

Abstract
Parental phenotype is known to influence the inheritance of atopic diseases, such as allergic asthma, with a maternal history being a more significant risk factor for progeny than paternal history. We hypothesized that recall Th1- or Th2-type immune responses during pregnancy would result in transfer of maternal factors that would differentially impact development of immune responsiveness in offspring. Following weaning, susceptibility and severity of allergic airway disease (a murine model of human asthma) was evaluated in progeny, disease being elicited by immunization with OVA-Al(OH)3 and challenge with aerosolized OVA. We found that progeny of mothers with Th1-biased immunity to OVA subjected to recall aerosol challenge during pregnancy had reduced levels of Ag-specific IgE and airway eosinophilia compared with progeny of mothers with Th2-biased immunity to OVA or naive mothers. Interestingly, progeny of mothers with Th1-type immunity to a heterologous albumin, BSA, were not protected from developing OVA-induced allergic airway disease. These findings demonstrated that maternal transfer of protection from development of allergic airway disease to offspring in this model of maternal Th1-type immunity was Ag specific.
Asthma is a chronic inflammatory disorder of the airways that is multifactorial in origin. It is known that CD4+ T lymphocytes play critical roles in disease pathogenesis. In individuals susceptible to allergic asthma, exposure to allergens results in the elicitation of allergen-specific CD4+ T cells capable of producing Th2 cytokines such as IL-4, IL-5, and IL-13. Th2 cytokines produced upon allergen re-exposure contribute to the effector phase of the allergic response in the lung via induction of IgE production by B cells, recruitment of eosinophils to the airway, bronchial hyperreactivity, and hyperplasia of epithelial goblet cells (1– 8). Although some pulmonary inflammation in response to inhaled Ag can occur in the absence of CD4+ cells, B cells, or Ig (9–14), their presence and effector functions are hallmarks of the allergic response in vivo in animal models of asthma and in humans with disease.
In contrast to adults, immune responses elicited in early life can be predominated by Ag-specific CD4+ T cells producing Th2 cytokines and a reduced frequency of IFN-γ-producing CD8+ cells. This tendency toward generation of Th2-biased immunological memory has been observed in neonatal mice and humans, even in response to infectious agents (15–17). The potential mechanisms underlying the immaturity of the immune system in early life are not completely understood, but have been suggested to be the result of developmental or functional deficiencies in APCs (18), neonatal T cells (19), or both (17).
It has been proposed that decreased exposure to stimuli capable of eliciting Th1-type immune responses during childhood is a mechanism to explain the increasing prevalence of allergic diseases such as asthma (20, 21). Although the concept as originally envisaged has been modified as the field evolved in the past 15 years, support remains for the idea that exposures to microbes could be important to direct maturation of the immune system in early life toward a healthy balance of Th1- and Th2-type immune responsiveness. Epidemiologic studies are supportive of this “hygiene hypothesis,” whereby decreased exposure to other children, infectious diseases, or microbial products is associated with increased risk of allergic disease (22–25). In addition, the ability of certain environmental conditions to increase or decrease asthma susceptibility, such as day care attendance or endotoxin exposure, can be transposed or eliminated based on parental history of atopy or asthma (26, 27). Parental phenotype is also known to influence the inheritance of atopic diseases, such as allergic asthma, with a maternal history being a more significant risk factor for progeny than paternal history. The mechanisms that explain parent-of-origin effects on this or other immune-mediated inflammatory diseases (such as diabetes, rheumatoid arthritis, or inflammatory bowel disease) is not known, but could be the result of immune interactions between mothers and their offspring in utero via the placenta or in early postnatal life via breast milk (28–32).
Understanding the mechanisms that mediate the maternal influence on attenuation or exacerbation of disease development in offspring is critical to prevention of childhood asthma, and perhaps other inflammatory diseases. We hypothesized that modulation of the maternal immune environment would affect development of immune responsiveness in offspring via transfer of factors to influence susceptibility or resistance to development of allergic airway disease. The idea that maternal immunity can affect development of immune responsiveness in children was not new. In fact, the role of passively transferred maternal Abs in protection of the fetus and newborn is a fundamental concept in immunology (33). Further, numerous studies demonstrate that maternal IgG can suppress IgE responses in offspring (34), although the mechanism by which the suppression occurs is not completely understood. We developed models of Th1- vs Th2-mediated airway disease and determined that following resolution of disease symptoms during 6– 8 wk, disease could be reinstated by re-exposure to aerosolized Ag. Parameters of disease severity, as indicated by Ag-specific Ig levels in serum and distribution of leukocyte populations in the airways, were virtually unaffected when the recall aerosol challenge was during pregnancy. Therefore, using this model, we produced offspring exposed to the effects of maternal-derived Th1- or Th2-type immune responses in utero and during nursing. We then determined the susceptibility or resistance of offspring to development of allergic airway disease following immunization with OVA-Al(OH)3 and aerosol challenge. We found that progeny of Th1-biased OVA-immune mothers subjected to recall aerosol challenge during pregnancy had reduced levels of Ag-specific IgE and airway eosinophilia compared with progeny of Th2-biased OVA immune or naive mothers. Interestingly, this effect was Ag specific in offspring because maternal Th1-biased immunity directed against a heterologous albumin (BSA) did not result in transmission of protection from OVA-induced allergic airway disease.
