bonferroni corrected P value becomes 0.00005. Broadhurst and Kell (2006) suggest some alternatives that take into account the internal correlation structure of the data. Denkert et al. (2006) validated differentiating peaks by means of random permutations of the classification vector, meaning that samples obtain a random classification as being a member of a certain class. This resulted in an expected number of false positive discoveries (n exp) and an observed number of discoveries (n obs).
Other methodologies to analyze megavariate data all try to combine the original variables into a set of newly defined variables that are linear combinations of the original variables. Such methods include principal-component analysis (PCA), principal-component discriminant analysis (PCDA) and partial-least-square discriminant analysis (PLS-DA). An overview of methodologies for data analysis in metabolomics research is given by Greef and Smilde (2005). Among these methodologies PCA is very popular and powerful. For example, Fiehn et al. (2000a), Jonsson et al. (2004) and Denkert et al. (2006) use PCA to find metabolites that differentiate between different samples. Pierce et al. (2006b) use PCA to find regions in GC × GC-TOFMS chromatograms that differentiate between samples and subsequently, use PARAFAC (Sinha et al. 2004b) to deconvolute these regions of interest. Another statistical method to find regions that differentiate between samples in GC × GC-MS data is the Fisher ratio method (Mohler et al. 2007; Pierce et al. 2006a). The Fisher ratio method can be applied directly on the four dimensional (4D) data structure from a GC × GC-TOF-MS instrument (no prior processing) and statistically differentiates regions of the signals containing large class-to-class variations from regions containing large within-class variations.
Another popular method to analyze megavariate data is hierarchical cluster analysis or HCA. HCA calculates Euclidian distances resulting in groups or clusters of samples that show multivariate similarity. The calculated distances are represented into dendograms where alike metabolic profiles are clustered together (e.g. Fiehn et al. 2000a).
In multivariate statistics methods have been introduced to reduce the probability to obtain correlations by chance. Methods such as cross validation, permutation tests and, most optimal, the use of separate datasets for training, validation and testing can be used for this purpose. Denkert et al. (2006) validated the markers that distinguish between ovarian carcinomas and borderline tumors by calculating the P-value for all significant metabolites after permuting or randomizing the classification factors. This led to the conclusion that all discovered metabolites perform better than chance. Dixon et al. (2007) used PLS-DA to find significant variables and proposed to use different strategies for either biomarker selection (discriminatory marker selection) or estimation of the predictive ability of a classification model. To determine potential biomarkers all samples were included in the model rather than using a separate training set. In case of a predictive model, the total dataset was split up into a training and a test set. Next, the training set was split up multiple times using a bootstrap methodology resulting in a bootstrap set and a validation set. A similar methodology is applied and explained by Westerhuis et al. (2008) whereby a class membership confidence measure can be estimated for each sample.
Method validation
Although GC-MS is considered a mature technique and is frequently used in metabolomics analysis, method validation has not had much attention in metabolomics research so far. Nearly all publications focus on the application and/or data preprocessing and data analysis rather than evaluating the method performance.
Some methods are described that use headspace sampling (HS) and/or solid-phase micro extraction of the headspace (HS-SPME) without a preceding sample workup, to introduce the metabolites in the analytical column (Table 4). Data on method validation of these methods is limited. Only Pauling et al. (1971) reported data on the application range (250–280 different metabolites measured) and intermediate precision (~10%). Because headspace sampling is used to introduce the sample, the application range of such methods is restricted to volatile metabolites.
The majority of the validation data available is on one-dimensional GC-MS methods using oximation and subsequently silylation as derivatization technique (OS-GC-MS). Due to the general applicability of silylation, these types of methods cover the largest range of different compound classes, including alcohols, aldehydes, amino acids, amines, (phospho-) organic acids, sugars, sugar acids, (acyl-) sugar amines, sugar phosphates, purines and pyrimidines. The repeatability is the validation parameter that is most assessed and reported. Only few papers report data on other validation parameters, such as selectivity, calibration model, accuracy (recovery), intermediate precision and LLOQ (Table 5). Typically, metabolite responses are linear over at least two orders of magnitude, recoveries between 70 and 140% are found. Furthermore, repeatability and intermediate precision of the sample work up and analysis are reported of 1–15% and 7–15%, respectively and detection limits in the range of 40–500 pg on-column. However, due to the derivatization, method performance is significantly influenced by the compound class or functional groups of a metabolite. The performance for critical (less stable) compounds (e.g. thiols and amides) highly depends on the inertness of the analytical system (Koek et al. 2006).
One extensively validated method with an alternative derivatization strategy is reported by Qiu et al. (2007) In this paper an ethyl-chloroformate derivatization is used prior to GC-MS analysis. Although chloroformate reagents only convert amine and carboxylic functional groups, limiting the application range to amines, amino acids and organic acids, the derivatization can be performed directly in the sample without prior removal of water and the reaction is very fast (60s) and the complete sample work up procedure takes less than 10 min. The results for linearity, accuracy, precision and detection limits are comparable to the results for oximation/silylation based methods. Although this method can be interesting when high throughput of samples is necessary, the application range of this method is limited compared to OS-GC-MS methods.
