

Abstract
Lysosomal cysteine protease cathepsin L (CTSL) is believed to play a role in tumor progression and is considered a marker for clinically invasive tumors. Studies from our laboratory using the classical mouse skin carcinogenesis model, with 7,12-dimethyl-benz[a]anthracene (DMBA) for initiation and 12-O-tetradecanoylphorbol-13-acetate (TPA) for promotion, showed that expression of CTSL is increased in papillomas and squamous cell carcinomas (SCC). We also carried out carcinogenesis studies using Ctsl-deficient nackt (nkt) mutant mice on three different inbred backgrounds. Unexpectedly, the multiplicity of papillomas were significantly higher in Ctsl-deficient than in wild-type mice on two unrelated backgrounds. Topical applications of TPA or DMBA alone to the skin of nkt/nkt mice did not induce papillomas, and there was no increase in spontaneous tumors in nkt/nkt mice on any of the three inbred backgrounds. Reduced epidermal cell proliferation in Ctsl-deficient nkt/nkt mice after TPA treatment suggested that they are not more sensitive than wild-type mice to TPA promotion. We also showed that deficiency of CTSL delays terminal differentiation of keratinocytes, and we propose that decreased elimination of initiated cells is at least partially responsible for the increased papilloma formation in the nackt model.
Keywords: mouse models, proteases, cysteine cathepsins, skin cancer, two-stage carcinogenesis
Introduction
Cysteine cathepsins, lysosomal proteases that are involved in extracellular and intracellular protein degradation, have multiple roles during cancer development and progression [1], including modulation of tumor cell proliferation, invasion, and apoptosis [2]. Cathepsin L (CTSL) is a widely expressed lysosomal protease with roles in epidermal homeostasis and hair follicle morphogenesis [3–5], tumor invasiveness, angiogenesis [6], and apoptosis [7–9]. Recently, the Nepveu laboratory identified a nuclear isoform of CTSL that regulates cell cycle progression through proteolytic processing of the CDP/Cux transcription factor [10–12]. Although mice and rats possess only one Ctsl gene, humans have two CTSL-like genes (CTSL and CTSL2); however, mouse and rat Ctsl genes are more closely related to human CTSL2 than to human CTSL gene [13] [14].
We previously described the recessive nackt (nkt) mutation, which is characterized by partial alopecia associated with CD4+ T cell deficiency. The nackt mutation is a 1.6 kilobases deletion in the Ctsl gene (Ctslnkt) [15] [16] that eliminates exon 7 from the Ctsl gene transcript (Benavides, unpublished data). Ctslnkt/Ctslnkt mice do not generate any of the mature active forms of CTSL, although a truncated unprocessed CTSL protein is present [17]. Homozygous nackt mutants exhibit phenotypes similar to Ctsl deficient mice [17], while heterozygous nackt mice are phenotypically indistinguishable from wild-type mice. CTSL background activity is comparably low in Ctslnkt/Ctslnkt and targeted knockout Ctsltm1Cptr/Ctsltm1Cptr mice. Together, these results indicate that nackt is a loss-of-function mutation of the Ctsl gene. Studies with the nackt model suggested a critical role for CTSL in hair follicle morphogenesis and cycling and in epidermal differentiation [4]. The nackt model was also used to demonstrate that CTSL influences the expression of extracellular matrix components in lymphoid organs [18].
Recent studies using the three other Ctsl-deficient mouse models currently available have shed light on the role of CTSL in a number of developmental and pathological processes. For example, the first reported Ctsl knockout mice (Ctsltm1Cptr) exhibited a 60–80% reduction in the number of CD4+ T cells in the thymus and periphery [19], gingival overgrowth [20], abnormal hair follicle morphogenesis and cycling [21] [5], late onset dilated cardiomyopathy [22] [23], and defects in adipogenesis and glucose metabolism [24]. A second independent Ctsl knockout line (Ctsltm1Alpk) has been described with abnormal skin and defective bone development [25]. The spontaneous recessive mutation furless (Ctslfs), carrying an enzymatically inactive CTSL, also exhibits a skin phenotype [3] and abnormal spermatogenesis [26].
We assessed the role of CTSL in multistage skin carcinogenesis by comparing Ctsl-deficient nackt and wild-type mice. Skin tumorigenesis was enhanced in nackt compared to wild-type mice, and this trait was influenced by the genetic background. However, loss of CTSL function did not act as an initiator or promoter, since topical 7,12-dimethyl-benz[a]anthracene (DMBA) or 12-O-tetradecanoylphorbol-13-acetate (TPA) alone did not induce papillomas in nackt mice. Our results also showed that the absence of CTSL enzymatic activity in mutant mice delays the onset of keratinocyte terminal differentiation and decreases proliferation after TPA treatment. These findings suggest that delayed keratinocyte transit and concomitant increased retention of initiated cells may be responsible for the increased tumor susceptibility in nackt mice.
Materials and Methods
Mouse Strains
SENCARB/Pt.Cg-nkt, DBA/2.Cg-nkt, BALB/c.Cg-nkt, and FVB/N.129S6-Cd4tm1Knw (CD4 knock-out) colonies were maintained under specific pathogen free (SPF) conditions in our AAALAC accredited Animal Facilities at M. D. Anderson Cancer Center, Smithville, Texas. All procedures were conducted in compliance with the Guide for the Care and Use of Laboratory Animals and under an IACUC-approved protocol. FVB/N-CD4 knockout congenic mice were a gift from Lisa M. Coussens from the University of California at San Francisco, San Francisco. We developed SENCARB/Pt.Cg-nkt and DBA/2.Cg-nkt congenic lines, thus placing the mutation on backgrounds with high and moderate skin tumor susceptibility, respectively. To develop these lines we used marker assisted “speed congenic” breeding strategies [27]. Microsatellite analysis of N6 mice from the two lines showed that more than 99% of the alleles in the genome scan were from the recipient strain. The BALB/c.Cg-nkt congenic (N12) line was developed previously [15].
Skin Carcinogenesis and Tissue Collection
Ctslnkt/Ctslnkt (nkt/nkt) age-matched mice plus wild-type littermate controls (male and female) were used to determine the susceptibility to chemically induced tumorigenesis in three inbred backgrounds (SENCARB/Pt, DBA/2, and BALB/c). In a separate experiment, FVB/N-Cd4 −/− and FVB/N wild-type mice were also challenged. The two-stage carcinogenesis protocol was performed by initiating mice (6–8 wk old) as previously described [28] with 10 nmol/200 µl acetone of DMBA (Sigma-Aldrich, St. Louis, MO) (SENCARB/Pt mice) or 100 nmol/200 µl acetone of DMBA (DBA/2, BALB/c, and FVB/N backgrounds). Promotion was started 2 wk after initiation with twice weekly applications of 1 µg of TPA (Sigma-Aldrich) (SENCARB/Pt), 2 µg (DBA/2), or 4 µg (BALB/c and FVB/N) in 200 µl of acetone on the dorsal skin. Tumors were counted weekly and statistical significance for tumor multiplicity was determined by calculation of p values using Wilcoxon Rank Sum test.