Materials and Methods
Animals
Male or female C57BL/6J mice were obtained from The Jackson Laboratory or bred in our colony at the University of Connecticut Health Center. All mice were fed sterile food and water and housed in microisolators under specific pathogen-free conditions. Their care was in accordance with institutional and Office of Laboratory Animal Welfare guidelines.
Ag exposures
The sensitization and challenge protocol used to establish maternal airway disease, summarized in Fig. 1, was modified from the 6-wk discontinuous model characterized by Schramm et al. (35). Six-week-old female C57BL/6J mice were immunized with OVA (25 μg, grade V; Sigma-Aldrich) emulsified in CFA containing 250 μg of killed Mycobacterium tuberculosis H37 Ra (BD Diagnostic Systems) (s.c.; Th1-type conditions) or adsorbed to 2 mg of Al(OH)3 (i.p.; Th2-type conditions) separated by at least 7 days. The second immunization of OVA-CFA-primed mice was with 25 μg of OVA emulsified in IFA (i.p.). Nine-35 days following the final immunization, animals were exposed daily to aerosolized Ag generated from 1% OVA in normal saline using a Bioaerosol Nebulizing Generator (BANG; CH Technologies). Exposures were 1 h for 7 consecutive days delivered via a nose-only inhalation exposure chamber with space for exposing 48 mice simultaneously (In-Tox Products). In some experiments, 6– 8 wk after the initial aerosol exposure, a time when the inflammatory response had resolved (35), females were bred with naive C57BL/6J males. Pregnant mice were subjected to a secondary challenge with aerosolized OVA daily, at embryonic day (E)3 E11–17 of pregnancy (day of vaginal plug is E0). Serum was collected 1 wk after the second i.p. immunization, 24 h after the 7 day primary aerosol challenge, and on day E18 (24 h after the 7 day secondary aerosol challenge). The gestational period in mice is 19–20 days and mice were not manipulated on those days. Some pregnant mice were sacrificed at E18, 24 h after the secondary aerosol challenge for analysis of airway leukocytes in bronchoalveolar lavage (BAL) fluid.
FIGURE 1.
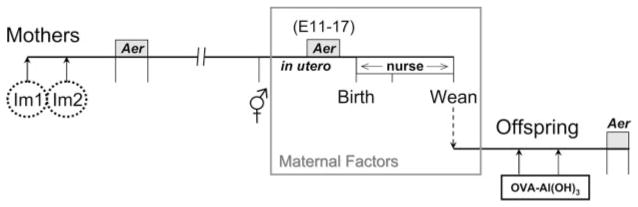
Model to determine the impact of maternal immunity on development of allergic airway disease in offspring. Imm, Immunization (OVA-CFA, OVA-Al(OH)3 or BSA-CFA); Aer, challenge with aerosolized Ag (always matched to immunizing Ag). Parameters of OVA-induced allergic airway disease severity assessed in offspring were OVA-specific Ig levels in serum and composition of leukocytes in the airways.
To evaluate the environmental effects of maternal derived Th1-type immune responses on the innate immune system of offspring, a parallel experiment was performed using 6-wk-old female C57BL/6J mice immunized with a heterologous Ag, BSA (25 μg, minimum 98%; Sigma-Aldrich). The protocol for immunization was identical with the Th1-type conditions described for OVA. Five weeks following the second immunization, animals were exposed to aerosolized Ag generated from 1% BSA in normal saline. Daily exposures were 1 h for 7 consecutive days. To avoid cross-contamination between OVA and BSA, it was necessary to generate aerosolized BSA using a separate BANG and to deliver the aerosolized Ag via a separate nose-only inhalation exposure chamber with space for exposing 12 mice simultaneously (In-Tox Products). Seven weeks after the initial aerosol exposure, females were bred to naive C57BL/6J males and subjected to secondary challenge with aerosolized BSA during pregnancy exactly as described above.
Susceptibility or resistance to development of OVA-induced allergic airway disease was assessed in offspring with different histories of exposure to the effects of maternal-derived immune responses. Pups were from mothers with Th1-type immunity to OVA (OVA-CFA) or BSA (BSA-CFA), or Th2-type immunity to OVA (OVA-Al(OH)3) subjected to recall challenge with aerosolized Ag during pregnancy. Control pups were from naive mothers never exposed to Ag. Pups 4–5 wk of age were immunized with 25 μg of OVA adsorbed to 2 mg of Al(OH)3 and 7 days later with 8 μg of OVA adsorbed to 2 mg of Al(OH)3. Three to 5 wk after the second immunization, mice were subjected to challenge with aerosolized 1% OVA daily for 4 or 7 days (1 h exposure time). Mice were sacrificed 24 h after the last aerosol exposure for assessment of disease severity as indicated by OVA-specific Ig levels in serum and distribution of leukocyte populations in the airways.
Analysis of airway leukocytes
BAL was performed 24 h after the last aerosol challenge, under terminal ketamine/xylazine anesthesia. Lungs from each animal were lavaged in situ with five 1-ml aliquots of sterile saline. Total leukocyte counts were performed with a hemacytometer using trypan blue dye exclusion as a measure of viability or with a Z2 Coulter Counter (6–20 μm; Beckman Coulter). The BAL cell differential was determined by analysis of cytocentrifuged slide preparations stained with Wright-Giemsa.