Apart from the one-dimensional GC-MS methods some two-dimensional gas chromatography (GC × GC) methods are described relying on oximation and silylation. The focus in these papers is mainly on data processing and data analysis but validation data are very limited. Some report the repeatability of injection (typically 5–10%) and/or the repeatability of the sample workup and analysis (typically 11–31%). Only Koek et al. (2008) describe a more elaborate validation covering selectivity, calibration model, repeatability and lower limit of detection (LLOD). Compared to one-dimensional OS-GC-MS higher peak capacity and lower detection limits (2–20 times improved) were obtained. Due to the use of a thicker film column (0.32 mm × 0.25 μm film thickness) in the second dimension the inertness of the analytical system improved, resulting in better linear ranges (lower intercepts) especially for critical compounds (Koek et al. 2008). In addition, the performance of the method for a set of test metabolites was stable in samples with varying matrix compositions, allowing accurate (relative) quantification. In view of these improvements OS-GC × GC-MS methods are capable of providing more detailed and improved information on sample compositions in metabolomics studies. However, data preprocessing and analysis is still very complex and time-consuming, due to limitations of the software currently available (Koek et al. 2010a).
Quality control
Only few paper were published on quality control in metabolomics studies. Sangster et al. (2006) suggested the use of a pooled sample from all study samples as a quality control sample. Initially, a small set of selected metabolites were visually inspected on peak shape, MS response, mass accuracy and retention time and subsequently the whole dataset was analyzed using PCA. In a good dataset QC samples were expected to cluster close together and show no time related trends. The use of PCA can be a very useful tool for a quick evaluation of the analytical data quality. However, it should be noted that even when quality control samples cluster together in PCA analysis, still deviations for individual metabolites can exists. Fiehn et al. (2008) used daily quality control samples. For this purpose, two method blanks and four calibration curve samples were used consisting of 31 pure reference compounds. Although this strategy gives information on the state of the analytical system at the beginning of a day, no information on the performance for individual samples is obtained. Furthermore Koek et al. (2006) describe a quality control strategy using isotopically labeled internal standards added prior to extraction and derivatization. The internal standards were used to control the quality of analysis and eventually to correct for disturbances during sample work-up.
Conclusions and perspectives
The purpose of metabolomics research is to gain understanding in the functioning of organisms and biological systems and to answer biological questions. Quantitative, reliable analytical data on (relative) metabolite concentrations is a prerequisite to achieve this goal.
GC-MS analysis is very suitable for comprehensive non-target analysis, and a large range of different metabolites can be measured when derivatization with a silylation reagent is used. Although derivatization coupled with GC-MS is frequently applied in today’s metabolomics research, method validation data described in literature is surprisingly limited. Especially, important information on intermediate precision and accuracy in GC × GC-MS is lacking. Reliable data is needed to validate the comparison of relative metabolite concentrations or ultimately to determine absolute metabolite concentrations in metabolomics samples, but this has to be based on satisfactory validation of linearity, intermediate precision and accuracy. In most metabolomics studies the relative concentrations of metabolites in different samples are compared, rather than comparing absolute concentrations, and in this approach the precision of a method is more important than the absence of bias. However, it still has to be investigated whether the relative quantification is correct, i.e. will the same amount of metabolite provide the same peak area in all matrices of interest? This is very critical, because differences in matrix compositions can significantly influence the recovery of metabolites.
The ultimate goal in metabolomics is to determine the absolute concentration levels for all metabolites in a sample. However, the largest bottleneck in absolute quantification is the difficulty in determining the accuracy of metabolomics methods. Presently no certified reference materials are available to determine the accuracies of metabolomics methods. In both setups, however, when the guidelines on method validation and quality control in this paper are followed (addition of extensive set of representative internal standards and determination of the calibration model by adding labeled standards to the matrix) relative or absolute quantification is possible. Still, the development of certified reference materials for metabolomics analysis is needed to achieve absolute quantification for all metabolites.
