

Abstract
Somatic mutations of FLT3 involving internal tandem duplication (ITD) of the juxtamembrane domainor point mutations in the kinase domain (TKD) appear to activate FLT3 in a FLT3 ligand (FL) - independent manner. To determine whether or not FLT3 mutants respond to FL for their activation, a FL-deficient (FL−/−) murine embryo fibroblast cell line (MEF) was established. Expression of FLT3/ITD and FLT3/TKD mutations in FL−/− MEF cells resulted in low levels of constitutive phosphorylation of FLT3.However, a more than 4-fold increase of FLT3 autophosphorylation was induced by exogenous FL. Rescue of endogenous FL expression in FL−/− MEF cells expressing FLT3 mutants led to more than a 3-fold increase of FLT3 phosphorylation. FL addition led to further activation of the FLT3 receptors and enhanced survival and/or decreased apoptosis in leukemia-derived cell lines and primary leukemic cells expressing FLT3 mutations. Functional studies revealed that exogenous FL promoted the colony-forming and recloning abilities of FLT3 mutant transduced primary bone marrow cells derived from FL−/− mice. Endogenous FL contributes in vivo to functional signaling through FLT3 as noted by the decreased survival of FL+/+ITD+/+ mice compared with FL−/−ITD+/+ mice. These data suggest that FL leads to further activation of FLT3 mutants and is especially important in light of recent findings of elevated FL levels in AML patients in response to chemotherapy.
Keywords: FLT3, Tyrosine kinase receptor, FLT3 ligand, Internal tandem duplication mutation, point mutation
Introduction
FLT3 is a member of the class III receptor tyrosine kinase family (Gilliland and Griffin, 2002). It is normally expressed by CD34 + hematopoietic stem/progenitor cells, dendritic cells, brain, placenta and gonads (Gilliland and Griffin). In leukemia, FLT3 expression is observed at the cells of most patients with acute myeloid leukemia (AML), B-precursor acute lymphoblastic leukemia (ALL), blast crisis chronic myelocytic leukemia (CML), and a smaller percentage of patients with T-ALL (Birg et al., 1992; Drexler et al., 1996; Carow et al., 1996). The ligand for FLT3 receptor is expressed by most leukemia/lymphoma-derived cell lines and primary AML cells (Drexler et al., 1996; Meierhoff et al., 1995; Brasel et al., 1995; Zheng et al, 2004). Interaction of wtFLT3 with FL causes receptor dimerization, leading to the activation of its tyrosine kinase domain and autophosphorylation (Lyman et al., 1993). Activated FLT3 transduces signals through phosphorylation of phospholipase C; casitas B-lineage lymphoma (CBL); Src homology 2 domain-containing phosphotyrosine phosphotase 2 (SHP2); Src homology 2 domain-containing inositol 5-phosphatase (SHIP); signal transducer and activator of transcription 5 (STAT5); and the Ras/Raf/MAPK and PI3 kinase pathways (Dosil, et al., 1993; Rosnet et al., 1996; Zhang, et al., 2000; Zhang, et al., 2000; Lavagna-Sevenier et al., 1998; Zhang et al., 1999; Marchetto et al., 1999). Activation of FLT3 by exogenous FL stimulates the proliferation of primary AML blasts (Lisovsky et al., 1996). FL also induces autocrine signaling in AML (Zheng, et al., 2004).
FLT3/ITD mutations are present in approximately 24% of patients with AML, while mutations in the activation loop of the tyrosine kinase domain (TKD) occurs in about 8% of patients with AML (Gilliland and Griffin, 2002). Both types of mutations have been reported to lead to constitutive activation of FLT3 in a ligand-independent manner with resultant transformation of hematopoietic cell lines (Fenski et al., 2000; Yamamoto et al.,2001; Tse et al., 2000). FLT3/ITD mutations are associated with leukocytosis in AML and with leukemic transformation of myelodysplasia (Kiyoi et al., 1997; Horrike et al., 1997; Meshinchi et al., 2001). The FLT3/ITD mutations, in particular, are associated with poor prognosis in patients with AML (Iwai et al., 1999; Kiyoi et al., 1999).
The current body of data regarding the FL-independent activation of FLT3 receptors by FLT3/ITD and FLT3/TKD mutations is derived from transfection of these mutant receptors into cell lines, most of which express FL. To investigate whether FL plays a role in the activation of FLT3 mutant receptors, we established FL deficient MEF cells from FL knockout mice. FLT3 mutants and wtFLT3 were introduced into the FL−/−cells to enable us to test the effects of FL on the different type of FLT3 receptors. FL addition augmented phosphorylation of FLT3 mutants and some downstream signaling proteins. FL stimulation also led to increased survival and anti-apoptosis in these cells. Consistent with in vitro data, FL+/+ITD+/+ mice have decreased survival compared with FL−/−ITD+/+ mice. Recent studies have demonstrated several log induction of FL levels in patients undergoing chemotherapy for AML (Sato et al., 2011). Our results suggest that FL augments the activation of FLT3 mutants, which is associated with increased proliferation and prolonged survival.
Results
FL addition leads to increased phosphorylation of FLT3 in FL−/− MEF cells expressing wtFLT3, FLT3/ITD, and FLT3/TKD mutants
Most of the previously published data establishing the paradigm that FLT3 mutants cause activation of FLT3 receptors in a ligand-independent manner were derived from experiments using cell lines expressing FL. In addition, all the experiments were performed in culture medium with serum which contains FL. This leaves open the possibilities that either ligand expressed by these cells could be having an effect through autocrine, paracrine, or intracrine activation of FLT3 mutants or that ligand in the serum could contribute to the activation of the mutant receptors. To eliminate the complicating factor of autocrine, paracrine, and intracrine FL expression on mutant FLT3 activation, FL−/− MEF cells were established from FL−/− mice. The FL deficiency in the established FL−/− MEF bulk cells was confirmed by RT-PCR analysis (Fig 1A). Absence of murine wtFLT3 expression was confirmed by Western blotting (data not shown). The FL−/− MEF cells were then transduced by a lentiviral vector containing the wt, ITD or TKD forms of FLT3 and clones were selected by limited dilution. Stable expression was confirmed by Western blotting (Fig 1B). Cells were then cultured in serum-free medium (to eliminate the effect of FL contained in serum) for 16 hours before stimulation with and without FL. The addition of FL induced more than a 4-fold increase in FLT3 phosphorylation in FL−/−MEF-FLT3, FL−/− MEF-ITD, and FL−/− MEF-TKD cells [quantitated by GS-800 Calibrated Densitometer (Bio-Rad) and Quantity One software (Bio-Rad)] (Fig 1B, lanes 4, 6, and 8 versus lanes 3, 5, and 7). There was no phosphorylation of FLT3 in FL−/−MEF-FLT3 and the phosphorylation of the FLT3 mutants in FL−/− MEF cells was significantly less in the absence of FL stimulation (Fig 1B, lanes 3, 5, and 7). Similar results were obtained from experiments using both bulk cells (Fig 1B) and additional clones (data not shown). These data suggest that the addition of FL leads to increased activation of FLT3/ITD and FLT3/TKD receptors.