Analysis of Cell Proliferation Following Treatments with TPA
For analysis of epidermal proliferation, groups of 4 DBA/2-nkt/nkt and 4 wild-type littermate controls mice were treated with a single topical application or twice-weekly applications of TPA (2 µg/200 µl acetone) on the dorsal skin for 2 wk and sacrificed 24 h after the final treatment. Mice received intraperitoneal (i.p.) injections of 5-bromo-2-deoxyuridine (BrdU; Sigma–Aldrich) at 100 µg/g body weight in PBS 30 min prior to euthanasia. Dorsal skin was fixed in formalin and embedded in paraffin prior to sectioning and staining with hematoxylin and eosin (H&E) and immunohistochemistry (IHC). BrdU incorporation was detected by standard 3-step immunoperoxidase detection using mouse anti-BrdU monoclonal antibody (Becton-Dickinson Immunocytometry System, Becton-Dickinson, San Jose, CA), biotin F(ab’) rabbit anti mouse IgG (Accurate Chemical, Westbury, NY), and Streptavidin Peroxidase (BioGenex, San Ramon, CA). Diaminobenzidine (BioGenex) was the chromagen used for visualization. Epidermal cell proliferation (labeling index) was determined by calculating the percentage of epidermal basal cells positive for BrdU and Ki67. A minimum of 4000 basal cells was counted using digital images of IHC slides (dorsal skin) captured with the Aperio ScanScope CS slide scanner (Aperio Technologies, Vista, CA). A fully automated nuclear algorithm provided with the instrument was adapted to count BrdU- and Ki67-positive cells in the epidermis of both wild-type and mutant phenotypes. Statistical significance was determined by calculation of p values using Wilcoxon Rank Sum test.
In Vivo Keratinocyte Transit Studies
Mice were injected i.p. with 100 µg/g body weight of BrdU (Sigma-Aldrich) in 200 µl of PBS 17 h after the last of four TPA applications (2 µg/200 µl acetone) over a two week period and sacrificed 1 h, 8 h, and 30 h after BrdU injection as described [29]. Dorsal skin was collected and processed for histopathology and IHC evaluation as described above. BrdU incorporation was quantified as described above with the Aperio ScanScope CS slide scanner. A total of 4 mice per genotype per time point were evaluated.
Immunohistochemistry
TPA-treated dorsal skin and papillomas from nkt/nkt and littermate controls were fixed in formalin overnight and then transferred to 70% ethanol. IHC analysis was performed with polyclonal antibodies directed against mouse keratins K8, K10, K13, and K14 (Covance Research Products, Richmond, CA), involucrin and profilaggrin/filaggrin (BabCo, Richmond, CA), Ki67 (Dako, Carpinteria, CA), Cd31 (PharMingen, San Diego, CA), p21, p27, VEGF, and active caspase-3 (R&D Systems, Minneapolis, MN) as previously described [4]. Control reactions without primary antibodies were routinely performed.
Protein Lysates and Western Blotting
Protein was isolated from TPA-treated dorsal epidermis and papillomas of mutant and littermate controls using RIPA lysis buffer. Protein lysates (25–50 µg) were electrophoresed on 10% Tris-Glycine Gels (Novex, San Diego, CA) and transferred onto nitrocellulose membranes. Western blot analysis was performed with the following primary antibodies: anti-mouse p21, p27, Stat3, VEGF, and caspase-3 (Santa Cruz Biotechnology, Santa Cruz, CA); CTSB, CTSD, and CTSL (R&D and Santa Cruz Biotechnology); and β-actin (Sigma-Aldrich). Binding of antibodies was detected using ECL+Plus Western blotting detection system (Amersham Pharmacia Biotech UK Limited, Buckinghamshire).
Genotyping of the Polymorphic Variant of the Mouse Patched (Ptch1) Gene
To differentiate the C57BL/6 (Ptch1B6) from the FVB (Ptch1FVB) allele [30] we used genomic DNA and primers designed to amplify a 179 bp segment from Ptch1 (accession number NT_039589.7) exon 23 under standard PCR conditions. The sequences of the primers were: Ptch1-FOR: 5’-GTGGCCGCAAGCCTTCTCTA-3’ and Ptch1-REV: 5’-ACCATCCTACCTCCCTGTGTTGAC-3’. The sequence of the amplification products was obtained using the ABI-PRISM™ Dye Terminator Cycle Sequencing Ready Reaction kit and an ABI 3130XL DNA sequencer (Perkin Elmer).
Results
CTSL Expression Increases During Two-stage Carcinogenesis
We analyzed the expression of CTSL in early-, mid- and late-stage papillomas and squamous cell carcinomas (SCC) from our archives and in normal dorsal skin using polyclonal goat anti mouse CTSL (carboxy terminal) antibody (M-19, sc-6500) for Western blotting. The tumors were generated in SENCARB/Pt mice using a two-stage skin carcinogenesis protocol [31]. Figure 1A shows that CTSL expression was markedly increased in papillomas and SCC compared to normal skin. These results suggested that CTSL might play a role in tumor development and progression during mouse skin tumorigenesis.
Figure 1. Two-stage carcinogenesis studies in Ctsl-deficient mice.

(A) Western blot of normal skin, early (11 wks), mid (21 wks), and late (35 wks) papillomas and squamous cell carcinomas from SENCARB/Pt mice showing how CTSL protein expression increases during papilloma development (antibody: M-19, sc-6500). (B) Tumor multiplicity of skin tumors after two-stage carcinogenesis in SENCARB/Pt-nkt/nkt mice (n = 9) was statistically highly significant from that of wild-type mice (n = 7) at 20 wk (P< 0.001, Wilcoxon Rank Sum test). (C) Tumor multiplicity in DBA/2-nkt/nkt mice (n = 18) was statistically highly significant from that of wild-type mice (n = 20) at 20 wk (P< 0.001). (D) Tumor incidence in DBA/2-nkt/nkt mice reached a plateau of 94% at 18 wk compared to 38% for wild-type mice (all the papillomas from this group regressed by wk 30). For two-stage carcinogenesis, 6–8 wk old mice were initiated with DMBA and after two weeks received repeated applications of TPA as described in Materials & Methods. The number of papillomas was determined weekly. The tumor incidence is defined as the percentage of mice with skin tumors and the tumor multiplicity is the average number of skin tumors per mouse. Mutant nkt/nkt mice (ν); wild-type mice (μ).