Besides the analytical method, data processing is essential in obtaining quantitative and unbiased analytical data. Deconvolution approaches, in principle, fit all requirements for metabolomics analysis, such as, handling of huge data sets and unbiased quantification. Nevertheless, the precision of deconvolution approaches compared to targeted approaches is still lower. Especially in GC × GC-MS, where target analysis is not a realistic option, improvement of the speed and precision of the processing tools is required to be able to handle large data sets (>30–50 samples).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Anastassiades M, Mastovska K, Lehotay SJ. Evaluation of analyte protectants to improve gas chromatographic analysis of pesticides. Journal of Chromatography. A. 2003;1015(1–2):163–184. doi: 10.1016/S0021-9673(03)01208-1. [DOI] [PubMed] [Google Scholar]
- Aura AM, Mattila I, nen-Laakso T, Miettinen J, Oksman-Caldentey KM, Oresic M. Microbial metabolism of catechin stereoisomers by human faecal microbiota: Comparison of targeted analysis and a non-targeted metabolomics method. Phytochemistry Letters. 2008;1(1):18–22. doi: 10.1016/j.phytol.2007.12.001. [DOI] [Google Scholar]
- Birkemeyer C, Luedemann A, Wagner C, Erban A, Kopka J. Metabolome analysis: The potential of in vivo labeling with stable isotopes for metabolite profiling. Trends in Biotechnology. 2005;23(1):28–33. doi: 10.1016/j.tibtech.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Blau K, Halket J. Handbook of derivatives for chromatography. New York: John Wiley & Sons Ltd.; 1993. [Google Scholar]
- Broadhurst DI, Kell DB. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics. 2006;2(4):171–196. doi: 10.1007/s11306-006-0037-z. [DOI] [Google Scholar]
- Chang, W. T., Thissen, U., Ehlert, K., Koek, M., Jellema, R., Hankemeier, T., et al. (2006). Effects of growth conditions and processing on rehmannia glutinosa using fingerprint strategy. Planta Medica, 72(5), 458–467. [DOI] [PubMed]
- Coucheney E, Daniell TJ, Chenu C, Nunan N. Gas chromatographic metabolic profiling: A sensitive tool for functional microbial ecology. Journal of Microbiologial Methods. 2008;75(3):491–500. doi: 10.1016/j.mimet.2008.07.029. [DOI] [PubMed] [Google Scholar]
- De Souza DP, Saunders EC, McConville MJ, Likic VA. Progressive peak clustering in GC-MS Metabolomic experiments applied to Leishmania parasites. Bioinformatics. 2006;22(11):1391–1396. doi: 10.1093/bioinformatics/btl085. [DOI] [PubMed] [Google Scholar]
- Denkert C, Budczies J, Kind T, Weichert W, Tablack P, Sehouli J, Niesporek S, Konsgen D, Dietel M, Fiehn O. Mass spectrometry-based metabolic profiling reveals different metabolite patterns in invasive ovarian carcinomas and ovarian borderline tumors. Cancer Research. 2006;66(22):10795–10804. doi: 10.1158/0008-5472.CAN-06-0755. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Xu Y, Brereton RG, Soini HA, Novotny MV, Oberzaucher E, Grammer K, Penn DJ. Pattern recognition of gas chromatography mass spectrometry of human volatiles in sweat to distinguish the sex of subjects and determine potential discriminatory marker peaks. Chemometrics and Intelligent Laboratory Systems. 2007;87(2):161–172. doi: 10.1016/j.chemolab.2006.12.004. [DOI] [Google Scholar]
- Dunn WB, Ellis D. Metabolomics: Current analytical platforms and methodologies. TrAC Trends in Analytical Chemistry. 2005;24(4):285–294. doi: 10.1016/j.trac.2004.11.021. [DOI] [Google Scholar]
- Fan TWM, Colmer TD, Lane AN, Higashi RM. Determination of metabolites by 1H NMR and GC: Analysis for organic osmolytes in crude tissue extracts. Analytical Biochemistry. 1993;214(1):260–271. doi: 10.1006/abio.1993.1486. [DOI] [PubMed] [Google Scholar]
- Fan TWM, Lane AN, Shenker M, Bartley JP, Crowley D, Higashi RM. Comprehensive chemical profiling of gramineous plant root exudates using high-resolution NMR and MS. Phytochemistry. 2001;57(2):209–221. doi: 10.1016/S0031-9422(01)00007-3. [DOI] [PubMed] [Google Scholar]
- Fiehn O. Metabolomics – the link between genotypes and phenotypes. Plant Molecular Biology. 2002;48:155–171. doi: 10.1023/A:1013713905833. [DOI] [PubMed] [Google Scholar]
- Fiehn O. Metabolic networks of Cucurbita maxima phloem. Phytochemistry. 2003;62(6):875–886. doi: 10.1016/S0031-9422(02)00715-X. [DOI] [PubMed] [Google Scholar]
- Fiehn O, Kopka J, Dormann P, Altmann T, Trethewey RN, Willmitzer L. Metabolite profiling for plant functional genomics. Nature Biotechnology. 2000;18(11):1157–1161. doi: 10.1038/81137. [DOI] [PubMed] [Google Scholar]
- Fiehn O, Kopka J, Trethewey RN, Willmitzer L. Identification of uncommon plant metabolites based on calculation of elemental compositions using gas chromatography and quadrupole mass spectrometry. Analytical Chemistry. 2000;72(15):3573–3580. doi: 10.1021/ac991142i. [DOI] [PubMed] [Google Scholar]