Fig 1. FL addition leads to increased phosphorylation of FLT3 in FL−/− MEF cells expressing FLT3/ITD or TKD mutants.


(A)Total cellular RNA was extracted from cell lines. RNA (50–100 ng) was reverse transcribed and amplified using primer pairs for murine FL and actin. PCR products were resolved on 1% agarose gel in the presence of ethidium bromide, and photographs were taken under UV transillumination.
(B) Cells were cultured in serum-free medium for 16 hours, washed once with PBS and stimulated with FL (100 ng/mL) for 15 minutes. Total cellular protein extracts derived from 1 × 107 cells were immunoprecipitated with anti-FLT3 antibody. The immunoprecipitates were resolved by 8% SDS-PAGE and subjected to immunoblot analysis with the antiphosphotyrosine antibody 4G10 (top panel). The same blot was stripped and reprobed with anti-FLT3 antibody (bottom panel).
We next studied the effect of endogenous FL expression on the activation of FLT3/ITD and FLT3/TKD mutant receptors. FL was introduced into different clones of FL−/− MEF-FLT3, FL−/− MEF-ITD, and FL−/− MEF-TKD cells by lentiviral transduction to simulate FL expression by leukemia blasts and cell lines. The expression of FL was assessed by the detection of the secreted form of FL in the cell culture supernatant. The level of secreted FL was comparable in FL−/− MEF-FLT3, FL−/− MEF-ITD, and FL−/− MEF-TKD cells expressing FL (Fig 2A). These cells were cultured in serum-free medium followed by stimulation with and without FL. Endogenous expression of FL resulted in a more than 3-fold increase in FLT3 phosphorylation in FL−/− MEF-FLT3, FL−/− MEF-ITD, and FL−/− MEF-TKD cells in the absence of exogenous FL (Fig 2B, lanes 3, 7, and 11 vs 1, 5, and 9). The addition of exogenous FL (100ng/mL) did not further augment FLT3 phosphorylation in FL−/− MEF-FLT3, FL−/− MEF-ITD, or FL−/− MEF-TKD cells expressing endogenous FL (Fig 2B, lanes 4, 8, and 12 vs lanes 3, 7, and 11). These results strongly suggest that endogenous FL expression strongly contributes to the observed activation of mutant FLT3 receptors.
Fig 2. Stable FL expression results in enhanced FLT3 phosphorylation in FL−/− MEF cells expressing FLT3/ITD and FLT3/TKD mutants.


(A) FL−/− MEF cells expressing wt and mutant forms of FLT3 with and without stable expression of FL were washed with PBS, plated at 5×105 cells/mL, and cultured for 24 hours in serum-free medium. Cell culture supernatant was then collected by centrifugation and FL concentration was assessed by ELISA assay.
(B) Cells were cultured in fresh serum free medium for 16 hours and washed with PBS once before stimulation with FL (100 ng/mL for 15 minutes). Total cellular protein extracts derived from 1 × 107 cells were immunoprecipitated with anti-FLT3 antibody. The immunoprecipitates were resolved by 8% SDS-PAGE and subjected to immunoblot analysis with the antiphosphotyrosine antibody 4G10 (top panel). The same blot was stripped and reprobed with anti-FLT3 antibody (bottom panel).
Exogenous FL augments the phosphorylation of FLT3 and enhances survival and resistance to apoptosis in cell lines expressing FLT3 mutants
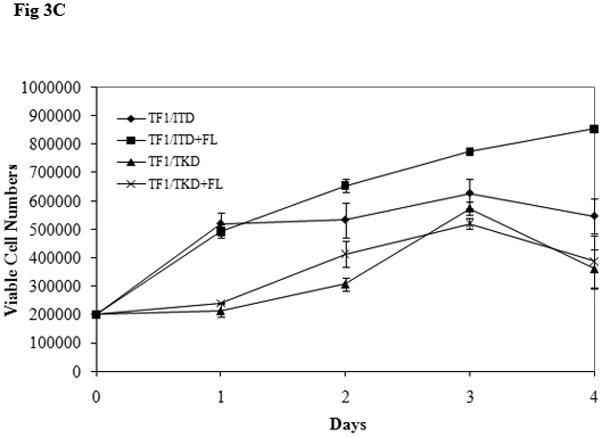
The possibility of further activation of FLT3 mutant receptors by exogenous FL was also tested in cell lines in which FL autocrine, paracrine, or intracrine stimulation of FLT3 receptor might exist. TF1 cells that do not express FLT3 receptor were transfected with constructs containing mutant forms of FLT3. TF1 cells expressing FLT3 mutant receptors were starved for 16 hours in serum-free medium and washed once with PBS to deplete soluble FL (either produced by the cells or contained in the serum). These cells were then stimulated with exogenous FL for 15 minutes. More than 2-fold enhancement of FLT3 phosphorylation was detected in both TF1/ITD and TF1/TKD cell lines (Fig 3A). Slightly increased STAT5 phosphorylation (1.8-fold increase in both TF1/ITD and TF1/TKD), AKT phosphorylation, and AKT protein expression was also observed after FL stimulation (Fig 3A). To assess the functional effect of exogenous FL, TF1/ITD and TF1/TKD cells were cultured in serum-free medium in the absence and presence of FL for 48 hours. Apoptosis was assessed by Annexin V and 7AAD staining followed by FACS analysis. The presence of FL decreased the apoptotic population from 61.27% to 49.54% and from 40.2% to 31.63% in TF1/ITD and TF1/TKD cells, respectively (Fig 3B). There was no change of cell cycle status in response to FL in these cells (data not shown). Survival assays performed by counting viable cell numbers revealed that exogenous FL increased the survival of TF1/ITD cells cultured in serum free medium (Fig 3C, P<0.05). However, no significant difference was observed in TF1/TKD cells in the presence and absence of FL (Fig 3C). This data suggests that the presence of FL augments the activation of FLT3/ITD mutant receptors and can contribute to resistance to apoptosis.
Fig 3. Addition of FL results in an increased phosphorylation of FLT3 and resistance to apoptosis in TF1 cells expressing FLT3/ITD and FLT3/TKD mutations.


(A) Cells were cultured in serum-free medium for 16 hours and washed once with PBS before stimulation with FL (100 ng/mL for 15 minutes). Total cellular protein extracts derived from 1 × 107 cells were immunoprecipitated with anti-FLT3 antibody. The immunoprecipitates were resolved by 8% SDS-PAGE and subjected to immunoblot analysis with the antiphosphotyrosine antibody 4G10. The same blot was stripped and reprobed with anti-FLT3 antibody. Total protein lysates (50 μg) were resolved by 10% SDS-PAGE and subjected to immunoblot analysis with anti-phosphoSTAT5, anti-phosphoAKT, and anti-phosphoMAPK antibody. The same blot was stripped and reprobed with anti-STAT5, anti-AKT, anti-MAPK, and anti-HSP90 antibodies.
(B) Cells were cultured in serum-free medium for 48 hours in the presence and absence of FL (100 ng/mL). The cells were then stained with Annexin V and 7AAD followed by FACS analysis. The result shown here is representative of three independent experiments.