Skin Tumorigenesis is Enhanced in the Absence of CTSL
To determine if the absence of CTSL enzymatic activity had an effect on skin tumor development, we conducted a two-stage skin carcinogenesis study with Ctsl-deficient nkt/nkt mice on SENCARB/Pt (very sensitive) and DBA/2 (sensitive) backgrounds and their wild-type littermates. Because SENCARB/Pt-nkt/nkt mice developed severe dermatitis, experiments using this inbred background were terminated at 20 wk and tumor multiplicity (tumors per mouse) was determined; tumor multiplicity in the DBA/2 group was scored at 30 wk [28].
In the SENCARB/Pt group, the first papillomas appeared in both nkt/nkt and wild-type littermates at 6 wk of promotion. The tumor incidence was similar between the two groups: (100% at 8 wk). However, the tumor multiplicity was higher in nkt/nkt mice compared to wild-type littermates. The number of papillomas increased rapidly in nkt/nkt mice, reaching an average of 18.1 papillomas per mouse (n = 9) at 20 wk, while littermate controls developed an average of 8.6 papillomas per mouse (n = 7, p < 0.001) at 20 wk (Figure 1B).
In the DBA/2 group, wild-type mice (n = 20; ungenotyped +/nkt or +/+ mice) developed 0.3 papillomas per mouse by 19 wk of TPA treatment, but all papillomas regressed after 20 wk. In DBA/2-nkt/nkt mice, the number of papillomas reached an average of 3.1 papillomas per mouse at 20 wk (n = 18, p < 0.001). These nkt/nkt papillomas never regressed and less than 1% progressed to SCC. In DBA/2-nkt/nkt mice, the papilloma incidence reached a plateau of 94% at 18 wk. For wild-type mice, before regression of papillomas, the incidence was never higher than 38% (Figure 1C and D). Tumors in these DBA/2-nkt/nkt mice generally displayed the histology typical of papillomas from a two-stage carcinogenesis protocol, however, we observed one atypical SCC with basaloid proliferation and follicular differentiation in a nkt/nkt mouse. This SCC expressed basal cell marker K14, but not differentiation marker K10, when determined by IHC (Figure 2). We compared the proliferation rate (by BrdU labeling) of theses Ctsl-deficient papillomas (at 30 weeks of promotion) with archive papillomas generated in wild-type DBA/2 mice using the same protocol and did not find statistical significant differences (Figure 2).
Figure 2. Typical papilloma and atypical SCC with basaloid proliferation and follicular differentiation in nkt/nkt mice.
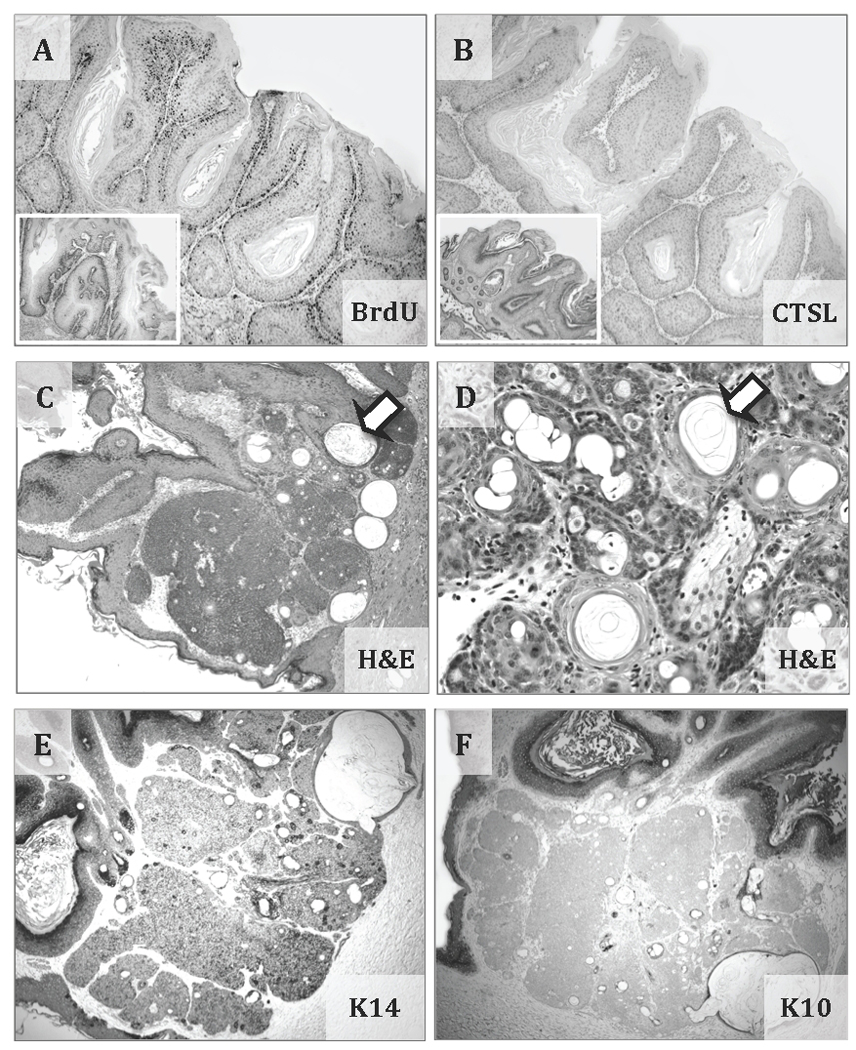
Representative immunohistochemistry images of a papilloma (at 30 weeks of promotion) from Ctsl-deficient DBA/2-nkt/nkt mice induced by the two-stage carcinogenesis protocol showing BrdU-labeled nuclei (A) and the absence of CTSL expression (B). Insets show wild-type papillomas from DBA/2 mice induced by the two-stage carcinogenesis (from archived samples). Representative hematoxylin and eosin staining images of an atypical SCC from Ctsl-deficient DBA/2-nkt/nkt mice showing epithelial proliferation composed of basaloid cells and the presence of keratin cysts (arrows) (C and D). Representative immunohistochemistry staining images showing the expression of basal cell marker K14 (E) and the absence of expression for the differentiation marker K10 (F) by the basaloid cells. Magnifications at ×40 (A, B, C, E and F) and ×100 (D).