Flow cytometry
mAbs used to identify airway leukocyte populations collected from BAL fluid were anti-CD45-FITC (30-F11), -TCRβ-PE (H57-597), -CD11b-PerCP-Cy5.5 (M1/70), -CD19-PE (1D3), -CD4-PerCP (RM4-5),- CD8α-allophycocyanin (53-6.7), and -CD90.2-allophycocyanin (53-2.1) purchased from BD Pharmingen. Cells (104–106) were incubated with 100 μl of appropriately diluted mAbs in PBS containing 0.2% BSA and 0.1% NaN3 for 30 min at 4°C, and then washed twice with the same buffer. Relative fluorescence intensities were determined on a 4-decade log scale by flow cytometric analysis using a FACSCalibur (BD Biosciences).
ELISPOT
Following sensitization with OVA-CFA or OVA-Al(OH)3, the frequencies of splenic CD4+ or CD8+ T cells capable to produce cytokines in response to stimulation with OVA peptide Ag were determined. Five days after the second i.p. immunization, suspensions of spleen cells were prepared using a ground glass homogenizer and passed through 100 μm of NITEX nylon mesh (Tetko) to remove connective tissue. RBCs were lysed via two sequential incubations in Tris-ammonium chloride (13 mM Tris, 135 nM NH4Cl (pH 7.2)) for 4 min at 37°C. Splenocytes were suspended in RPMI 1640 containing 10% FBS and 2 × 10−5 M 2-ME. ELISPOTs were performed essentially as described (36). Cells were plated in duplicate (as four, 2-fold serial dilutions starting at 106 cells/well) onto opaque membrane-sealed 96-well ELISPOT plates (Millipore) previously coated with Abs to capture individual cytokines (anti-IL-4 or -IFN-γ, obtained from BD Pharmingen). After 33 h of culture at 37°C with or without OVA peptides, cells were removed by washing with distilled water. Cytokines captured from CD4+ or CD8+ T cells were visualized by sequential incubations of the membrane-coated plates using biotinylated Abs directed against a distinct epitope in the homologous cytokine, followed by HRP-conjugated anti-biotin (Vector Laboratories). The substrate for development was 3-amino-9-ethylcarbazole and the signature generated by the cytokine-producing cell in this assay was a spot. The ImmunoSpot Image Analyzer (Cellular Technology) was used to enumerate cytokine-producing cells on the basis of the comparison of control (T cells cultured without OVA peptides) and experimental wells (T cells cultured in the presence of cognate MHC class II- (OVA323–339 and OVA265–280) or MHC class I-(OVA257–264) restricted OVA peptides) to detect OVA-specific CD4+ or CD8+ T cells, respectively.
Determination of serum Ig levels
Serum Ig levels were measured by ELISA using isotype-specific capture Abs. Specifically, Costar 3590 microtiter plates (Corning) were coated with rat anti-mouse IgE (R35-72) or IgG1 (A85-3) (BD Pharmingen), goat anti-mouse IgG2a or IgA (Southern Biotechnology Associates) at 2 μg/ml in PBS for 16 h at 4°C. After blocking nonspecific binding, isotype-specific Abs were captured in duplicate, as three to four, 2-fold serial dilutions of serum (within established linear ranges of the standard for each individual isotype). Detection of Ag-specific Igs was with OVA-digoxigenin or BSA-digoxigenin conjugates followed by anti-digoxigenin-peroxidase (Roche Diagnostics) essentially as described (37). Development was with the TMB microwell peroxidase substrate system (Kirkegaard & Perry Laboratories) and A450 measured with a Bio-Rad model 480 microplate reader.
Noninvasive whole body plethysmography
Changes in breathing pattern were measured in unrestrained animals by barometric plethysmography using the whole body plethysmograph (Buxco). The body plethysmograph detects volume and resultant pressure changes during the respiratory cycle of the animal (38, 39). Mice were placed in a main chamber of the whole body plethysmograph, which was previously calibrated with a rapid injection of 1 ml of air. After a 5-min adjustment to the body box, baseline readings were taken and averaged over 1 min. Aerosolized normal saline (control) or methacholine in increasing concentrations (3–100 mg/ml) were nebulized through an inlet of the chamber for 2 min, and readings are taken every 10 s for 4 min after the nebulization. The readings 10 and 20 s before and after the peak response were averaged with the peak response for the enhanced pause (Penh) value associated with that nebulization. Data are reported as the percent change in Penh value from the normal saline challenge, for each mouse, for each increasing dose of methacholine.
Statistical analysis
Results are expressed as mean ± SEM. Differences in Ab levels and airway inflammatory cells between groups were determined using nonparametric Mann-Whitney U or Kruskal-Wallis tests. Differences in breathing pattern in response to the bronchoconstricting agent methacholine between sensitized and challenged mice were compared using repeated measures ANOVA. All statistical comparisons were performed using Prism 4 (GraphPad Software). Statistical significance was defined as a p value ≤0.05.
Results
Allergic airway disease was similar in pregnant and nonpregnant mice
It has been demonstrated that adult mice subjected to a discontinuous (interrupted) model of OVA-induced allergic airway disease develop secondary allergic airway inflammation with all the hallmarks of the primary disease including airway eosinophilia, OVA-specific Ig production, and airway hyperresponsiveness (35). In this model, adult mice are sensitized with OVA-Al(OH)3 (three weekly immunizations) and 1 wk later are challenged daily with aerosolized OVA. At 4 wk following the primary challenge, mice are subjected to a secondary challenge daily with aerosolized OVA. Parameters of disease severity are virtually indistinguishable following the primary (acute) or secondary (discontinuous) airway challenge. These results indicate that immunological memory specific for OVA exists in mice at 4 wk following the primary challenge, as has been shown by others (40), and that the OVA-specific memory lymphocytes are able to orchestrate the secondary airway inflammatory response upon re-exposure to aerosolized Ag (35).