(C) Cells were cultured in serum free medium for 4 days in the presence and absence of FL (100 ng/mL). Viable cell numbers were assessed by trypan blue exclusion assay. The result shown here is representative of three independent experiments (Bars represent standard deviation in the figure).
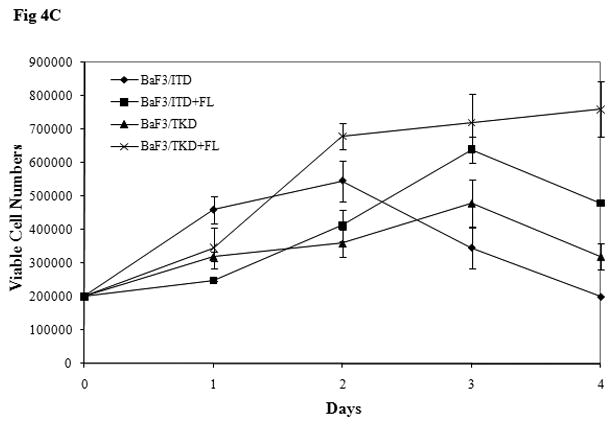
BaF3 is another cell line lacking expression of endogenous FLT3. BaF3 cells transfected with mutant forms of FLT3 were starved in serum-free medium for 16 hours and washed with PBS before FL stimulation. The addition of FL increased FLT3 phosphorylation in both BaF3/ITD and BaF3/TKD cells. An increase of MAPK phosphorylation was also observed (Fig 4A). In both BaF3/ITD and BaF3/TKD cells, exogenous FL also led to slightly increased phosphorylation of AKT (1.3 and 2.2-fold increase in BaF3/ITD and BaF3/TKD, respectively, Fig 4A). Changes of other FLT3 downstream signals were not observed. An MTT survival assay was performed in BaF3/ITD and BaF3/TKD cells cultured in serum-free medium in the presence and absence of FL for 4 days. FL addition resulted in increased survival of BaF3/ITD and BaF3/TKD cells (Fig 4B, P < 0.05). The effect was also confirmed by counting viable cell numbers by trypan blue exclusion (Fig 4C, P<0.05). These findings suggest that FL stimulation prolongs the survival of cells expressing FLT3 mutants.
Fig 4. Exogenous FL augments FLT3 phosphorylation and provides a survival advantage to BaF3 cells expressing FLT3 mutants.


(A) Cells were cultured in serum-free medium for 16 hours and washed once with PBS once before stimulation with FL (100 ng/mL for 15 miutes). Total cellular protein extracts derived from 1 × 107 cells were immunoprecipitated with anti-FLT3 antibody. The immunoprecipitates were resolved by 8% SDS-PAGE and subjected to immunoblot analysis with antiphosphotyrosine antibody 4G10. The same blot was stripped and reprobed with anti-FLT3 antibody. Total protein lysates (50 μg) were resolved by 10% SDS-PAGE and subjected to immunoblot analysis with anti-phosphoSTAT5, anti-phosphoAKT, and anti-phosphoMAPK antibody. The same blot was stripped and reprobed with anti-STAT5, anti-AKT, anti-MAPK, and anti-HSP90 antibodies.
(B) Cells were cultured in serum free medium for 4 days in the presence and absence of FL (100 ng/mL). Survival was assessed by MTT assay. The result shown here is representative of three independent experiments (Bars represent standard deviation in the figure).
(C) Cells were cultured in serum free medium for 4 days in the presence and absence of FL (100 ng/mL). Viable cell numbers were assessed by trypan blue exclusion assay. The result shown here is representative of three independent experiments (Bars represent standard deviation in the figure).
The MV411 cell line is a human leukemia-derived cell line expressing a FLT3/ITD mutation and has lost the wild-type allele. In agreement with the results seen in TF1 and BaF3 transfectants, exogenous FL resulted in increased phosphorylation of FLT3 in MV411 cells (Fig 5A). Slightly enhanced phosphorylation of FLT3 downstream signaling, including STAT5 and AKT, was also detected after FL stimulation (Fig 5A). MV411 cells express endogenous FL that might diminish the degree of the effect of exogenous FL on FLT3/ITD mutant receptor. To evaluate the effect of FL on the survival of MV411 cells, MV411 cells were cultured in serum-free medium in the presence and absence of FL for 4 days. FL addition led to prolonged survival of MV411 cells (Fig 5B, P < 0.05).
Fig 5. Addition of FL leads to full activation of FLT3 receptors and increased survival of MV411 cells.


(A) Cells were cultured in serum-free medium for 16 hours and washed once with PBS once before stimulation with FL (100 ng/mL for 15 minutes). Total cellular protein extracts derived from 1 × 107 cells were immunoprecipitated with anti-FLT3 antibody. The immunoprecipitates were resolved by 8% SDS-PAGE and subjected to immunoblot analysis with antiphosphotyrosine antibody 4G10. The same blot was stripped and reprobed with anti-FLT3 antibody. Total protein lysates (50 μg) were resolved by 10% SDS-PAGE and subjected to immunoblot analysis with anti-phosphoSTAT5, anti-phosphoAKT, and anti-phosphoMAPK antibody. The same blot was stripped and reprobed with anti-STAT5, anti-AKT, anti-MAPK, and anti-HSP90 antibodies.
(B) Cells were cultured in serum free medium for 4 days in the presence and absence of FL (100 ng/mL). The survival of the cells was assessed by MTT assay. The result shown here is representative of three independent experiments (Bars represent standard deviation in the figure).
FL addition results in increased phosphorylation of mutant FLT3 receptors and prolongs survival of primary AML blasts expressing homozygous FLT3/ITD mutations
To study whether further activation of mutant FLT3 receptors in response to FL also occurs in primary cells, experiments were carried on primary AML blasts expressing homozygous FLT3/ITD mutations (i.e., no detectable wild type FLT3 by PCR). The rate of homozygous FLT3/ITD mutations is very low (<3% of AML cases) and we had access to only 4 samples that were in sufficient quantities to thaw with adequate viability and recovery of sufficient protein to carry out the Western blots. The blasts were stimulated with 100 ng/mL FL. Consistent with what we observed in the FLT3 mutant cell lines, exogenous FL caused enhanced FLT3 phosphorylation (Fig 6A). Increased MAPK phosphorylation was also detected in two out of four samples (patient #1 and 2). The survival of AML blasts was assessed by culturing them in complete medium with and without FL for 4 days. The presence of FL increased the survival of AML blasts derived from three of four patient samples (Fig 6B, P < 0.05 in patient #2, 3 and 4). Possible mutations involving genes other than FLT3 could occur in patient #1 and overshadow the biologic effect of FL. Relatively high level expression of endogenous FL might be another possibility that could diminish the effect of added FL.
Fig 6. FL addition results in increased phosphorylation of FLT3 receptors and prolongs survival of primary AML blasts expressing homozygous FLT3/ITD mutations.


(A) Frozen AML blasts were thawed and stimulated with 100 ng/mL FL for 15 minutes in serum-free medium. Total cellular protein extracts were immunoprecipitated with anti-FLT3 antibody. The immunoprecipitates were resolved by 8% SDS-PAGE and subjected to immunoblot analysis with the antiphosphotyrosine antibody 4G10. The same blot was stripped and reprobed with anti-FLT3 antibody. Total protein lysates (50 μg) were resolved by 10% SDS-PAGE and subjected to immunoblot analysis with anti-phosphoSTAT5, anti-phosphoAKT, and anti-phosphoMAPK antibodies. The same blot was stripped and reprobed with anti-STAT5, anti-AKT, anti-MAPK, and anti-HSP90 antibodies.