Topical applications of TPA (2 µg twice-weekly for 20 wk) or DMBA (100 nmol, single dose) alone did not induce papillomas in DBA/2-nkt/nkt mice (data not shown). No differences were found between tumors in nkt/nkt and wild-type mice (SENCARB/Pt and DBA/2 background) when expression of p21, caspase-3, and VEGF, or BrdU-labeling index was compared by IHC (data not shown).
To determine whether the absence of CTSL was able to overcome the resistance of an inbred strain to two-stage skin carcinogenesis, we carried out a similar study using nkt/nkt mice on the relatively skin tumor resistant BALB/c background. However, BALB/c-nkt/nkt congenic mice (n = 10) and control littermates (n = 10) did not develop papillomas by 20 wk of promotion (data not shown). In addition, aging studies in SENCARB/Pt, BALB/c and DBA/2 mice showed no increase in spontaneous skin tumors during an observation period of 20 months when nkt/nkt mutants were compared with their wild-type counterparts (approximately 30 mice for each mutant and control group) (data not shown). This suggests that Ctsl is not acting as a typical tumor suppressor gene in our model.
Susceptible nkt/nkt Mice Carry the SCC-resistant Allele of Ptch1
Considering the close proximity of the patched homolog 1 (Ptch1) gene (63.6 Mb) and Ctsl (64.4 Mb) on mouse chromosome 13, we genotyped all our congenic strains carrying the Ctslnkt mutant allele for the SCC susceptible Ptch1 polymorphism described by Wakabayashi and colleagues for FVB/N mice [30]. Homozygous nkt/nkt mice from all nackt congenic strains carried the resistant B6 allele (PtchB6/threonine), ruling out an enhancement effect on the two-stage carcinogenesis due to this carboxy-terminal polymorphism in the Ptch1 gene. In contrast, the standard BALB/c, DBA/2, and SENCARB/Pt inbred strains shared the susceptible allele (PtchFVB/asparagine).
CD4 T-cell Deficiency Cannot Explain the Increased Skin Tumorigenesis in nackt Mice
To determine whether low levels of CD4 T-cells in nackt mice affected skin tumor development in our model, we conducted a two-stage skin carcinogenesis protocol using FVB/N-Cd4 −/− and wild-type littermates and assessed the number of papillomas per mouse for each genotype. Interestingly, tumor multiplicity at 24 wk was significantly higher in FVB/N wild-type, with an average of 14.2 papillomas per mouse (n = 8), than in CD4 T-cell-deficient FVB/N mice, with an average of 5.9 papillomas per mouse (n = 10; p<0.001) (Figure 3). Despite the difference in tumor multiplicity, no histological differences were found between Cd4 −/− and wild-type papillomas.
Figure 3. Two-stage carcinogenesis study in CD4 T-cell deficient mice.
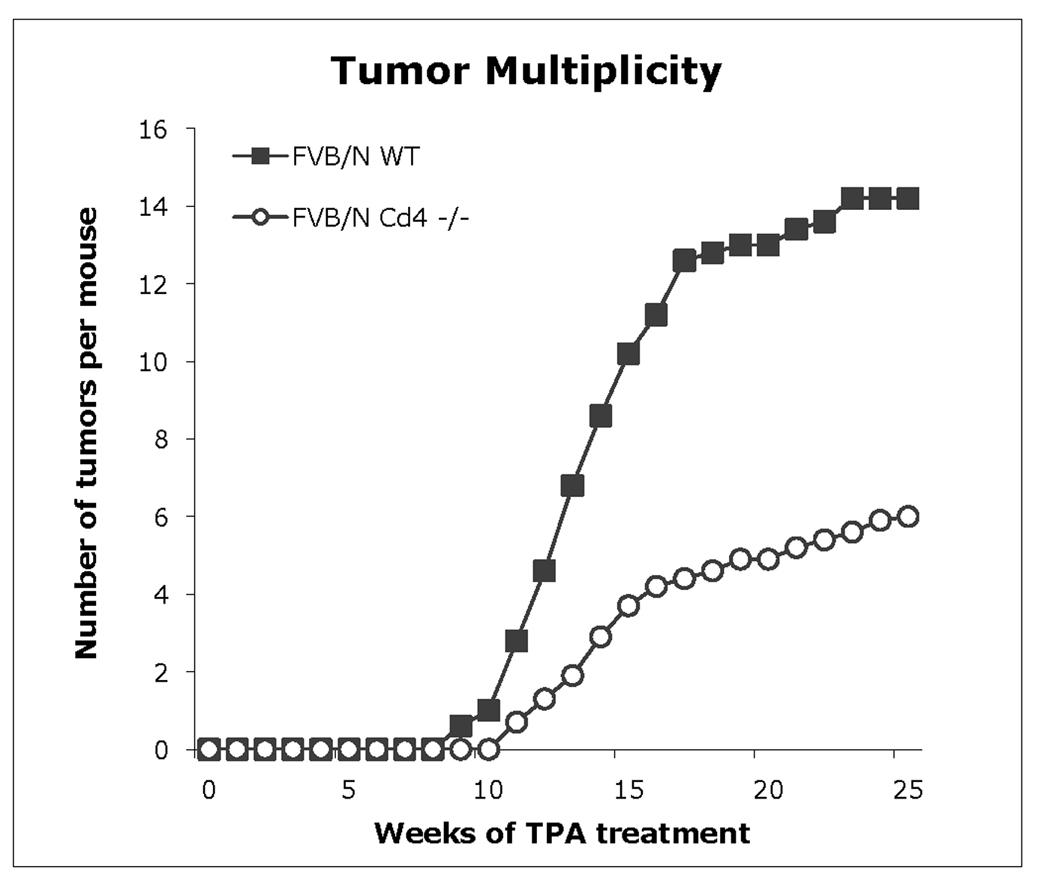
Tumor multiplicity of FVB/N-Cd4 −/− mice (n = 10) was statistically highly significant from that of FVB/N wild-type mice (n = 8) at 20 wk (P< 0.001, Wilcoxon Rank Sum test). Mice 6–8 wk old were initiated with DMBA and after two weeks received repeated applications of TPA as described in Materials & Methods. The number of papillomas was determined weekly. Tumor multiplicity is the average number of skin tumors per mouse. FVB/N wild-type mice (ν); Cd4 −/− mice (μ).