We modified this discontinuous model for our studies designed to determine the impact of maternal-derived Th1- or Th2-type immune responses during pregnancy on maternal transmission of asthma susceptibility (Fig. 1). Female C57BL/6J mice were immunized with Ag in adjuvant and then were challenged daily with aerosolized Ag. Eight weeks later, following resolution of airway inflammation (35), mice were mated, and at E11–17 of pregnancy were subjected to secondary aerosol challenge. The time for allergen re-exposure was chosen based on the potential to impact development of the innate and adaptive immune system (41– 45), as well as T cell selection events occurring in the embryonic thymus (46, 47). Following weaning, offspring were evaluated for susceptibility or resistance to allergic airway disease elicited by two immunizations with OVA-Al(OH)3 and challenge daily with aerosolized OVA (48–50).
Because the hormonal environment in pregnancy alters the magnitude and character of the immune response (51–53), we first examined whether previously sensitized and aerosol-challenged C57BL/6J mice would develop recall allergic airway disease during pregnancy. Following two i.p. immunizations with OVA- Al(OH)3 and primary OVA aerosol exposure (~ 8 wk prior), pregnant or nonpregnant female mice were subjected to secondary OVA aerosol challenge on days equivalent to E11–17 of pregnancy, then sacrificed 24 h after the last aerosol exposure. Comparable concentrations of OVA-specific IgG1 and IgE Abs were present in serum from pregnant or nonpregnant female mice (Fig. 2). The cellular composition of the BAL fluid was also similar between groups, with eosinophils being the major leukocyte population in both pregnant (78 ± 3%) and nonpregnant (73 ± 7%) mice (data not shown). Thus, pregnancy had no detectable impact on immunologic parameters indicative of allergic airway disease in this model.
FIGURE 2.
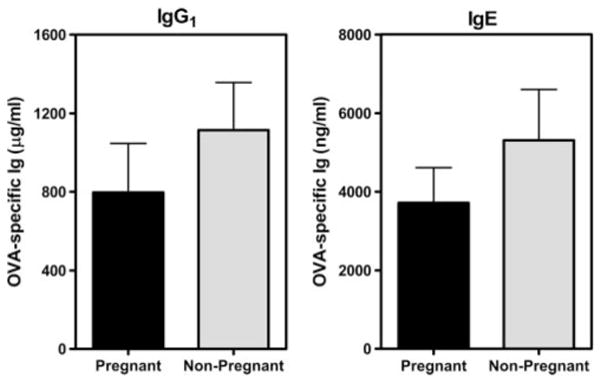
Pregnancy does not affect serum levels of OVA-specific IgG1 and IgE. C57BL/6J female mice were immunized three times with OVA-Al(OH)3 (at weekly intervals) and 9 days later were challenged daily, for 7 days, with aerosolized OVA as described in Materials and Methods. Seven weeks later, select females were bred with naive C57BL/6J males and both pregnant and nonpregnant females were re-exposed to aerosolized OVA on days corresponding to E11–17 of pregnancy. Serum was collected 24 h after the last aerosol exposure (E18) and concentrations of OVA-specific IgG1 or IgE in serum were determined by ELISA. Results are from one experiment (three mice per group) and the absence of detectable differences between pregnant and nonpregnant mice was consistent with analyses of eight other mice in separate experiments.
Adjuvant determined the Th1- vs Th2-bias of systemic immunity and airway inflammation
Female C57BL/6J mice were immunized twice (separated by 10 days) with 25 μg of OVA emulsified in CFA (s.c.; Th1-inducing conditions) or Al(OH)3 (i.p.; Th2-inducing conditions). Secondary immunization of OVA-CFA-primed mice was OVA in IFA (i.p.). Five days after the second immunization, the frequency of splenic T cells producing IL-4 or IFN-γ in response to cognate OVA peptide stimulation was determined by ELISPOT (Fig. 3). OVA-specific CD4+ or CD8+ IFN-γ-producing cells were unique to mice primed with OVA-CFA, a signature cytokine of Th1-polarized cells, and were virtually undetectable in mice immunized with OVA-Al(OH)3 (frequency < 10 per 105 CD4+ or CD8+ cells). In contrast, an equivalent frequency (~45– 60 per 105 CD4+ cells) of IL-4-producing CD4+ cells (a signature cytokine of Th2-polarized cells) was detected in splenocytes from mice immunized with either OVA-CFA or OVA-Al(OH)3. Similar concurrent Th1- and Th2-type cytokine production by Ag-specific CD4+ cells is also observed in response to infection with Listeria monocytogenes (54).
FIGURE 3.