- Fiehn O, Kristal B, Ommen BV, Sumner LW, Sansone SA, Taylor C, Hardy N, Kaddurah-Daouk R. Establishing reporting standards for metabolomic and metabonomic studies: A call for participation. OMICS. 2006;10(2):158–163. doi: 10.1089/omi.2006.10.158. [DOI] [PubMed] [Google Scholar]
- Fiehn O, Wohlgemuth G, Scholz M, Kind T, Lee DY, Lu Y, Moon S, Nikolau B. Quality control for plant metabolomics: Reporting MSI-compliant studies. The Plant Journal. 2008;53(4):691–704. doi: 10.1111/j.1365-313X.2007.03387.x. [DOI] [PubMed] [Google Scholar]
- Forster, J., Famili, I., Fu, P., Palsson, B. O., & Nielsen, J. (2003). Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Research, 13(2), 244–253. [DOI] [PMC free article] [PubMed]
- Gullberg J, Jonsson P, Nordstrom A, Sjostrom M, Moritz T. Design of experiments: An efficient strategy to identify factors influencing extraction and derivatization ao Arabidopsis thaliana samples in metabolic studies with gas chromatography/mass spectrometry. Analytical Biochemistry. 2004;331:283–295. doi: 10.1016/j.ab.2004.04.037. [DOI] [PubMed] [Google Scholar]
- Guo X, Lidstrom ME. Metabolite profiling analysis of Methylobacterium extorquensAM1 by comprehensive two-dimensional gas chromatography coupled with time-of-flight mass spectrometry. Biotechnology and Bioengineering. 2008;99(4):929–940. doi: 10.1002/bit.21652. [DOI] [PubMed] [Google Scholar]
- Hajslova J, Zrostlikova J. Matrix effects in (ultra)trace analysis of pesticide residues in food and biotic matrices. Journal of Chromatography. A. 2003;1000(1–2):181–197. doi: 10.1016/S0021-9673(03)00539-9. [DOI] [PubMed] [Google Scholar]
- Hall R, Beale M, Fiehn O, Hardy N, Sumner L, Bino RJ. Plant metabolomics: The missing link in functional genomics strategies. Plant Cell. 2002;14(7):1437–1440. doi: 10.1105/tpc.140720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harshman RA. Foundations of the PARAFAC procedure: Models and conditions for an “explanatory” multimodal factor analysis. UCLA Working Papers in Phonetics. 1970;16:1–84. [Google Scholar]
- Hartmann C, Smeyers-Verbeke J, Massart DL, McDowall RD. Validation of bioanalytical chromatographic methods. Journal of Pharmaceutical and Biomedical Analysis. 1998;17(2):193–218. doi: 10.1016/S0731-7085(97)00198-2. [DOI] [PubMed] [Google Scholar]
- Hiller K, Hangebrauk J, Jager C, Spura J, Schreiber K, Schomburg D. MetaboliteDetector: Comprehensive analysis tool for targeted and nontargeted GC/MS based metabolome analysis. Analytical Chemistry. 2009;81(9):3429–3439. doi: 10.1021/ac802689c. [DOI] [PubMed] [Google Scholar]
- Hoggard JC, Synovec RE. Automated resolution of nontarget analyte signals in GC x GC-TOFMS data using parallel factor analysis. Analytical Chemistry. 2008;80(17):6677–6688. doi: 10.1021/ac800624e. [DOI] [PubMed] [Google Scholar]
- Horning EC, Horning MG. Metabolic profiles: Gas-phase methods for analysis of metabolites. Clinical Chemistry. 1971;17:802–809. [PubMed] [Google Scholar]
- Hope JL, Prazen BJ, Nilsson EJ, Lidstrom ME, Synovec RE. Comprehensive two-dimensional gas chromatography with time-of-flight mass spectrometry detection: Analysis of amino acid and organic acid trimethylsilyl derivatives, with application to the analysis of metabolites in rye grass samples. Talanta. 2005;65(2):380–388. doi: 10.1016/j.talanta.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Hope JL, Sinha AE, Prazen BJ, Synovec RE. Evaluation of the DotMap algorithm for locating analytes of interest based on mass spectral similarity in data collected using comprehensive two-dimensional gas chromatography coupled with time-of-flight mass spectrometry. Journal of Chromatography. A. 2005;1086(1–2):185–192. doi: 10.1016/j.chroma.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Huang X, Regnier FE. Differential metabolomics using stable isotope labeling and two-dimensional gas chromatography with time-of-flight mass spectrometry. Analytical Chemistry. 2008;80(1):107–114. doi: 10.1021/ac071263f. [DOI] [PubMed] [Google Scholar]
- Humston EM, Dombek KM, Hoggard JC, Young ET, Synovec RE. Time-dependent profiling of metabolites from Snf1 mutant and wild type yeast cells. Analytical Chemistry. 2008;80(21):8002–8011. doi: 10.1021/ac800998j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humston EM, Knowles JD, McShea A, Synovec RE. Quantitative assessment of moisture damage for cacao bean quality using two-dimensional gas chromatography combined with time-of-flight mass spectrometry and chemometrics. Journal of Chromatography. A. 2010;1217(12):1963–1970. doi: 10.1016/j.chroma.2010.01.069. [DOI] [PubMed] [Google Scholar]
- ICH (International conference on harmonisation) (2005). Q2(R1).Validation of analytical procedures: Text and methodology.
- Jellema RH. Variable shift and alignment. In: Stephen DB, Tauler R, Beata W, editors. Comprehensive chemometrics. Oxford: Elsevier; 2009. pp. 85–108. [Google Scholar]
- Jellema, R. H., Krishnan, S., Hendriks, M. M. W. B., Muilwijk, B., & Vogels, J. T. W. E. (2010). Deconvolution using signal segmentation. Chemometics and Intelligent Laboratory Systems, Article in press, corrected proof (doi:10.1016/j.chemolab.2010.07.007).