Epidermal Proliferation After TPA Treatment is Decreased in nkt/nkt Mice Compared to Wild-Type Littermates
To examine the role of CTSL in tumor promoter-induced epidermal proliferation in vivo, DBA/2-nkt/nkt mutant mice and littermate controls were treated topically with TPA. Twenty-four hours after a single application of 3.4 nmol of TPA, the epidermis of wild-type mice showed increased keratinocyte proliferation (compared to untreated skin), while the epidermis of Ctsl-deficient mice exhibited reduced proliferation compared to treated wild-type mice (data not shown). Following four topical treatments of 3.4 nmol of TPA over a 2-wk period, the epidermis of Ctsl-deficient mice still exhibited significantly reduced keratinocyte proliferation compared to wild-type mice (Figure 4A and B). The number of BrdU and Ki67 positive cells were reduced approximately 50% in the skin of DBA/2-nkt/nkt mice when compared to wild-type mice (Figure 4C and D). The epidermal thickness was also reduced in Ctsl-deficient mice compared with control littermates (Figure 4E). Western blot analysis of epidermal lysates from these TPA-treated skin samples showed a small increase in p21 protein expression (Figure 4F), which may explain, in part, the decreased epidermal proliferation. Interestingly, epidermal cell proliferation measured by BrdU labeling in untouched nkt/nkt skin did not reveal significant differences with wild-type skin, even though mild epidermal hyperplasia is one of the pathological features of the mutant skin [4].
Figure 4. Epidermal proliferation after TPA treatment.

Representative BrdU immunostaining of TPA-treated dorsal skin from a DBA/2-nkt/nkt mouse (A) and a wild-type littermate (B) (both magnifications ×100). Mice were treated with four topical applications of 3.4 nmol of TPA over a 2-wk period and sacrificed 24 h after the final treatment. Bar graphs show cell proliferation level as measured by Ki67 (C) and BrdU (D) indexes, as well as epidermal thickness (E). The determination of epidermal thickness and labeling index was performed as described in Materials and Methods. Values represent mean ± SD (*P<.05). Black bars, mutant mice; grey bars, wild-type littermates. (F) Western blot analysis of p21 in the epidermis of TPA-treated dorsal skin from DBA/2-nkt/nkt mice and littermate controls. Protein was normalized to β-actin.
Western blotting and IHC from TPA-treated skin did not reveal differences in expression of p27, Stat3, or VEGF between wild-type and mutant mice (data not shown). We also examined expression of CTSB and CTSD to rule out compensatory overexpression of these lysosomal proteases in Ctsl-deficient mice. Moderate overexpression of CTSD was observed by Western blotting (1.5-fold) and by two-dimensional gel electrophoresis followed by mass spectrometry (2.8-fold up-regulation) (data not shown). No significant differences were found in the expression levels of CTSB.
Deficiency of CTSL in the Skin Alters Keratinocyte Transit After TPA Treatment
We analyzed keratinocyte transit through the epidermis by following the fate of BrdU-labeled cells at 1, 8, and 30 h after BrdU injection (17 h after the last TPA application). The transit of keratinocytes was delayed in DBA/2-nkt/nkt compared with wild-type skin in the three time points analyzed. After 30 h, many labeled keratinocytes of the wild-type skin had reached the granular and cornified layers of the epidermis, whereas in the mutant skin, basal and suprabasal keratinocytes retained most of the BrdU-labeled nuclei (Figure 5). Our analysis showed that the absence of CTSL resulted in delayed terminal differentiation of keratinocytes and decreased epidermal cell turnover. Because ordered differentiation, upward migration, and exfoliation of peripheral keratinocytes are essential for timely removal of cells harboring oncogenic mutations, the increased transit time in nkt/nkt mice may lead to retention of DMBA-initiated keratinocytes and increased tumorigenesis.
Figure 5. Keratinocyte transit after TPA treatment.
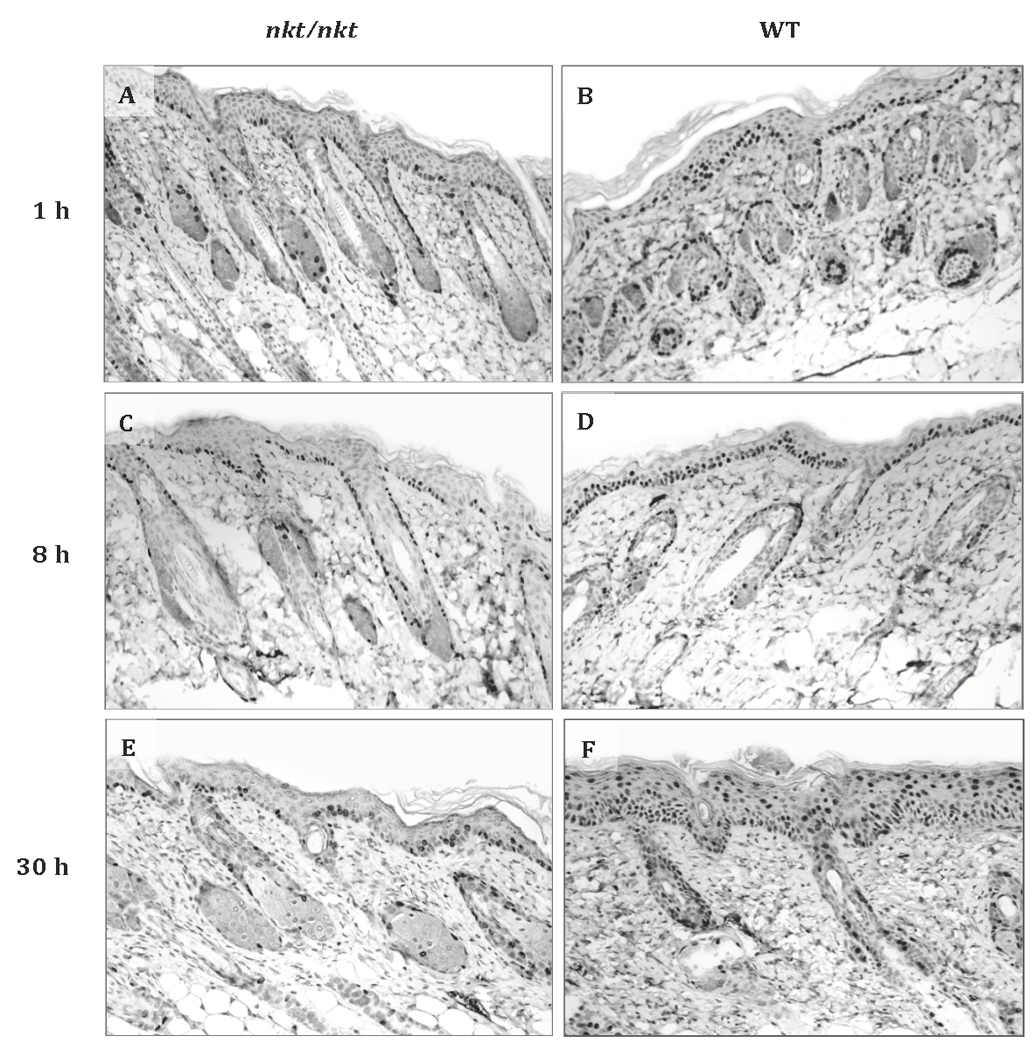
Representative BrdU immunostaining of TPA-treated dorsal skin from a DBA/2-nkt/nkt mouse and a wild-type littermate after 1 h (A and B), 8 h (C and D), and 30 h (E and F) of BrdU injection (all magnifications ×100). Mice were injected i.p. with BrdU 17 h after the last of four TPA applications (3.4 nmol) over a two week period and sacrificed 1, 8, and 30 h after BrdU injection. A total of 4 mice per genotype per time point were evaluated. At 8 h and 30 h after BrdU injection, progression of BrdU-labeled keratinocytes towards the surface is clearly slower in the mutant epidermis. Percentages of BrdU-labeled nuclei in the basal and suprabasal layer for the three time points are shown in Table 1.