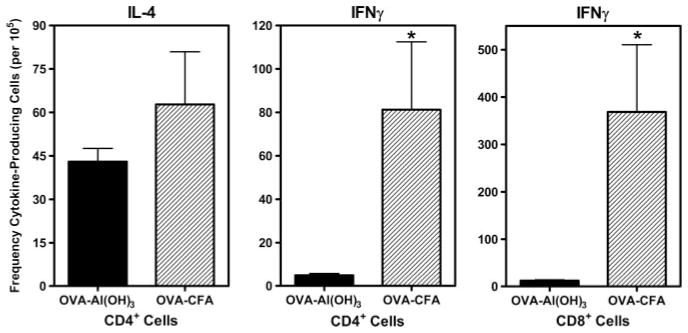
The profile of OVA-specific cytokine production was determined by the adjuvant used for immunization. C57BL/6J female mice were immunized with OVA-CFA or OVA-Al(OH)3 as described in Materials and Methods. Five days following the second immunization, spleen cells were prepared and OVA-specific cytokine production was determined by ELISPOT. Graphs represent the frequency of cytokine-producing cells per 105 class II-(CD4) or class I-restricted (CD8) T cells, determined from comparisons between T cells cultured in wells containing cognate OVA peptides and control wells (without OVA peptide). Results are expressed as mean ± SE and represent four mice per group analyzed from wells containing three to four dilutions of T cells in duplicate wells. *, p ≤ 0.05 when compared between groups.
The airway inflammatory response in OVA-CFA- or OVA-Al(OH)3-sensitized mice was assessed following challenge with aerosolized OVA. Cells were harvested from the airway by BAL and the differential leukocyte count was determined by analysis of cytocentrifuged slide preparations stained with Wright-Giemsa and by fluorescence flow cytometry (Fig. 4, A and B). The number of airway leukocytes was significantly less in mice primed with OVA-CFA (79.8 ± 10.6 × 104) than with OVA-Al(OH)3 (172.9 ± 24.2 × 104). In contrast to robust airway eosinophilia (76% of leukocytes) in aerosol challenged OVA-Al(OH)3-sensitized mice, a hallmark of allergic airway disease (2, 4, 55), eosinophils represented only a minority (<1% of leukocytes) of the airway inflammatory response in aerosol-challenged OVA-CFA-sensitized mice. The majority of leukocytes infiltrating the airways upon aerosol challenge of OVA-CFA-sensitized mice were lymphocytes, primarily CD8+ T cells (~25-fold greater numbers than OVA-Al(OH)3-sensitized mice). Differences in lung histology were consistent with the pathology that typifies Th1- vs Th2-biased lung disease. Th1-biased lung disease pathology was dominated in the late stages by lymphocytes and macrophages in the absence of mucus (L. Puddington and R. S. Thrall, unpublished results). This was opposed to the pathology of Th2-biased lung disease which is dominated by eosinophils, lymphocytes, and mucus plugs (56). Furthermore, changes in breathing pattern in response to inhaled methacholine were evident during OVA-Al(OH)3-, but not OVA-CFA-, induced airway inflammatory disease as measured by unrestrained whole body plethysmography (Fig. 4C). Although there are limitations in assessment of physiological parameters using this method (39), the increased sensitivity to methacholine observed was consistent with our invasive measurements of lung resistance during OVA-Al(OH)3-induced airway inflammatory disease (56). Similarly, our results corresponded to data from OVA aerosol-challenged mice following adoptive transfer of in vitro-differentiated OVA-specific CD4+ cells. Whereas both Th1 or Th2 cells migrate to the airways following aerosol challenge, only transferred Th2 cells induce eosin-ophilic inflammation and exhibit altered sensitivity to methacholine when examined using whole body plethysmography (57, 58).
FIGURE 4.
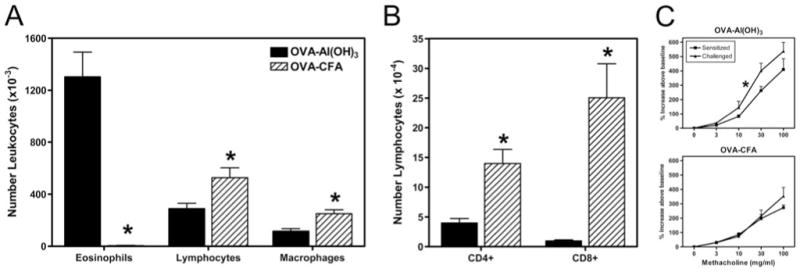
The airway inflammatory response and sensitivity to methacholine observed following aerosol challenge was dependent on the adjuvant used for sensitization. C57BL/6J female mice were immunized with OVA-Al(OH)3 or OVA-CFA and challenged for 10 days with 1% aerosolized OVA (daily exposure time 60 min). A, The differential leukocyte counts in BAL fluid collected 24 h following the last aerosol challenge as determined from analysis of cytocentrifuged slide preparations stained with Wright-Giemsa; B, distribution of T lymphocyte subsets determined by fluorescence flow cytometry; and C, changes in breathing pattern following aerosol challenge with methacholine determined by unrestrained whole body plethysmography in sensitized only (■) or sensitized and challenged mice (▲). Results are expressed as mean ± SE and represent 6 mice per group (A and B) with the exception of the physiology which represent 9–10 mice per group (C). *, p ≤ 0.05 when compared between groups.