(B) Frozen AML blasts were thawed and cultured in complete medium for 16 hours, separated by Ficoll-Hypague density gradient centrifugation, and incubated in fresh medium containing 10% FBS with and without FL (100ng/mL) for 4 days. Survival of the cells was assessed by MTT assay. The result shown here is representative of two independent experiments (Bars represent standard deviation in the figure).
FL stimulation increases the clonogenic potential of FL−/− primary hematopoietic cells expressing FLT3/ITD or FLT3/TKD mutations
To evaluate the effect of FL stimulation on primary hematopoietic progenitor cells expressing FLT3 mutants, lineage negative progenitors were harvested from FL−/−mice and transduced with FLT3/ITD-GFP, FLT3/TKD-GFP, or GFP control lentivirus. GFP-positive cells were selected by FACS, cultured in methylcellulose with and without FL, and the colony numbers were counted after 9 days. The number of first–generation colonies did not differ significantly between ITD, TKD and control vector transduced bone marrow (BM) cells when cultured in the absence of FL stimulation (Fig 7). Exogenous FL slightly increased the number of first-generation colonies derived from BM cells transduced with FLT3 mutants (Fig 7).
Fig 7. Exogenous FL increases the clonogenic potential of FL−/− primary hematopoietic stem/progenitor cells expressing FLT3/ITD and FLT3/TKD mutations.

Bone marrow was harvested from FL−/− mice, lineage depleted, and transduced with EF1-ITD-UBC-GFP, EF1-TKD-UBC-GFP, or EF1-UBC-GFP control virus by spin-infection. GFP positive cells were then sorted by FACS. 103 cells were plated into triplicate 35 mm Petri dishes in 1 ml of Methocult M3434. Colonies consisting of >35 cells were scored after 9 days. For colony-replating all cells from the previous culture were collected and replated at 1×104 cells/plate in fresh methylcellulose medium. Cultures were replated three times and designated as 2nd plating, 3rd plating and 4th plating in the figure. The result shown here is representative of two independent experiments (Bars represent standard deviation in the figure).
After colonies were counted, all of the cells in each culture dish were harvested from the methylcellulose, washed and re-plated in fresh methylcellulose medium to examine their potential to form colonies upon secondary plating; this process was repeated for two more passages to determine the frequency of tertiary and quaternary colonies. Control FL−/− BM cells transduced with GFP control vector alone cultured in the absence and presence of FL exhausted virtually all of their proliferative potential by the fourth passage (Fig 7). Upon serial re-plating, the number of colonies from FLT3/ITD and FLT3/TKD transduced FL−/− BM cells cultured in the absence of FL also decreased with each passage (Fig 7). In contrast, BM cells from FL−/− mice transduced with FLT3 mutants and cultured in the presence of FL retained their re-plating activity and continued to produce colonies despite repeated passages (Fig 7, P < 0.05). Besides the increase in colony numbers, there was also a significant increase in colony size from FL−/− BM cells expressing FLT3 mutants stimulated with FL (data not shown).
These results indicate that FL stimulation is able to confer a proliferative advantage and immortalized potential on hematopoietic progenitors expressing FLT3 mutants.
Endogenous FL shortens the survival of FL+/+ITD+/+ mice in comparison with FL−/−ITD+/+ mice
The work by Li, et al demonstrated that FLT3/ITD knock-in mice developed myeloproliferative disease (MPD) which eventually progressed to mortality by 6 to 20 months (Li et al., 2008). To determine whether or not expression of FL would have any functional effect on survival, the FL−/−FLT3wt/wt mice were crossed with FL+/+FLT3wt/ITD mice followed by cross of the second generation double heterozygotes to generate FL+/+ITD+/+, and FL−/−ITD+/+ mice. 51 FL+/+ITD+/+ mice and 32 FL−/−ITD+/+ mice were followed for the development of fatal MPD and the median survival of the two groups were 187 and 256 days, respectively (P<0.05, Fig 8). Thus, the presence of endogenous FL significantly decreased the survival of homozygous ITD mice (FL+/+ITD+/+ mice vs FL−/−ITD+/+ mice, P<0.05). The majority of the mice in both groups developed splenomegaly and leukocytosis (data not shown). However, the spleen weight and white blood cell count did not differ significantly between the two groups (P<0.05). By determination of the phenotype by FACS analysis of the bone marrow, in conjunction with blood counts, spleen weight and bone marrow morphology, 88% of the FL−/−ITD+/+ mice and 94% of the FL+/+ITD+/+ mice developed myeloproliferative disease (Table 1). The in vivo data strongly support a functional role for FL in that it significantly accelerated the progress of fatal MPD.
Fig 8. Endogenous FL shortens the survival of FL+/+ITD+/+ mice in comparison with FL−/−ITD+/+ mice.
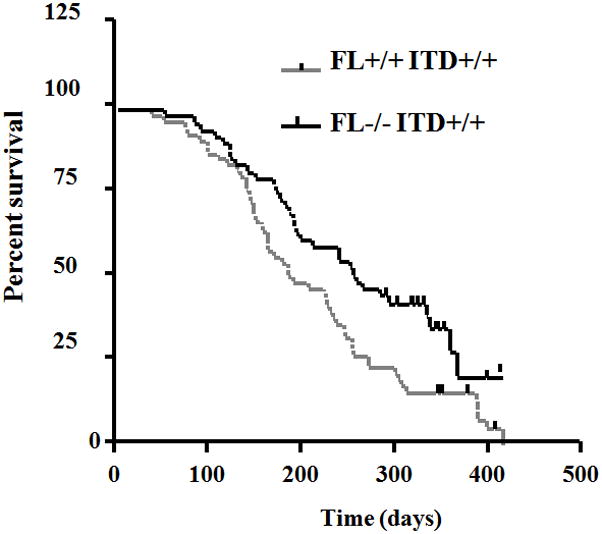
Kaplan-Meier plot of survival of FL+/+ITD+/+ mice (n=51) and FL−/−ITD+/+ mice (n=32).
Table 1.