Discussion
Cellular proteases like CTSL promote tumor growth, invasion, and metastasis [32], and CTSL is considered a clinical marker for invasive tumors [33] [34]. High levels of CTSL have been found in nearly all human epithelial tumors in comparison with normal tissues, including breast [35], prostate [36], colorectal tumors [37], and in head and neck SCC [38]. Our finding that CTSL expression increased during two-stage skin carcinogenesis is consistent with previous reports of the strong association between high levels of cathepsins and cancer. Thus our finding that nkt/nkt mice, which do not express functional CTSL, have enhanced susceptibility to two-stage skin carcinogenesis compared to wild-type mice was unexpected. However, a protective role for CTSL in skin cancer was previously demonstrated using Ctsl −/− mice crossed with the HPV16-transgenic mice [39] [40]. In this in vivo model, Ctsl deficiency promotes skin carcinogenesis and lymph node metastasis, with HPV16; Ctsl −/− mice showing early onset of dysplastic and neoplastic lesions and poorly differentiated carcinomas [40]. Our findings are also supported by recent reports showing increased DMBA/TPA-induced skin carcinogenesis in transgenic mice overexpressing the CTSL inhibitor hurpin [41] as well as increased intestinal neoplasia in ApcMin;furless (Ctsl deficient) mice [42]. Similar unexpected results in two-stage skin carcinogenesis were obtained with collagenase-2 (MMP8) [43] and Id1 [44] knockout mice.
On the other hand, Ctsl deficiency in a mouse model of pancreatic cancer reduces tumor burden and invasiveness [2], suggesting that the protective role of CTSL is organ- or tissue-specific. In agreement with this, our experiments with MNU-induced thymic lymphomas revealed no differences in tumor incidence between DBA/2-nkt/nkt and littermate controls. Both groups exhibited approximately 92% tumor incidence 160 days after a single injection of 75 mg/kilogram body weight of MNU (Benavides, unpublished).
It was recently reported that keratinocytes from Ctsl knock-out mice show enhanced proliferation, suggesting that CTSL regulates keratinocyte proliferation by controlling growth factor recycling [39]. However, in our Ctsl-deficient nackt model there was decreased keratinocyte proliferation in TPA-treated skin. The difference in keratinocyte proliferation may be attributable to the different genetic backgrounds in these models. The Ctsl knock-out model was described mainly in 129P2;C57BL/6 or 129P2;C57BL/6;FVB (when crossed with HPV16 transgenic mice) mixed genetic backgrounds, whereas the nackt model was studied using defined congenic strains in SENCARB/Pt, DBA/2, and BALB/c inbred backgrounds. Additionally, when compared with archived papillomas generated in wild-type DBA/2 mice, the proliferation rate of the Ctsl-deficient papillomas was similar, suggesting that the mutant keratinocytes overcome their resistance to TPA.
How CTSL deficiency leads to enhanced skin tumorigenesis is unclear. As previously reported by Dennem‰rker et al [40], the absence of CTSL did not affect angiogenesis or apoptosis during two-stage carcinogenesis in our model, since IHC and Western blot analysis using antibodies against CD31 and activated caspase-3 revealed no significant differences between mutant and wild-type papillomas. On the other hand, our data obtained from the TPA-induced epidermal proliferation studies support the idea that disrupted terminal differentiation of the epidermis, together with decreased turnover of initiated keratinocytes, could be responsible for the enhanced skin tumorigenesis. The influence of keratinocyte turnover on skin carcinogenesis was previously described in K10 [45] and in Cox-1 and Cox-2 knockout [29] mice. In these cases, the exit of keratinocytes from the basal layer was accelerated and increased turnover resulted in decreased tumor formation after two-stage carcinogenesis. Since therapeutics inhibiting CTSL are being developed to treat human cancer, it is very important to carefully evaluate the potential of CTSL to promote rather than prevent tumorigenesis in different organs.
Table 1.
Epidermal turnover after TPA*
| BrdU-labeled nuclei/200 nuclei | ||||
|---|---|---|---|---|
| nkt/nkt mice | Wild-type mice | |||
| Time after BrdU† | Basal | Suprabasal | Basal | Suprabasal |
| 1h | 29.8 ± 3.2 | 1.1 ± 0.8 | 33.0 ± 8.8 | 3.2 ± 1.4 |
| 8h | 33.3 ± 7.0 | 1.8 ± 0.9 | 40.6 ± 4.9 | 14.1 ± 5.4 |
| 30h | 25.6 ± 4.7 | 14.9 ± 3.7 | 25.9 ± 7.5 | 30.9 ± 4.7 |
12-O-tetradecanoylphorbol-13-acetate
5-bromo-2-deoxyuridine
Acknowledgments
We thank Lezlee Coghlan, Pamela S. Huskey, April Weiss, Nancy Doradau, Dale Weiss, and Donna Schutz for their assistance with maintenance of the mouse strains. We are grateful to Marcela Franco, MÛnica Flores, and John Bartonico for technical assistance and Melissa Bracher for administrative assistance. We also wish to thank Kevin Lin for statistical analyses and the Histology and Tissue Processing Facility Core for the processing of samples. This work was supported by Pilot Project from NIEHS Center Grant ES07784 to FB and NIH Grant CA90922 to CJC. This study also made use of the Research Animal Support Facility-Smithville, including Genetic Services and Mutant Mouse Pathology Service, supported by P30 CA16672-30 DHHS/NCI Cancer Center Support Grant (CCSG). FVB/N-CD4 knock-out congenic mice where a gift from Lisa M. Coussens from the University of California at San Francisco, San Francisco, CA.