The recall response during pregnancy was Th1 or Th2 biased based on adjuvant used for initial immunization
Six weeks after the primary aerosol exposure (a time when the airway inflammatory response had resolved in OVA-Al(OH)3-sensitized and aerosol-challenged mice (35)), OVA-CFA- or OVA-Al(OH)3-sensitized female mice were bred with naive C57BL/6J male mice. Pregnant mice were subjected to secondary challenge with aerosolized OVA daily, at E11–17 (see Fig. 1). Fig. 5 shows ELISA results for OVA-specific IgG1, IgE, IgG2a, or IgA in the serum from pregnant females at gestational day E18, 24 h after the seventh daily aerosol exposure. Interestingly, challenge with aerosolized OVA was absolutely required to generate OVA-specific IgA, but not IgG1 or IgE in OVA-CFA or OVA-Al(OH)3-immunized mice (D. M. Selander, E. G. Lingenheld, and L. Puddington, unpublished results). Pregnant mice previously primed with OVA-CFA and subjected to recall OVA aerosol exhibited higher serum concentrations of OVA-specific IgG2a (~25-fold, p < 0.05) and IgA when compared with similarly challenged pregnant mice primed with OVA-Al(OH)3. No differences were observed between serum concentrations of OVA-specific IgG1 in OVA-CFA-vs OVA-Al(OH)3-sensitized mice. These results, in conjunction with the ELISPOT data (see Fig. 3), suggested that mice immunized with OVA-CFA- or OVA-Al(OH)3-generated populations of OVA-specific memory CD4+ cells capable of providing T cell help to direct OVA-specific B cell isotype switching. The character of the Ab response was consistent with the recall OVA-specific CD4+ cell cytokine production observed by ELISPOT following OVA-CFA or OVA-Al(OH)3 immunization. It is known that IFN-γ produced by CD4+ cells directs class switch recombination during cognate interactions with Ag-specific B cells, resulting in IgG2a-producing plasma cells, thus OVA-specific IgG2a was detected primarily in the serum of OVA-CFA-sensitized mice. Similarly, IL-4 directs class switch recombination to IgG1 (and IgE) (59, 60). Similar frequencies of CD4+ cells producing IL-4 were elicited following OVA-CFA or OVA-Al(OH)3 immunization, and similar levels of OVA-specific IgG1 were detected in OVA aerosol-challenged pregnant mice, irrespective of the adjuvant used for maternal sensitization.
FIGURE 5.
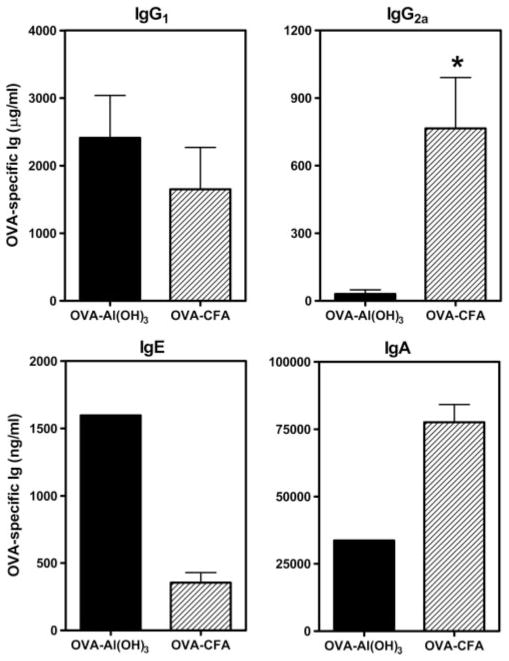
The profiles of maternal OVA-specific Ig isotypes following secondary aerosol challenge during pregnancy were Th1- or Th2-biased dependent on the adjuvant used for sensitization. C57BL/6J female mice were immunized with OVA-Al(OH)3 or OVA-CFA and challenged for 7 days with 1% aerosolized OVA (daily exposure time 60 min). Six weeks later, females were bred with naive C57BL/6J males and pregnant mice were re-exposed to aerosolized OVA on E11–17 of pregnancy. Serum was collected 24 h after the last aerosol exposure (E18) and OVA-specific Igs were measured by ELISA as described in Materials and Methods. Results represent the mean ± SE from four to eight mice per group. Statistical analysis could not be performed for OVA-specific IgE or IgA as analysis of sera was from the OVA-Al(OH)3 mother corresponding to pups shown for IgE and IgA in Fig. 6. *, p ≤ 0.05 when compared between groups.
Maternal OVA-specific Abs transferred passively to offspring were Th1- or Th2-biased based on maternal immunization
Sera were obtained from 2-wk-old progeny with different histories of exposure to the effects of maternal derived immune responses. Pups were born to and nursing on mothers with Th1-type or Th2-type immunity to OVA (OVA-CFA- vs OVA-Al(OH)3-primed mothers, respectively) subjected to recall challenge with aerosolized OVA during pregnancy. OVA-specific Igs were detected in sera from mice born to OVA immune (Fig. 6) but not naive control mothers. Therefore, the acquisition of OVA-specific Igs by offspring was via passive transfer in utero or during nursing. All isotypes of maternal-derived OVA-specific Igs were present in pups of OVA-immune mothers, with the relative concentrations of each isotype reflective of their distribution in maternal sera (see Fig. 5). Thus, offspring whose mothers were primed with OVA-CFA and subjected to recall OVA aerosol challenge during pregnancy (at E11–17) had significantly greater serum concentrations of OVA-specific IgG2a than offspring of similarly challenged mothers primed with OVA-Al(OH)3. Interestingly, maternal OVA-specific IgE was present at similar levels in all 2-wk-old progeny nursed on OVA-immune mothers, irrespective of the adjuvant used for maternal sensitization (Fig. 6 and data not shown).
FIGURE 6.