Disease phenotype of FL+/+ITD+/+ and FL−/−ITD+/+ mice
| Disease phenotype | FL+/+ ITD+/+ (%) | FL−/−ITD+/+ (%) |
|---|---|---|
| Myeloproliferative disease | 94 | 88 |
| Acute myeloid leukemia | 2 | 4.8 |
| Lymphoid proliferative disease | 2 | 2.4 |
| Unclassifiable | 2 | 4.8 |
Discussion
The biochemical characteristics of FLT3/ITD mutations were initially investigated by expressing FLT3/ITD mutations in Cos7 cells (Kiyoi et al., 1998). These initial studies suggested that FLT3/ITD receptors dimerize in a ligand-independent manner, leading to autophosphorylation of the receptor through constitutive activation of the tyrosine kinase domain. FL-independent tyrosine phosphorylation of FLT3 was also observed in Cos7 and 32D cells transfected with FLT3/TKD mutations (Yamamoto et al., 2001). However, most cell lines express FL and thus the possible contribution of their endogenous FL expression to the activation of FLT3 mutant receptors was not clear. It was previously shown that autophosphorylation of wtFLT3 receptors can occur without exogenous FL stimulation in 32D and BaF3 cells expressing wtFLT3 (Zhang et al., 2000; Zheng et al.,2002). FL has been shown to stimulate autocrine signaling in primary AML blasts expressing wtFLT3 (Zheng et al.,2004). In the present study, our use of FL−/− MEF cells eliminated the possibility of autocrine, paracrine, or intracrine stimulation of FLT3 receptors by FL. FL−/− MEF cells transduced with FLT3/ITD and FLT3/TKD mutants were further cultured in serum-free medium before addition of FL to exclude the possible effect of FL contained in the serum. Increased tyrosine phosphorylation of both FLT3/ITD and FLT3/TKD receptors in response to FL addition was observed, thus indicating that activation was augmented by FL. Furthermore, when endogenous FL expression was rescued in these cell lines by retroviral transduction, there was no further enhancement of FLT3 phosphorylation by added exogenous FL, implying that autocrine, paracrine, and/or intracrine FL may lead to complete activation of mutant FLT3 receptors. We also saw evidence, in TF1 cells expressing FLT3/ITD and FLT3/TKD mutants, MV4-11 cells and primary AML samples with homozygous FLT3/ITD mutations, that maximal activation of FLT3 mutant receptors was only observed in response to addition of FL. The effect of concurrent stimulation of wtFLT3 receptors was excluded because these cells exclusively express mutant versions of FLT3. The fact that these cell lines did not show full activation by endogenous FL, in contrast to the FL transfected FL−/− MEF cell lines, could be reflective of the higher level of FL expression driven by the retroviral promoter. The average FL level was approximately 6000 pg/mL in the FL transfected FL−/− MEF cell lines (Fig 2A), which is similar to the significant surge of FL in plasma measured in patients with AML after chemotherapy. The surge of FL results in a blunting of the inhibitory activity of FLT3 kinase inhibitors in vitro (Sato et al., 2011).
Thus, it seems likely that part of the phosphorylation of FLT3 mutants observed in Cos7 and 32D cells is at least partially explained by autocrine, paracrine, and/or intracrine FL stimulation and/or stimulation by FL contained in the serum. This hypothesis is further supported by the findings that an anti-FLT3 antibody that blocks FL binding to the receptors is able to at least partially block the phosphorylation of FLT3 mutants along with its downstream signaling in BaF3 and 32D cells (Piloto et al., 2005). Our findings suggest that FL might be required for complete activation of FLT3 mutants. As to the mechanism that explains this observed stimulation, the possibility that increased stability and/or decreased turnover of phosphorylated forms in response to FL addition was raised. However, the possibility of decreased turnover is less likely since the FL stimulation was for a short period (15 minutes) in our experiments. Further studies will be required to investigate the exact mechanism.
Our data suggests that FLT3 mutant receptors still respond to FL with further activation. However, the increase of activation of downstream signaling of FLT3 in response to FL addition is relatively modest in comparison to that of the FLT3 mutant receptor itself. Whether FL mediated activation of FLT3 mutant receptors induces activation of other unknown signaling pathways requires further investigation. The functional effect (survival and apoptosis) of exogenous FL on FLT3 mutant receptors is of modest effect in the experiment with some of the cell lines and one of four primary AML samples. One possible explanation is the minimization of further effect of added FL by the presence of endogenous FL expression.
In this report, we demonstrate that FL stimulation of mutants of FLT3 contributes to the immortalization and proliferation of FL−/− primary hematopoietic progenitors. In addition, FL promoted increased survival of TF1 and BaF3 cells expressing FLT3 mutants as well as MV411 cells. FL also prolonged the survival of primary AML blasts expressing homozygous FLT3/ITD mutations. Further evidence also comes from an anti-FLT3 antibody that blocks FL binding to FLT3. This antibody partially inhibits the proliferation of BaF3/ITD cells (Li et al., 2004). Finally, endogenous FL decreased the survival of homozygous ITD mice (FL+/+ ITD+/+) compared with FL knock-out mice (FL−/− ITD+/+). Thus, FL plays a role in survival and proliferative signaling mediated by FLT3 mutants. The role of FL in stimulating FLT3 mutants is also consistent with a recent report showing FL shifts the dose-response of FLT3/ITD mutants in response to a number of FLT3 tyrosine kinase inhibitors (TKIs), many of which are currently in clinical trials (Sato et al., 2011). It is noted that FL levels are induced by >50 folds in patients undergoing chemotherapy for AML (Levis M, 2011). The induction of high levels of ligand and resultant stimulation of mutant receptors might also account for the failure to achieve thorough inhibition of FLT3 signaling in some FLT3 tyrosine kinase inhibitor clinical trials. As FLT3/ITD and TKD mutants have been identified in approximately 24% and 8%of AML patients, respectively, FL mediated activation of FLT3 mutant receptors might be contributing to FLT3-mediated leukemogenesis (Yamamoto et al., 2001; Nakao et al., 1996; Yokota et al., 1997; Stirewalt et al., 2001; Abu-Duhier et al., 2001). For all of the above reasons, FL targeted therapy, either antibodies against FL itself or those that would block the ability of FL to activate FLT3, may be indicated as an adjuvant therapy for the treatment of FLT3 mutant AML.
These findings also potentially have important implications regarding other growth factor receptor pathways. Autostimulated growth of leukemia cells has been associated with expression of GM-CSF, SCF, and VEGF (Freedman et al.,1993; Pietsch, 1993; Rogers et al., 1994; Bellamy et al., 2001). Ligand-independent activation of the KIT tyrosine kinase domain induced by the KIT G560 and KIT V816 activating mutations has been reported (Furitsu et al., 1993). EGFR kinase domain mutations resulting in constitutive phosphorylation and activation of EGFR have been identified in lung cancer (Pao and Miller, 2005). Based on the data presented here, it is possible that the activation of these KIT and EGFR mutant receptors still respond to SCF and EGF, respectively, to some degree. Generation of SCF and EGF deficient cell lines with expression of KIT and EGFR mutants, respectively, would shed light on the generality of these findings.
Materials and Methods
Reagents
Recombinant human FL was purchased from PeproTech (Rocky Hill, NJ). Rabbit antihuman FLT3 and anti-STAT5 antibody were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal mouse antiphosphotyrosine antibody 4G10 and recombinant protein A-agarose were purchased from Upstate Biotechnology (Lake Placid, NY). Antiphospho-STAT5, antiphospho-AKT, antiphospho-MAPK, anti-AKT, and anti-MAPK antibodies were purchased from Cell Signaling Technology(Cell Signaling, Beverly, MA). Horseradish peroxidase-conjugated secondary antibody and the enhanced chemiluminescence (ECL) detection system were from Amersham (Arlington Heights, IL).