Abbreviations
- BrdU
5-bromo-2-deoxyuridine
- TPA
12-O-tetradecanoylphorbol-13-acetate
- DMBA
7,12-dimethylbenz[a]anthracene
- i.p.
intraperitoneal
- PBS
phosphate-buffered saline
- SCC
squamous cell carcinoma
- CTSL
cathepsin L
References
- 1.Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer. 2006;6(10):764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- 2.Gocheva V, Zeng W, Ke D, et al. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006;20(5):543–556. doi: 10.1101/gad.1407406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roth W, Deussing J, Botchkarev VA, et al. Cathepsin L deficiency as molecular defect of furless: hyperproliferation of keratinocytes and pertubation of hair follicle cycling. Faseb J. 2000;14(13):2075–2086. doi: 10.1096/fj.99-0970com. [DOI] [PubMed] [Google Scholar]
- 4.Benavides F, Starost MF, Flores M, Gimenez-Conti IB, Guenet JL, Conti CJ. Impaired hair follicle morphogenesis and cycling with abnormal epidermal differentiation in nackt mice, a cathepsin L-deficient mutation. Am J Pathol. 2002;161(2):693–703. doi: 10.1016/S0002-9440(10)64225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tobin DJ, Foitzik K, Reinheckel T, et al. The lysosomal protease cathepsin L is an important regulator of keratinocyte and melanocyte differentiation during hair follicle morphogenesis and cycling. Am J Pathol. 2002;160(5):1807–1821. doi: 10.1016/S0002-9440(10)61127-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joyce JA, Baruch A, Chehade K, et al. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004;5(5):443–453. doi: 10.1016/s1535-6108(04)00111-4. [DOI] [PubMed] [Google Scholar]
- 7.Levicar N, Dewey RA, Daley E, et al. Selective suppression of cathepsin L by antisense cDNA impairs human brain tumor cell invasion in vitro and promotes apoptosis. Cancer Gene Ther. 2003;10(2):141–151. doi: 10.1038/sj.cgt.7700546. [DOI] [PubMed] [Google Scholar]
- 8.Stoka V, Turk B, Turk V. Lysosomal cysteine proteases: structural features and their role in apoptosis. IUBMB Life. 2005;57(4–5):347–353. doi: 10.1080/15216540500154920. [DOI] [PubMed] [Google Scholar]
- 9.Stoka V, Turk V, Turk B. Lysosomal cysteine cathepsins: signaling pathways in apoptosis. Biol Chem. 2007;388(6):555–560. doi: 10.1515/BC.2007.064. [DOI] [PubMed] [Google Scholar]
- 10.Goulet B, Baruch A, Moon NS, et al. A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol Cell. 2004;14(2):207–219. doi: 10.1016/s1097-2765(04)00209-6. [DOI] [PubMed] [Google Scholar]
- 11.Goulet B, Sansregret L, Leduy L, et al. Increased expression and activity of nuclear cathepsin L in cancer cells suggests a novel mechanism of cell transformation. Mol Cancer Res. 2007;5(9):899–907. doi: 10.1158/1541-7786.MCR-07-0160. [DOI] [PubMed] [Google Scholar]
- 12.Sullivan S, Tosetto M, Kevans D, et al. Localization of nuclear cathepsin L and its association with disease progression and poor outcome in colorectal cancer. Int J Cancer. 2009;125(1):54–61. doi: 10.1002/ijc.24275. [DOI] [PubMed] [Google Scholar]
- 13.Santamaria I, Velasco G, Cazorla M, Fueyo A, Campo E, Lopez-Otin C. Cathepsin L2, a novel human cysteine proteinase produced by breast and colorectal carcinomas. Cancer Res. 1998;58(8):1624–1630. [PubMed] [Google Scholar]
- 14.Puente XS, Lopez-Otin C. A genomic analysis of rat proteases and protease inhibitors. Genome Res. 2004;14(4):609–622. doi: 10.1101/gr.1946304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benavides F, Giordano M, Fiette L, et al. Nackt (nkt), a new hair loss mutation of the mouse with associated CD4 deficiency. Immunogenetics. 1999;49(5):413–419. doi: 10.1007/s002510050514. [DOI] [PubMed] [Google Scholar]
- 16.Benavides F, Venables A, Poetschke Klug H, et al. The CD4 T cell-deficient mouse mutation nackt (nkt) involves a deletion in the cathepsin L (CtsI) gene. Immunogenetics. 2001;53(3):233–242. doi: 10.1007/s002510100320. [DOI] [PubMed] [Google Scholar]
- 17.Benavides F, Perez C, Blando J, Guenet JL, Conti CJ. The radiation-induced nackt (nkt) allele is a loss-of-function mutation of the mouse cathepsin L gene. J Immunol. 2006;176(2):702–703. doi: 10.4049/jimmunol.176.2.702. [DOI] [PubMed] [Google Scholar]
- 18.Lombardi G, Burzyn D, Mundinano J, et al. Cathepsin-L influences the expression of extracellular matrix in lymphoid organs and plays a role in the regulation of thymic output and of peripheral T cell number. J Immunol. 2005;174(11):7022–7032. doi: 10.4049/jimmunol.174.11.7022. [DOI] [PubMed] [Google Scholar]
- 19.Nakagawa T, Roth W, Wong P, et al. Cathepsin L: critical role in Ii degradation and CD4 T cell selection in the thymus. Science. 1998;280(5362):450–453. doi: 10.1126/science.280.5362.450. [DOI] [PubMed] [Google Scholar]
- 20.Nishimura F, Naruishi H, Naruishi K, et al. Cathepsin-L, a key molecule in the pathogenesis of drug-induced and I-cell disease-mediated gingival overgrowth: a study with cathepsin-L-deficient mice. Am J Pathol. 2002;161(6):2047–2052. doi: 10.1016/S0002-9440(10)64483-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reinheckel T, Deussing J, Roth W, Peters C. Towards specific functions of lysosomal cysteine peptidases: phenotypes of mice deficient for cathepsin B or cathepsin L. Biol Chem. 2001;382(5):735–741. doi: 10.1515/BC.2001.089. [DOI] [PubMed] [Google Scholar]
- 22.Stypmann J, Glaser K, Roth W, et al. Dilated cardiomyopathy in mice deficient for the lysosomal cysteine peptidase cathepsin L. Proc Natl Acad Sci U S A. 2002;99(9):6234–6239. doi: 10.1073/pnas.092637699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petermann I, Mayer C, Stypmann J, et al. Lysosomal, cytoskeletal, and metabolic alterations in cardiomyopathy of cathepsin L knockout mice. Faseb J. 2006;20(8):1266–1268. doi: 10.1096/fj.05-5517fje. [DOI] [PubMed] [Google Scholar]