- Jeong ML, Jiang H, Chen HS, Tsai CJ, Harding SA. Metabolic profiling of the sink-to-source transition in developing leaves of quaking aspen. Plant Physiology. 2004;136(2):3364–3375. doi: 10.1104/pp.104.044776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiye A, Huang Q, Wang G, Zha W, Yan B, Ren H, Gu S, Zhang Y, Zhang Q, Shao F, Sheng L, Sun J. Global analysis of metabolites in rat and human urine based on gas chromatography/time-of-flight mass spectrometry. Analytical Biochemistry. 2008;379(1):20–26. doi: 10.1016/j.ab.2008.04.025. [DOI] [PubMed] [Google Scholar]
- Jonsson N, Gullberg J, Nordstrom A, Kusano M, Kowalczyk M, Sjöström M, Moritz T. A strategy for identifying differences in large series of metabolomic samples analyzed by GC/MS. Analytical Chemistry. 2004;76(6):1738–1745. doi: 10.1021/ac0352427. [DOI] [PubMed] [Google Scholar]
- Jonsson, P., Johansson, A. I., Gullberg, J., Trygg, J., Jiye, A, Grung, B., et al. (2005). High-throughput data analysis for detecting and identifying differences between samples in GC/MS-based metabolomic analyses. Analytical Chemistry, 77(17), 5635–5642. [DOI] [PubMed]
- Jonsson P, Stenlund H, Moritz T, Trygg J, Sjöström M, Verheij ER, Lindberg J, Schuppe-Koistinen I, Antti H. A strategy for modelling dynamic responses in metabolic samples characterized by GC/MS. Metabolomics. 2006;2(3):135–143. doi: 10.1007/s11306-006-0027-1. [DOI] [Google Scholar]
- Knapp DR. Handbook of analytical derivatization reactions. New York: John Wiley & Sons, Inc; 1979. [Google Scholar]
- Koek MM, Bakels F, Engel W, van den Maagdenberg A, Ferrari MD, Coulier L, Hankemeier T. Metabolic profiling of ultrasmall sample volumes with GC/MS: From microliter to nanoliter samples. Analytical Chemistry. 2010;82(1):156–162. doi: 10.1021/ac9015787. [DOI] [PubMed] [Google Scholar]
- Koek MM, Muilwijk B, van Stee LLP, Hankemeier T. Higher mass loadability in comprehensive two-dimensional gas chromatography-mass spectrometry for improved analytical performance in metabolomics analysis. Journal of Chromatography. A. 2008;1186(1–2):420–429. doi: 10.1016/j.chroma.2007.11.107. [DOI] [PubMed] [Google Scholar]
- Koek MM, Muilwijk B, vander Werf MJ, Hankemeier T. Microbial metabolomics with gas chromatography/mass spectrometry. Analytical Chemistry. 2006;78(4):1272–1281. doi: 10.1021/ac051683+. [DOI] [PubMed] [Google Scholar]
- Koek, M., van der Kloet, F., Kleemann, R., Kooistra, T., Verheij, E., & Hankemeier, T. (2010b). Semi-automated non-target processing in GCxGC-MS metabolomics analysis: Applicability for biomedical studies. Metabolomics, online first article, doi:10.1007/s11306-010-0219-6. [DOI] [PMC free article] [PubMed]
- Kuhara T. Diagnosis of inborn errors of metabolism using filter paper urine, urease treatment, isotope dilution and gas chromatography-mass spectrometry: Review. Journal of Chromatography B. 2001;758(1):3–25. doi: 10.1016/S0378-4347(01)00138-4. [DOI] [PubMed] [Google Scholar]
- Kusano M, Fukushima A, Kobayashi M, Hayashi N, Jonsson P, Moritz T, Ebana K, Saito K. Application of a metabolomic method combining one-dimensional and two-dimensional gas chromatography-time-of-flight/mass spectrometry to metabolic phenotyping of natural variants in rice. Journal of Chromatography B. 2007;855(1):71–79. doi: 10.1016/j.jchromb.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Lee D, Fiehn O. High quality metabolomic data for Chlamydomonas reinhardtii. Plant Methods. 2008;4(1):7. doi: 10.1186/1746-4811-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Xu Z, Lu X, Yang X, Yin P, Kong H, Yu Y, Xu G. Comprehensive two-dimensional gas chromatography/time-of-flight mass spectrometry for metabonomics: Biomarker discovery for diabetes mellitus. Analytica Chimica Acta. 2009;633(2):257–262. doi: 10.1016/j.aca.2008.11.058. [DOI] [PubMed] [Google Scholar]
- Lindon CL, Nichelson JK, Holmes E. The handbook of metabonomics and metabolomics. Amsterdam: Elsevier; 2007. [Google Scholar]
- Lommen A. MetAlign: Interface-driven, versatile metabolomics tool for hyphenated full-scan mass spectrometry data preprocessing. Analytical Chemistry. 2009;81(8):3079–3086. doi: 10.1021/ac900036d. [DOI] [PubMed] [Google Scholar]
- Lu, Y., A. J., Wang, G., Hao, H., Huang, Q., Yan, B., et al. (2008). Gas chromatography/time-of-flight mass spectrometry based metabonomic approach to differentiating hypertension- and age-related metabolic variation in spontaneously hypertensive rats. Rapid Communications in Mass Spectrometry, 22(18), 2882–2888. [DOI] [PubMed]
- Ma C, Wang H, Lu X, Xu G, Liu B. Metabolic fingerprinting investigation of Artemisia annua L. in different stages of development by gas chromatography and gas chromatography-mass spectrometry. Journal of Chromatography. A. 2008;1186(1–2):412–419. doi: 10.1016/j.chroma.2007.09.023. [DOI] [PubMed] [Google Scholar]
- Martins AM, Camacho D, Shuman J, Sha W, Mendes P, Shulaev V. A systems biology study of two distinct growth phases of Saccharomyces cerevisiae cultures. Current Genomics. 2004;5(8):649–663. doi: 10.2174/1389202043348643. [DOI] [Google Scholar]