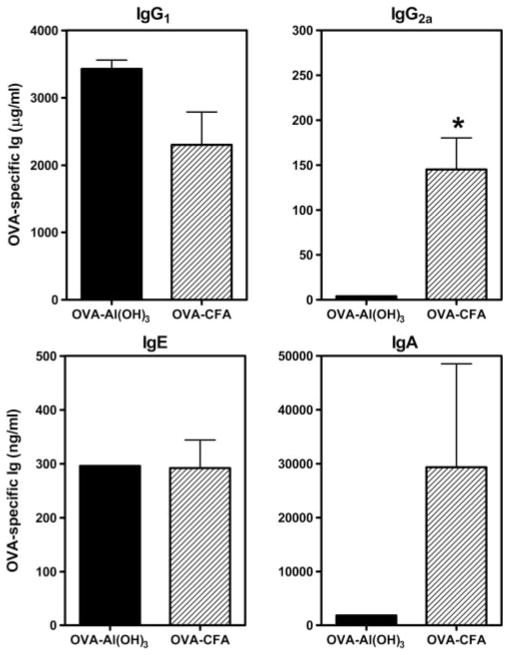
Transmission of Th1- or Th2-biased maternal Abs to offspring was determined by the adjuvant used for immunization. Females were immunized with OVA-Al(OH)3 or OVA-CFA and challenged for 7 days with 1% aerosolized OVA (daily exposure time 60 min). Six weeks later, females were bred with naive C57BL/6J males and pregnant mice were re-exposed to aerosolized OVA on E11–17 of pregnancy. Serum was collected from naive 2-wk-old pups (1 wk before weaning) and concentrations of OVA-specific Igs were measured by ELISA. OVA-specific Igs were absent from serum of pups born to naive C57BL/6J mothers (data not shown). Bar labels refer to conditions of maternal sensitization. Results represent the mean ± SE from three to four determinations per group. Each determination was made using sera from individual mice for IgG1 and IgG2a or sera combined from two mice for IgE and IgA. Statistical analysis could not be performed for OVA-specific IgE or IgA as analysis of sera from progeny of OVA-Al(OH)3 immune mothers was a pooled sample from two mice. *, p ≤ 0.05 when compared between groups.
Maternal Th1-biased OVA-specific immunity transferred resistance to development of OVA-induced allergic airway disease
Susceptibility or resistance to development of OVA-induced allergic airway disease was assessed in offspring with different histories of exposure to the effects of maternal-derived immune responses. Pups were from mothers with Th1- or Th2-type immunity to OVA (sensitized with OVA-CFA or OVA-Al(OH)3 and OVA aerosol-challenged 7– 8 wk prior) subjected to secondary challenge with aerosolized OVA during pregnancy (see Fig. 1). Control pups were from naive mothers never exposed to OVA. Following weaning and before the first immunization, maternal OVA-specific Ig levels in pups of immune mothers decline significantly (Ref. 61 and data not shown). Likewise, passively transferred maternal OVA-specific IgE present in mice nursing on OVA-immune mothers (see Fig. 6) declined to levels virtually undetectable by ELISA (<25–50 ng of OVA-specific IgE/ml) before the sensitization (data not shown). Allergic airway disease was elicited in 4- to 5-wk-old progeny by two immunizations with OVA-Al(OH)3 and challenge with aerosolized OVA (48–50). Parameters of disease severity measured in progeny of OVA-immune or naive mothers were Ag-specific Ig levels in serum and distribution of leukocyte populations in the airways.
Reduced allergic airway disease was evident in offspring of OVA-CFA-sensitized mothers subjected to secondary challenge with aerosolized OVA during pregnancy when compared with off-spring of OVA-Al(OH)3-sensitized or naive mothers (Fig. 7). Serum OVA-specific IgE levels in progeny of OVA-CFA mothers (776 ± 278 ng/ml) were 2- or 4-fold less than in progeny of OVA-Al(OH)3 mothers (1312 ± 632 ng/ml) or naive control mothers (2853 ± 728 ng/ml), respectively. Similarly, the magnitude of the OVA-induced airway inflammatory response was reduced, indicated by lower numbers of leukocytes recovered in BAL fluid from offspring of OVA-CFA mothers (54.4 ± 6.0 × 104) in comparison to offspring of OVA-Al(OH)3 or naive mothers (115.7 ± 13.8 × 104 or 133.6 ± 12.7 × 104). In addition, differential leukocyte counts of cytocentrifuged slide preparations stained with Wright-Giemsa, performed in conjunction with fluorescence flow cytometric analyses, demonstrated significantly reduced numbers of eosinophils and T cells (both CD4+ and CD8+) in the airways of progeny of OVA-CFA mothers compared with progeny of OVA-or naive mothers. Thus, major determinants of OVA-Al(OH)3 induced allergic airway disease severity were attenuated in offspring as a consequence of being born to and nursed on mothers with Th1-biased immunity to OVA.
FIGURE 7.