- 24.Yang M, Zhang Y, Pan J, et al. Cathepsin L activity controls adipogenesis and glucose tolerance. Nat Cell Biol. 2007;9(8):970–977. doi: 10.1038/ncb1623. [DOI] [PubMed] [Google Scholar]
- 25.Potts W, Bowyer J, Jones H, et al. Cathepsin L-deficient mice exhibit abnormal skin and bone development and show increased resistance to osteoporosis following ovariectomy. Int J Exp Pathol. 2004;85(2):85–96. doi: 10.1111/j.0959-9673.2004.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wright WW, Smith L, Kerr C, Charron M. Mice that express enzymatically inactive cathepsin L exhibit abnormal spermatogenesis. Biol Reprod. 2003;68(2):680–687. doi: 10.1095/biolreprod.102.006726. [DOI] [PubMed] [Google Scholar]
- 27.Wakeland E, Morel L, Achey K, Yui M, Longmate J. Speed congenics: a classic technique in the fast lane (relatively speaking) Immunol Today. 1997;18(10):472–477. doi: 10.1016/s0167-5699(97)01126-2. [DOI] [PubMed] [Google Scholar]
- 28.Stern MC, Benavides F, LaCava M, Conti CJ. Genetic analyses of mouse skin tumor progression susceptibility using SENCAR inbred derived strains. Mol Carcinog. 2002;35(1):13–20. doi: 10.1002/mc.10067. [DOI] [PubMed] [Google Scholar]
- 29.Tiano HF, Loftin CD, Akunda J, et al. Deficiency of either cyclooxygenase (COX)-1 or COX-2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer Res. 2002;62(12):3395–3401. [PubMed] [Google Scholar]
- 30.Wakabayashi Y, Mao JH, Brown K, Girardi M, Balmain A. Promotion of Hras-induced squamous carcinomas by a polymorphic variant of the Patched gene in FVB mice. Nature. 2007;445(7129):761–765. doi: 10.1038/nature05489. [DOI] [PubMed] [Google Scholar]
- 31.Stern MC, Benavides F, Klingelberger EA, Conti CJ. Allelotype analysis of chemically induced squamous cell carcinomas in F(1) hybrids of two inbred mouse strains with different susceptibility to tumor progression. Carcinogenesis. 2000;21(7):1297–1301. [PubMed] [Google Scholar]
- 32.Kane SE, Gottesman MM. The role of cathepsin L in malignant transformation. Semin Cancer Biol. 1990;1(2):127–136. [PubMed] [Google Scholar]
- 33.Tumminello FM, Leto G, Pizzolanti G, et al. Cathepsin D, B and L circulating levels as prognostic markers of malignant progression. Anticancer Res. 1996;16(4B):2315–2319. [PubMed] [Google Scholar]
- 34.Cuvier C, Jang A, Hill RP. Exposure to hypoxia, glucose starvation and acidosis: effect on invasive capacity of murine tumor cells and correlation with cathepsin (L + B) secretion. Clin Exp Metastasis. 1997;15(1):19–25. doi: 10.1023/a:1018428105463. [DOI] [PubMed] [Google Scholar]
- 35.Lah TT, Kalman E, Najjar D, et al. Cells producing cathepsins D, B, and L in human breast carcinoma and their association with prognosis. Hum Pathol. 2000;31(2):149–160. doi: 10.1016/s0046-8177(00)80214-2. [DOI] [PubMed] [Google Scholar]
- 36.Friedrich B, Jung K, Lein M, et al. Cathepsins B, H, L and cysteine protease inhibitors in malignant prostate cell lines, primary cultured prostatic cells and prostatic tissue. Eur J Cancer. 1999;35(1):138–144. doi: 10.1016/s0959-8049(98)00273-1. [DOI] [PubMed] [Google Scholar]
- 37.Herszenyi L, Plebani M, Carraro P, et al. The role of cysteine and serine proteases in colorectal carcinoma. Cancer. 1999;86(7):1135–1142. doi: 10.1002/(sici)1097-0142(19991001)86:7<1135::aid-cncr6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 38.Strojan P, Budihna M, Smid L, et al. Prognostic significance of cysteine proteinases cathepsins B and L and their endogenous inhibitors stefins A and B in patients with squamous cell carcinoma of the head and neck. Clin Cancer Res. 2000;6(3):1052–1062. [PubMed] [Google Scholar]
- 39.Reinheckel T, Hagemann S, Dollwet-Mack S, et al. The lysosomal cysteine protease cathepsin L regulates keratinocyte proliferation by control of growth factor recycling. J Cell Sci. 2005;118(Pt 15):3387–3395. doi: 10.1242/jcs.02469. [DOI] [PubMed] [Google Scholar]
- 40.Dennemarker J, Lohmuller T, Mayerle J, et al. Deficiency for the cysteine protease cathepsin L promotes tumor progression in mouse epidermis. Oncogene. 29(11):1611–1621. doi: 10.1038/onc.2009.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walz M, Kellermann S, Bylaite M, et al. Expression of the human Cathepsin L inhibitor hurpin in mice: skin alterations and increased carcinogenesis. Exp Dermatol. 2007;16(9):715–723. doi: 10.1111/j.1600-0625.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 42.Boudreau F, Lussier CR, Mongrain S, et al. Loss of cathepsin L activity promotes claudin-1 overexpression and intestinal neoplasia. Faseb J. 2007 doi: 10.1096/fj.07-8113com. [DOI] [PubMed] [Google Scholar]
- 43.Balbin M, Fueyo A, Tester AM, et al. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet. 2003;35(3):252–257. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- 44.Sikder H, Huso DL, Zhang H, et al. Disruption of Id1 reveals major differences in angiogenesis between transplanted and autochthonous tumors. Cancer Cell. 2003;4(4):291–299. doi: 10.1016/s1535-6108(03)00245-9. [DOI] [PubMed] [Google Scholar]
- 45.Reichelt J, Furstenberger G, Magin TM. Loss of keratin 10 leads to mitogen-activated protein kinase (MAPK) activation, increased keratinocyte turnover, and decreased tumor formation in mice. J Invest Dermatol. 2004;123(5):973–981. doi: 10.1111/j.0022-202X.2004.23426.x. [DOI] [PubMed] [Google Scholar]