Establishment of FL−/− MEF cells
Embryos were removed from female FL−/− mice (purchased from Taconic, Hudson, NY.) on day 13 or 14 of pregnancy. Internal organs and heads were removed. The remainder of the embryos were washed, minced, digested with Trypsin/EDTA, and then resuspended in DMEM containing 10% FBS followed by centrifuging at 1000g for 5 minutes. The cell pellet was resuspended in DMEM containing 10% FBS and cultured at 37°C with 5% CO2. After 3 passages, the cells were transfected with CMV-SV40-large T antigen (kindly provided by Dr. Linzhao Cheng, Johns Hopkins University) to immortalize FL−/− MEF cells. The FL−/− MEF cells represent bulk cells after transformation.
Reverse transcription–polymerase chain reaction (RT-PCR)
Total cellular RNA was extracted from cultured cell lines (RNeasy columns; Qiagen, Valencia, CA). RNA (50 ng–100 ng) was reverse transcribed and amplified(One-Step RT-PCR kit; Invitrogen, Carlsbad, CA). PCR was performed in the presence of the following primer pairs:5′-CAAATTCCTCCCTGTTGCTG -3′ and 5′-AGGTGGGAGATGTTGGTCTG -3′, specific for murine FL and amplifies a 363–base pair (bp) sequence; 5′-GCTGGTCGTCGACAACGGCTC-3′and 5′-CAAACATGATCTGGGTCATCTTTTC-3′, specific for β-actin and amplifies a 353-bp sequence. Reverse transcription was performed at 50°C for 30 minutes. Thirty-five cycles of amplification were then performed at 94°C for 30 seconds for denaturation, at 55°C for 30 seconds for annealing, and finally at 72°Cfor 1 minute for extension (DNA iCycler; Bio-Rad, Hercules, CA). PCR products were resolved on a 1% agarose gel in the presence of ethidium bromide and photographs were taken under UV transillumination.
DNA constructs and retroviral transduction
The mutant FLT3/ITD sequences were isolated from bone marrow samples from AML patients at our institution by RT-PCR. The ITD fragments were sequenced and used to replace the corresponding wild-type region in full-length FLT3 cDNA. This ITD resulted in an addition of 6 amino acids, RTDFRE, after amino acid No. 596. The FLT3/TKD mutation was obtained by mutagenesis of wtFLT3 to substitute nucleotide G of D835 with T resulting in an Aspartic acid to Tyrosine change (QuickChange Site-Directed Mutagenesis Kit; Stratagene, La Jolla, CA). Both types of mutants were then cloned into the pBabePuro retroviral vector (kindly provided by Dr. Alan D Friedman) and pCIneo expression vector (Promega, Madison, WI).
Retrovirus was obtained by harvesting supernatant 48 hours after transfection of 293T cells with pBabePuro constructs. FL−/− MEF cells were cultured with virus in the presence of polybrene (8 μg/mL) for 2 days. Stable transfectants were then selected by limiting dilution in 96-well plates with 2 μg/mL puromycin. Experiments were performed on at least two clones selected from the transfectants.
The fragment containing the secreted form of FL coding sequence was amplified from the pUMVC3-hFLex plasmid (kindly provided by Dr. Katharine A. Whartenby, Johns Hopkins University) by PCR and cloned into the EF1-CMV-GFP lentiviral vector (kindly provided by Dr. Linchao Cheng, Johns Hopkins University). Viral stocks were generated and transduction of cells was conducted as described above. GFP positive cells were sorted by flow cytometry.
TF1 and BaF3 cells were transfected with pCINeo constructs containing either FLT3/ITD or FLT3/TKD mutation by electroporation (Bio-Rad, Hercules, CA). Stable transfectants were then selected by limiting dilution in 96-well plates with 1 mg/mL G418 (Invitrogen, Carlsbad, California).
Colony-replating assay
Bone marrow was harvested from FL−/− mice, lineage depleted (Lineage Cell Depletion kit; Militenyibi Biotec, Auburn CA), and transduced with EF1-ITD-UBC-GFP, EF1-TKD-UBC-GFP, or EF1-UBC-GFP lentivirus by 3 rounds of spin-infection on days 1, 2 and 3. The EF1-ITD-UBC-GFP vector was constructed by inserting full-length FLT3/ITD in EF1-UBC-GFP lentiviral vector (kindly provided by Dr. Linzhao Cheng). The EF1-TKD-UBC-GFP was constructed by inserting wtFLT3 into the EF1-UBC-GFP followed by mutagenesis of wtFLT3 to substitute nucleotide G of D835 with T resulting in an Aspartic acid to Tyrosine change (QuickChange Site-Directed Mutagenesis Kit; Stratagene, La Jolla, CA). GFP positive cells were then sorted by FACS. 103 cells were plated into triplicate 35 mm Petri dishes (Nunc) in 1 ml of Methocult M3434 (StemCell Technologies Inc., Vancouver) containing 50 ng/mL rm SCF, 10 ng/mL rm IL-3, 10 ng/mL rh IL-6, and 3 units/mL rh Erythropoietin. Cultures were incubated at 37°C in a humidified atmosphere of 5% CO2, and colonies consisting of >35 cells were scored after 9–11 days. For the colony-replating assay all cells from the previous culture were collected and re-plated at 1×104 cells/plate in fresh methylcellulose medium. All cells were replated three times.
Generation of mice with FL−/−ITD+/+ and FL+/+/ITD+/+ genotype
FL−/− FLT3wt/wt C57/Bl6 mice were crossed with FL+/+FLT3wt/ITD C57/Bl6 knock-in mice previously generated in the lab (Li et al., 2008). Heterozygotes with the genotype of FL+/−FLT3wt/ITD were then crossed to generate the desired FL−/−ITD+/+ and FL+/+/ITD+/+ mice. Genotypes were analyzed by PCR with the following primers: ITD, 5′-CTCTCGGGA ACTCCCACTTA-3′, 5′-TGCAGATGATCCAGGTGACT-3; FL, 5′-GCCCAA ATGTGCGTATACCT-3′, 5′-CCCAGCACAGTATGGGAACT-3′; NeoFL, 5′-ACACTTCGAAGCTGGAAAGC-3′, 5′-GGGGAACTTCCTGACTAGGG-3′. Survival of FL−/−ITD+/+ and FL+/+/ITD+/+ mice were followed up to 525 days. White blood cell (WBC) differential counting was performed by collecting 50 μL peripheral blood and subjecting it to analysis using a Hemavet950 Hematology system (Drew Scientific, Oxford, CT). Murine internal organs were dissected from moribund mice and subjected to gross examination. Bone marrow was collected and subjected to histologic and flow cytometry analysis. All animal procedures were conducted in conformity with institutional guidelines.