- Matsumoto I, Kuhara T. A new chemical diagnostic method for inborn errors of metabolism by mass spectrometry—Rapid, practical, and simultaneous urinary metabolites analysis. Mass Spectrometry Reviews. 1996;15(1):43–57. doi: 10.1002/(SICI)1098-2787(1996)15:1<43::AID-MAS3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Mills GA, Walker V. Headspace solid-phase microextractio profiling of volatile compounds in urine: Application to metabolic investigations. Journal of Chromatography B. 2001;2001(753):259–268. doi: 10.1016/S0378-4347(00)00554-5. [DOI] [PubMed] [Google Scholar]
- Mohler RE, Dombek KM, Hoggard JC, Pierce KM, Young ET, Synovec RE. Comprehensive analysis of yeast metabolite GC x GC-TOFMS data: Combining discovery-mode and deconvolution chemometric software. Analyst. 2007;132(8):756–767. doi: 10.1039/b700061h. [DOI] [PubMed] [Google Scholar]
- Mohler RE, Dombek KM, Hoggard JC, Young ET, Synovec RE. Comprehensive two-dimensional gas chromatography time-of-flight mass spectrometry analysis of metabolites in fermenting and respiring yeast cells. Analytical Chemistry. 2006;78(8):2700–2709. doi: 10.1021/ac052106o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler RE, Tu BP, Dombek KM, Hoggard JC, Young ET, Synovec RE. Identification and evaluation of cycling yeast metabolites in two-dimensional comprehensive gas chromatography-time-of-flight-mass spectrometry data. Journal of Chromatography. A. 2008;1186(1–2):401–411. doi: 10.1016/j.chroma.2007.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenthal K, Wienkoop S, Scholz M, Selbig J, Weckwerth W. Correlative GC-TOF-MS-based metabolite profiling and LC-MS-based protein profiling reveal time-related systemic regulation of metabolite-protein networks and improve pattern recognition for multiple biomarker selection. Metabolomics. 2005;1(2):109–121. doi: 10.1007/s11306-005-4430-9. [DOI] [Google Scholar]
- Nicholson JK, Lindon JC. Systems biology: Metabonomics. Nature. 2008;455(7216):1054–1056. doi: 10.1038/4551054a. [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Lindon JC, Holmes E. ‘Metabonomics’: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica. 1999;29(11):1181–1189. doi: 10.1080/004982599238047. [DOI] [PubMed] [Google Scholar]
- NIST. (2010). Development of a Standard Reference Material for Metabolites in Plasma, NIST, viewed 14 March 2010, http://www.nist.gov/cstl/analytical/organic/metabolitesinserum.cfm
- O’Hagan S, Dunn WB, Brown M, Knowles JD, Kell DB. Closed-loop, multiobjective optimization of analytical instrumentation: Gas chromatography/time-of-flight mass spectrometry of the metabolomes of human serum and of yeast fermentations. Analytical Chemistry. 2005;77(1):290–303. doi: 10.1021/ac049146x. [DOI] [PubMed] [Google Scholar]
- O’Hagan S, Dunn WB, Knowles JD, Broadhurst D, Williams R, Ashworth JJ, Cameron M, Kell DB. Closed-Loop, multiobjective optimization of two-dimensional gas chromatography/mass spectrometry for serum metabolomics. Analytical Chemistry. 2007;79(2):464–476. doi: 10.1021/ac061443+. [DOI] [PubMed] [Google Scholar]
- Oh C, Huang X, Regnier FE, Buck C, Zhang X. Comprehensive two-dimensional gas chromatography/time-of-flight mass spectrometry peak sorting algorithm. Journal of Chromatography. A. 2008;1179(2):205–215. doi: 10.1016/j.chroma.2007.11.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong ES, Chor CF, Zou L, Ong CN. A multi-analytical approach for metabolomic profiling of zebrafish (Danio rerio) livers. Molecular bioSystems. 2009;5:288–298. doi: 10.1039/b811850g. [DOI] [PubMed] [Google Scholar]
- Pan L, Qiu Y, Chen T, Lin J, Chi Y, Su M, Zhao A, Jia W. An optimized procedure for metabonomic analysis of rat liver tissue using gas chromatography/time-of-flight mass spectrometry. Journal of Pharmaceutical and Biomedical Analysis. 2010;52(4):589–596. doi: 10.1016/j.jpba.2010.01.046. [DOI] [PubMed] [Google Scholar]
- Pasikanti KK, Ho PC, Chan EC. Development and validation of a gas chromatography/mass spectrometry metabonomic platform for the global profiling of urinary metabolites. Rapid Communications in Mass Spectrometry. 2008;22:2984–2992. doi: 10.1002/rcm.3699. [DOI] [PubMed] [Google Scholar]
- Pauling L, Robinson AB, Teranishi R, Cary P. Quantitative analysis of urine vapor and breath by gas-liquid partition chromatography. Proceedings of the National Academy of Sciences of the United States of America. 1971;68(10):2374–2376. doi: 10.1073/pnas.68.10.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters FT, Maurer HH. Bioanalytical method validation and its implications for forensic and clinical toxicology—A review. Accreditation and Quality Assurance. 2002;7(11):441–449. doi: 10.1007/s00769-002-0516-5. [DOI] [Google Scholar]
- Pierce KM, Hoggard JC, Hope JL, Rainey PM, Hoofnagle AN, Jack RM, Wright BW, Synovec RE. Fisher ratio method applied to third-order separation data to identify significant chemical components of metabolite extracts. Analytical Chemistry. 2006;78(14):5068–5075. doi: 10.1021/ac0602625. [DOI] [PubMed] [Google Scholar]
- Pierce KM, Hope JL, Hoggard JC, Synovec RE. A principal component analysis based method to discover chemical differences in comprehensive two-dimensional gas chromatography with time-of-flight mass spectrometry (GC x GC-TOFMS) separations of metabolites in plant samples. Talanta. 2006;70(4):797–804. doi: 10.1016/j.talanta.2006.01.038. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Su M, Liu Y, Chen M, Gu J, Zhang J, Jia W. Application of ethyl chloroformate derivatization for gas chromatography-mass spectrometry based metabonomic profiling. Analytica Chimica Acta. 2007;583(2):277–283. doi: 10.1016/j.aca.2006.10.025. [DOI] [PubMed] [Google Scholar]