Immunoprecipitation and Western blot analysis
Cells were washed twice with ice-cold phosphate-buffered saline(PBS) and lysed for 30 minutes in ice-cold NP-40 lysis buffer(20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 100 mM NaF, 10% glycerol,1% NP-40, and 10 mM EDTA [ethylenediaminetetraacetic acid])containing protease inhibitors (Roche, Indianapolis, IN). Clarified lysates (500 μg) were incubated with rabbit polyclonal antibody to human FLT3followed by incubation with protein A–agarose at 4°C.The immunoprecipitates were washed 3 times with ice-cold TBS-T(10 mM Tris-HCl, pH 7.4, 100 mM NaCl, 0.1% Tween-20), resuspended in sodium dodecyl sulfate (SDS) sample buffer, heated, and separated by SDS–polyacrylamide gel electrophoresis (PAGE) in 8%gels. Gels were blotted onto polyvinylidene fluoride (PVDF) microporous membrane (Millipore, Bedford, MA) and stained with the indicated antibody. Antibody binding was detected by incubation with a horseradish peroxidase–conjugated secondary antibody, followed by chemiluminescence detection. For detection of phosphorylation or protein expression of STAT5, AKT, or MAPK, total protein lysates (50 μg) were loaded, separated by PAGE, and transferred to PVDF membrane. The blot was first probed with anti-phosphoSTAT5, anti-phosphoAKT, and anti-phosphoMAPK antibody. The same blot was stripped and reprobed with anti-STAT5, anti-AKT, anti-MAPK, and anti-HSP90 antibody.
Apoptosis and survival assay
Evaluation of apoptosis was performed by annexin V/7-AAD staining according to the recommendations of the manufacturer (Becton Dickinson, San Jose, CA). After serum starvation, 5 × 105 cells were washed with cold PBS and re-suspended in 100 μL of binding buffer. Following incubation with 5 μL of annexin V-PE and 5 μL of 7-AAD in the dark for 15 minutes at room temperature, cells were analyzed by flow cytometry (FACSort, Becton Dickinson)with Cell Quest software (Becton Dickinson). Compensation was performed with samples stained with either annexin V-PE or 7-AAD alone.
2×104 cells in 100 μl serum-free medium were plated into triplicate wells of 96-wellplates. Survival was assessed by MTT assay (Roche, Indianapolis, IN) and viable cell number counting by trypan blue (GIBCO, Rockville, MD) exclusion according to the manufacturer’s instructions.
Detection of FL concentration by ELISA assay
Cells were washed with PBS once, plated at 5×105 cells/mL, and cultured for 24 hours in serum-free medium. Cell culture supernatant was collected by centrifugation and FL concentration was assessed according to the manufacturer’s instruction (R&D systems, Minneapolis, MN).
Primary AML samples
Diagnostic bone marrow samples were obtained under institutional review board–approved protocols from patients with AML at the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University (Baltimore, MD) prior to therapy. Leukemic blast cells were separated by Ficoll-Hypaque (Amersham, Piscataway, NJ) density gradient centrifugation. Cells were cultured in RPMI 1640 medium containing 10% FBS (Invitrogen, Carlsbad, CA).
Acknowledgments
We thank Dr. Katharine A. Whartenby, Alan Friedman and Linzhao Cheng for their gift of reagents. D.S. is also supported by the Kyle Haydock Professorship in Oncology and the Giant Food Pediatric Cancer Research Fund.
Support: This work was supported by grants from the NIH (CA90668, CA70970, and CA095600–01) and the Leukemia and Lymphoma Society (DS).
It was also supported by grants from the NCI (NCI Leukemia SPORE P50 CA100632–06, R01 CA128864) and the American Society of Clinical Oncology (ML). ML is a Clinical Scholar of the Leukemia and Lymphoma Society.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Abu-Duhier FM, Goodeve AC, Wilson GA, Care RS, Peake IR, Reilly JT. Genomic structure of human FLT3: implications for mutational analysis. Br J Haematol. 2001;113:1076–1077. doi: 10.1046/j.1365-2141.2001.02821.x. [DOI] [PubMed] [Google Scholar]
- Bellamy WT, Richter L, Sirjani D, Roxas C, Glinsmann-Gibson B, Frutiger Y, et al. Vascular endothelial cell growth factor is an autocrine promoter of abnormal localized immature myeloid precursors and leukemia progenitor formation in myelodysplastic syndromes. Blood. 2001;97:1427–1434. doi: 10.1182/blood.v97.5.1427. [DOI] [PubMed] [Google Scholar]
- Birg F, Courcoul M, Rosnet O, Bardin F, Pebusque MJ, Marchetto S, et al. Expression of the FMS/KIT-like gene FLT3 in human acute leukemias of the myeloid and lymphoid lineages. Blood. 1992;80:2584–2593. [PubMed] [Google Scholar]
- Brasel K, Escobar S, Anderberg R, de Vries P, Gruss HJ, Lyman SD. Expression of the flt3 receptor and its ligand on hematopoietic cells. Leukemia. 1995;9:1212–1218. [PubMed] [Google Scholar]
- Carow CE, Levenstein M, Kaufmann SH, Chen J, Amin S, Rockwell P, et al. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood. 1996;87:1089–1096. [PubMed] [Google Scholar]
- Dosil M, Wang S, Lemischka IR. Mitogenic signalling and substrate specificity of the Flk2/Flt3 receptor tyrosine kinase in fibroblasts and interleukin 3-dependent hematopoietic cells. Mol Cell Biol. 1993;13:6572–6585. doi: 10.1128/mcb.13.10.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler HG. Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia. 1996;10:588–599. [PubMed] [Google Scholar]
- Fenski R, Flesch K, Serve S, Mizuki M, Oelmann E, Kratz-Albers K, et al. Constitutive activation of FLT3 in acute myeloid leukaemia and its consequences for growth of 32D cells. Br J Haematol. 2000;108:322–330. doi: 10.1046/j.1365-2141.2000.01831.x. [DOI] [PubMed] [Google Scholar]
- Freedman MH, Grunberger T, Correa P, Axelrad AA, Dube ID, Cohen A. Autocrine and paracrine growth control by granulocyte-monocyte colony-stimulating factor of acute lymphoblastic leukemia cells. Blood. 1993;81:3068–3075. [PubMed] [Google Scholar]
- Furitsu T, Tsujimura T, Tono T, Ikeda H, Kitayama H, Koshimizu U, et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest. 1993;92:1736–1744. doi: 10.1172/JCI116761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532–1542. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- Horiike S, Yokota S, Nakao M, Iwai T, Sasai Y, Kaneko H, et al. Tandem duplications of the FLT3 receptor gene are associated with leukemic transformation of myelodysplasia. Leukemia. 1997;11:1442–1446. doi: 10.1038/sj.leu.2400770. [DOI] [PubMed] [Google Scholar]
- Iwai T, Yokota S, Nakao M, Okamoto T, Taniwaki M, Onodera N, et al. Internal tandem duplication of the FLT3 gene and clinical evaluation in childhood acute myeloid leukemia. The Children’s Cancer and Leukemia Study Group, Japan. Leukemia. 1999;13:38–43. doi: 10.1038/sj.leu.2401241. [DOI] [PubMed] [Google Scholar]
- Kiyoi H, Naoe T, Nakano Y, Yokota S, Minami S, Miyawaki S, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood. 1999;93:3074–3080. [PubMed] [Google Scholar]
- Kiyoi H, Naoe T, Yokota S, Nakao M, Minami S, Kuriyama K, et al. Internal tandem duplication of FLT3 associated with leukocytosis in acute promyelocytic leukemia. Leukemia. 1997;11:1447–1452. doi: 10.1038/sj.leu.2400756. [DOI] [PubMed] [Google Scholar]
- Kiyoi H, Towatari M, Yokota S, Hamaguchi M, Ohno R, Saito H, et al. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia. 1998;12:1333–1337. doi: 10.1038/sj.leu.2401130. [DOI] [PubMed] [Google Scholar]
- Lavagna-Sevenier C, Marchetto S, Birnbaum D, Rosnet O. The CBL-related protein CBLB participates in FLT3 and interleukin-7 receptor signal transduction in pro-B cells. J Biol Chem. 1998;273:14962–14967. doi: 10.1074/jbc.273.24.14962. [DOI] [PubMed] [Google Scholar]
- Levis M, Ravandi F, Wang ES, Baer MR, Perl A, Coutre S, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011 doi: 10.1182/blood-2010-08-301796. (Epub) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Pioloto O, Nguyen HB, Greenberg K, Takamiya K, Racke F, et al. Knock-in of an internal tandem duplication mutation into murine FLT3 confers myeloproliferative disease in a mouse model. Blood. 2008;111:3849–3858. doi: 10.1182/blood-2007-08-109942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li H, Wang MN, Lu D, Bassi R, Wu Y, et al. Suppression of leukemia expressing wild-type or ITD-mutant FLT3 receptor by a fully human anti-FLT3 neutralizing antibody. Blood. 2004;104:1137–1144. doi: 10.1182/blood-2003-07-2585. [DOI] [PubMed] [Google Scholar]
- Lisovsky M, Estrov Z, Zhang X, Sanchez-Williams G, Snell V, et al. Flt3 ligand stimulates proliferation and inhibits apoptosis of acute myeloid leukemia cells: regulation of Bcl-2 and Bax. Blood. 1996;88:3987–3997. [PubMed] [Google Scholar]
- Lyman S, James L, Vanden Bos T, de Vries P, Brasel K, Gliniak B, et al. Molecular cloning of a ligand for the flt3/flk-2 tyrosine kinase receptor: a proliferative factor for primitive hematopoietic cells. Cell. 1993;75:1157–1167. doi: 10.1016/0092-8674(93)90325-k. [DOI] [PubMed] [Google Scholar]
- Meierhoff G, Dehmel U, Gruss HJ, Rosnet O, Birnbaum D, Quentmeier H, et al. Expression of FLT3 receptor and FLT3-ligand in human leukemia-lymphoma cell lines. Leukemia. 1995;9:1368–1372. [PubMed] [Google Scholar]
- Meshinchi S, Woods WG, Stirewalt DL, Sweetser DA, Buckley JD, Tjoa TK, et al. Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood. 2001;97:89–94. doi: 10.1182/blood.v97.1.89. [DOI] [PubMed] [Google Scholar]
- Marchetto S, Fournier E, Beslu N, Aurran-Schleinitz T, Dubreuil P, Borg JP, et al. SHC and SHIP phosphorylation and interaction in response to activation of the FLT3 receptor. Leukemia. 1999;13:1374–1382. doi: 10.1038/sj.leu.2401527. [DOI] [PubMed] [Google Scholar]
- Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–1918. [PubMed] [Google Scholar]
- Pao W, Miller VA. Epidermal growth factor receptor mutations, small-molecule kinase inhibitors, and non-small-cell lung cancer: current knowledge and future directions. J Clin Oncol. 2005;23:2556–2568. doi: 10.1200/JCO.2005.07.799. [DOI] [PubMed] [Google Scholar]
- Pietsch T. Paracrine and autocrine growth mechanisms of human stem cell factor (c-kit ligand) in myeloid leukemia. Nouv Rev Fr Hematol. 1993;35:285–286. [PubMed] [Google Scholar]
- Piloto O, Levis M, Huso D, Li Y, Li H, Wang MN, et al. Inhibitory anti-FLT3 antibodies are capable of mediating antibody-dependent cell-mediated cytotoxicity and reducing engraftment of acute myelogenous leukemia blasts in nonobese diabetic/severe combined immunodeficient mice. Cancer Res. 2005;65:1514–1522. doi: 10.1158/0008-5472.CAN-04-3081. [DOI] [PubMed] [Google Scholar]
- Rogers SY, Bradbury D, Kozlowski R, Russell NH. Evidence for internal autocrine regulation of growth in acute myeloblastic leukemia cells. Exp Hematol. 1994;22:593–598. [PubMed] [Google Scholar]
- Rosnet O, Buhring HJ, deLapeyriere O, Beslu N, Lavagna C, Marchetto S, et al. Expression and signal transduction of the FLT3 tyrosine kinase receptor. Acta Haematol. 1996;95:218–223. doi: 10.1159/000203881. [DOI] [PubMed] [Google Scholar]
- Sato T, Yang X, Knapper S, White P, Smith BD, Galkin S, et al. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood. 2011 doi: 10.1182/blood-2010-01-266742. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirewalt DL, Kopecky KJ, Meshinchi S, Appelbaum FR, Slovak ML, Willman CL, et al. FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood. 2001;97:3589–3595. doi: 10.1182/blood.v97.11.3589. [DOI] [PubMed] [Google Scholar]
- Tse KF, Mukherjee G, Small D. Constitutive activation of FLT3 stimulates multiple intracellular signal transducers and results in transformation. Leukemia. 2000;14:1766–1776. doi: 10.1038/sj.leu.2401905. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97:2434–2439. doi: 10.1182/blood.v97.8.2434. [DOI] [PubMed] [Google Scholar]
- Yokota S, Kiyoi H, Nakao M, Iwai T, Misawa S, Okuda T, et al. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines. Leukemia. 1997;11:1605–1609. doi: 10.1038/sj.leu.2400812. [DOI] [PubMed] [Google Scholar]
- Zhang S, Broxmeyer H. Flt3 ligand induces tyrosine phosphorylation of gab1 and gab2 and their association with shp-2, grb2, and pi3 kinase. Biochem Biophys Res Commun. 2000;277:195–199. doi: 10.1006/bbrc.2000.3662. [DOI] [PubMed] [Google Scholar]
- Zhang S, Fukuda S, Lee Y, Hangoc G, Cooper S, Spolski R, et al. Essential role of signal transducer and activator of transcription (Stat)5a but not Stat5b for Flt3-dependent signaling. J Exp Med. 2000;192:719–728. doi: 10.1084/jem.192.5.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Mantel C, Broxmeyer HE. Flt3 signaling involves tyrosyl-phosphorylation of SHP-2 and SHIP and their association with Grb2 and Shc in Baf3/Flt3 cells. J Leukoc Biol. 1999;65:372–380. doi: 10.1002/jlb.65.3.372. [DOI] [PubMed] [Google Scholar]
- Zheng R, Friedman AD, Small D. Targeted inhibition of FLT3 overcomes the block to myeloid differentiation in 32Dcl3 cells caused by expression of FLT3/ITD mutations. Blood. 2002;100:4154–4161. doi: 10.1182/blood-2002-03-0936. [DOI] [PubMed] [Google Scholar]
- Zheng R, Levis M, Piloto O, Brown P, Baldwin BR, Gorin NC, et al. FLT3 ligand causes autocrine signaling in acute myeloid leukemia cells. Blood. 2004;103:267–274. doi: 10.1182/blood-2003-06-1969. [DOI] [PubMed] [Google Scholar]