- Ralston-Hooper K, Hopf A, Oh C, Zhang X, Adamec J, lveda MS. Development of GCxGC/TOF-MS metabolomics for use in ecotoxicological studies with invertebrates. Aquatic Toxicology. 2008;88(1):48–52. doi: 10.1016/j.aquatox.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Roessner U, Luedemann A, Brust D, Fiehn O, Linke T, Willmitzer L, Fernie AR. Metabolic profiling allows comprehensive phenotyping of genetically or environmentally modified plant systems. Plant Cell. 2001;13(1):11–29. doi: 10.1105/tpc.13.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessner U, Wagner C, Kopka J, Trethewey RN, Willmitzer L. Simultaneous analysis of metabolites in potato tuber by gas chromatography-mass spectrometry. The Plant Journal. 2000;23(1):131–142. doi: 10.1046/j.1365-313x.2000.00774.x. [DOI] [PubMed] [Google Scholar]
- Roessner U, Willmitzer L, Fernie AR. High-resolution metabolic phenotyping of genetically and environmentally diverse potato tuber systems. Identification of phenocopies. Plant Physiology. 2001;127(3):749–764. doi: 10.1104/pp.010316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubingh CM, Bijlsma S, Jellema RH, Overkamp KM, van der Werf MJ, Smilde AK. Analyzing longitudinal microbial metabolomics data. Journal of Proteome Research. 2009;8(9):4319–4327. doi: 10.1021/pr900126e. [DOI] [PubMed] [Google Scholar]
- Sangster T, Major H, Plumb R, Wilson AJ, Wilson ID. A pragmatic and readily implemented quality control strategy for HPLC-MS and GC-MS-based metabonomic analysis. Analyst. 2006;131(10):1075–1078. doi: 10.1039/b604498k. [DOI] [PubMed] [Google Scholar]
- Schauer N, Steinhauser D, Strelkov S, Schomburg D, Allison G, Moritz T, Lundgren K, Roessner-Tunali U, Forbes MG, Willmitzer L, Fernie AR, Kopka J. GC-MS libraries for the rapid identification of metabolites in complex biological samples. FEBS Letters. 2005;579(6):1332–1337. doi: 10.1016/j.febslet.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Schmarr HG, Bernhardt J. Profiling analysis of volatile compounds from fruits using comprehensive two-dimensional gas chromatography and image processing techniques. Journal of Chromatography. A. 2010;1217(4):565–574. doi: 10.1016/j.chroma.2009.11.063. [DOI] [PubMed] [Google Scholar]
- Shellie RA, Welthagen W, Zrostlikova J, Spranger J, Ristow M, Fiehn O, Zimmermann R. Statistical methods for comparing comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry results: Metabolomic analysis of mouse tissue extracts. Journal of Chromatography. A. 2005;1086(1–2):83–90. doi: 10.1016/j.chroma.2005.05.088. [DOI] [PubMed] [Google Scholar]
- Sinha AE, Hope JL, Prazen BJ, Fraga CG, Nilsson EJ, Synovec RE. Multivariate selectivity as a metric for evaluating comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry subjected to chemometric peak deconvolution. Journal of Chromatography. A. 2004;1056(1–2):145–154. [PubMed] [Google Scholar]
- Sinha AE, Hope JL, Prazen BJ, Nilsson EJ, Jack RM, Synovec RE. Algorithm for locating analytes of interest based on mass spectral similarity in GC x GC-TOF-MS data: Analysis of metabolites in human infant urine. Journal of Chromatography. A. 2004;1058(1–2):209–215. [PubMed] [Google Scholar]
- Strelkov S, von Elstermann M, Schomburg D. Comprehensive analysis of metabolites in Corynebacterium glutamicum by gas chromatography/mass spectrometry. Biological Chemistry. 2004;385(9):853–861. doi: 10.1515/BC.2004.111. [DOI] [PubMed] [Google Scholar]
- Styczynski MP, Moxley JF, Tong LV, Walther JL, Jensen KL, Stephanopoulos GN. Systematic identification of conserved metabolites in gc/ms data for metabolomics and biomarker discovery. Analytical Chemistry. 2007;79(3):966–973. doi: 10.1021/ac0614846. [DOI] [PubMed] [Google Scholar]
- Thompson M, Ellison SLR, Wood R. Harmonized guidlines for single-laboratory validation of methods of analysis (IUPAC Technical Report) Pure and Applied Chemistry. 2002;74(5):835–855. doi: 10.1351/pac200274050835. [DOI] [Google Scholar]
- Tian J, Shi C, Gao P, Yuan K, Yang D, Lu X, Xu G. Phenotype differentiation of three E. coli strains by GC-FID and GC-MS based metabolomics. Journal of chromatography. B, Analytical Technologies in the Biomedical and Life Sciences. 2008;871(2):220–226. doi: 10.1016/j.jchromb.2008.06.031. [DOI] [PubMed] [Google Scholar]
- Tianniam S, Tarachiwin L, Bamba T, Kobayashi A, Fukusaki E. Metabolic profiling of Angelica acutiloba roots utilizing gas chromatography-time-of-flight-mass spectrometry for quality assessment based on cultivation area and cultivar via multivariate pattern recognition. Journal of Bioscience and Bioengineering. 2008;105(6):655–659. doi: 10.1263/jbb.105.655. [DOI] [PubMed] [Google Scholar]
- Tikunov Y, Lommen A, de Vos CH, Verhoeven HA, Bino RJ, Hall RD, Bovy AG. A novel approach for nontargeted data analysis for metabolomics. Large-scale profiling of tomato fruit volatiles. Plant Physiology. 2005;139(3):1125–1137. doi: 10.1104/pp.105.068130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), & Center for Veterinary Medicine (CVM). (2001). Guidance for industry; bioanalytical method validation.
- van der Greef J, Martin S, Juhasz P, Adourian A, Plasterer T, Verheij ER, McBurney RN. The art and practice of systems biology in medicine: Mapping patterns of relationships. Journal of Proteome Research. 2007;6(4):1540–1559. doi: 10.1021/pr0606530. [DOI] [PubMed] [Google Scholar]
- van der Greef J, Smilde AK. Symbiosis of chemometrics and metabolomics: Past, present, and future. Journal of Chemometrics. 2005;19(5–7):376–386. doi: 10.1002/cem.941. [DOI] [Google Scholar]
- van der Greef J, Stroobant P, van der Heijden R. The role of analytical sciences in medical systems biology. Current Opinion in Chemical Biology. 2004;8(5):559–565. doi: 10.1016/j.cbpa.2004.08.013. [DOI] [PubMed] [Google Scholar]
- van der Greef, J., Vogels, J. T. W. E., Wulfert, F., & Tas, A. C. (2004b). Method and system for identifying and quantifying chemical components of a mixture, 2004267459 (patent).
- van der Kloet FM, Bobeldijk I, Verheij ER, Jellema RH. Analytical error reduction using single point calibration for accurate and precise metabolomic phenotyping. Journal of Proteome Research. 2009;8(11):5132–5141. doi: 10.1021/pr900499r. [DOI] [PubMed] [Google Scholar]
- Vikram A, Prithiviraj B, Hamzehzarghani H, Kushalappa A. Volatile metabolite profiling to discriminate diseases of McIntosh apple inoculated with fungal pathogens. Journal of the Science of Food and Agriculture. 2004;84(11):1333–1340. doi: 10.1002/jsfa.1828. [DOI] [Google Scholar]
- Villas-Bôas JM, Delicado DG, kesson M, Nielsen J. Simultaneous analysis of amino and nonamino organic acids as methyl chloroformate derivatives using gas chromatography-mass spectrometry. Analytical Biochemistry. 2003;322(1):134–138. doi: 10.1016/j.ab.2003.07.018. [DOI] [PubMed] [Google Scholar]
- Villas-Bôas JM, kesson M, Smedsgaard J, Nielsen J. Global metabolite analysis of yeast: Evaluation of sample preparation methods. Yeast. 2005;22(14):1155–1169. doi: 10.1002/yea.1308. [DOI] [PubMed] [Google Scholar]
- Wagner C, Sefkow M, Kopka J. Construction and application of a mass spectral and retention time index database generated from plant GC/EI-TOF-MS metabolite profiles. Phytochemistry. 2003;62(6):887–900. doi: 10.1016/S0031-9422(02)00703-3. [DOI] [PubMed] [Google Scholar]
- Weckwerth W, Loureiro ME, Wenzel K, Fiehn O. Differential metabolic networks unravel the effects of silent plant phenotypes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(20):7809–7814. doi: 10.1073/pnas.0303415101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckwerth W, Wenzel K, Fiehn O. Process for the integrated extraction identification, and quantification of metabolites, proteins and RNA to reveal their co-regulation in biochemical networks. Proteomics. 2004;4(1):78–83. doi: 10.1002/pmic.200200500. [DOI] [PubMed] [Google Scholar]
- Welthagen W, Shellie RA, Spranger J, Ristow M, Zimmermann R, Fiehn O. Comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry (GC x GC-TOF) for high resolution metabolomics: Biomarker discovery on spleen tissue extracts of obese NZO compared to lean C57BL/6 mice. Metabolomics. 2005;1(1):65–73. doi: 10.1007/s11306-005-1108-2. [DOI] [Google Scholar]
- Westerhuis JA, Hoefsloot HCJ, Smit S, Vis DJ, Smilde AK, van Velzen EJJ, van Duijnhoven JPM, van Dorsten FA. Assessment of PLSDA cross validation. Metabolomics. 2008;4(1):81–89. doi: 10.1007/s11306-007-0099-6. [DOI] [Google Scholar]
- Windig W, Smith WF. Chemometric analysis of complex hyphenated data: Improvements of the component detection algorithm. Journal of Chromatography. A. 2007;1158(1–2):251–257. doi: 10.1016/j.chroma.2007.03.081. [DOI] [PubMed] [Google Scholar]
- Wishart, D. S., Knox, C., Guo, A. C., Eisner, R., Young, N., Gautam, B., et al. (2009). HMDB: A knowledgebase for the human metabolome. Nucleic Acids Research, 37(Database issue), D603–D610. [DOI] [PMC free article] [PubMed]
- Wishart DS, Lewis MJ, Morrissey JA, Flegel MD, Jeroncic K, Xiong Y, Cheng D, Eisner R, Gautam B, Tzur D, Sawhney S, Bamforth F, Greiner R, Li L. The human cerebrospinal fluid metabolome. Journal of Chromatography B. 2008;871(2):164–173. doi: 10.1016/j.jchromb.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Wishart, D. S., Tzur, D., Knox, C., Eisner, R., Guo, A. C., Young, N., et al. (2007). HMDB: The human metabolome database. Nucleic Acids Research,35(Suppl. 1), D521–D526. [DOI] [PMC free article] [PubMed]
- Zhang Q, Wang G, Du Y, Zhu L, Jiye A. GC/MS analysis of the rat urine for metabonomic research. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences. 2007;854(1–2):20–25. doi: 10.1016/j.jchromb.2007.03.048. [DOI] [PubMed] [Google Scholar]