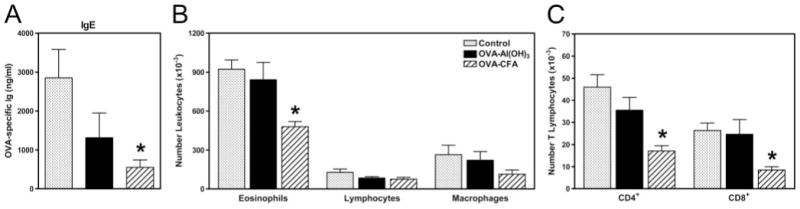
Decreased severity of allergic airway disease in offspring of mothers with Th1- but not Th2-type immunity to OVA. Pups were from mothers with Th1- or Th2-type immunity to OVA (sensitized with OVA-CFA or OVA-Al(OH)3 and OVA aerosol challenged 7– 8 wk prior) subjected to secondary challenge with aerosolized OVA during pregnancy. Control pups were from naive mothers never exposed to OVA. Following weaning, allergic airway disease was elicited in 4- to 5-wk-old progeny by two immunizations with OVA-Al(OH)3 followed by challenge for 7 days with 1% aerosolized OVA (daily exposure time 60 min); all pups were immunized and challenged identically. The differences between groups of offspring were restricted solely to factors transmitted as a result of maternal sensitization and exposure to recall OVA during pregnancy. Parameters of disease severity measured in progeny of OVA-immune or naive mothers were (A) OVA-specific IgE levels in serum determined by ELISA; (B) distribution of leukocyte populations in the airways determined from analysis of cytocentrifuged slide preparations stained with Wright-Giemsa; and (C) distribution of T lymphocyte subsets determined by fluorescence flow cytometry. Bar labels refer to conditions of maternal sensitization. Results are expressed as mean ± SE and represent five to eight mice per group. *, p ≤ 0.05 when compared between groups.
Th1-biased immunity to BSA was not sufficient for maternal transfer of resistance to development of OVA-induced allergic airway disease
It was possible that maternal transfer of resistance to development of OVA-induced allergic airway disease (in offspring of OVA-CFA mothers) was the result of Ag-independent effects on development or maturation of the innate immune system in offspring. This would be consistent with the concept that early life exposures to pathogens that elicit Th1-biased immune responses protect offspring from development of allergic airway disease (21–25). To evaluate whether maternal transfer of Ag-independent factors contributed to the resistance of progeny of OVA-CFA mothers to OVA-induced allergic airway disease (see Fig. 7), an analogous experiment was performed using a heterologous albumin, BSA. Mothers with Th1-type immunity to OVA or to BSA (C57BL/6J females mice sensitized with OVA-CFA or BSA-CFA and aerosol challenged 7– 8 wk prior) were subjected to secondary challenge with aerosolized OVA or BSA, respectively, during pregnancy (see Fig. 1). The character and magnitude of the anti-BSA response (following sensitization with BSA-CFA and challenge with aerosolized BSA) was comparable to the anti-OVA response (following sensitization with OVA-CFA and challenge with aerosolized OVA). For example, serum levels of IgG2a were virtually identical in BSA-CFA- or OVA-CFA-primed mice after BSA or OVA aerosol challenge (1921 ± 254 μg/ml or 2218 ± 397 μg/ml, respectively) and were significantly greater than levels in naive mice (879 ± 306 μg/ml). Furthermore, there was no evidence of cross-reactivity between BSA- and OVA-specific Igs of any isotype, as determined by ELISA analyses of serum Ig from immune mice (data not shown).
Susceptibility or resistance to development of OVA-induced allergic airway disease was assessed in offspring with different histories of exposure to the effects of maternal-derived immune responses. Pups were from mothers with Th1-type immunity to OVA or BSA subjected to secondary challenge with the respective aerosolized OVA or BSA during pregnancy. Control pups were from naive mothers never exposed to OVA or BSA. Allergic airway disease was elicited in 4- to 5-wk-old progeny by two immunizations with OVA-Al(OH)3 and challenge with aerosolized OVA (48–50). Parameters of disease severity measured in progeny of OVA- or BSA-immune or naive mothers were OVA-specific Ig levels in serum and distribution of leukocyte populations in the airways. Reduced allergic airway disease was evident in offspring of OVA-CFA-sensitized mothers subjected to secondary challenge with aerosolized OVA during pregnancy when compared with offspring of BSA-CFA-sensitized or naive mothers never exposed to OVA or BSA (Fig. 8). Interestingly, essentially no protection from development of OVA-induced allergic airway disease was transmitted to progeny of Th1-biased immune mothers when Ag specificity was directed against heterologous albumin, BSA. In this experiment, serum OVA-specific IgE in progeny of OVA-CFA mothers was undetectable, whereas in progeny of BSA-CFA mothers or naive control mothers OVA-specific IgE levels were virtually identical (474 ± 117 ng/ml and 528 ± 203 ng/ml, respectively). Similarly, the magnitude of the OVA-induced airway inflammatory response was reduced, indicated by lower numbers of leukocytes recovered in BAL fluid from offspring of OVA-CFA mothers (12.9 ± 3.9 × 104) in comparison to offspring of BSA-CFA or naive mothers (144.2 ± 61.9 × 104 or 178.0 ± 58.2 × 104). In addition, differential leukocyte counts of cytocentrifuged slide preparations stained with Wright-Giemsa, performed in conjunction with fluorescence flow cytometric analyses, demonstrated 7- to 60-fold reduced numbers of eosinophils and lymphocytes (both CD4+ T cells and CD19+ B cells) in the airways of progeny of OVA-CFA mothers compared with progeny of BSA-CFA or naive mothers. Thus, major determinants of OVA-induced allergic airway disease severity were attenuated in offspring as a consequence of being born to and nursed on mothers with Th1-biased immunity to OVA, but not when maternal Th1-biased immunity was directed against a heterologous albumin, BSA.
FIGURE 8.
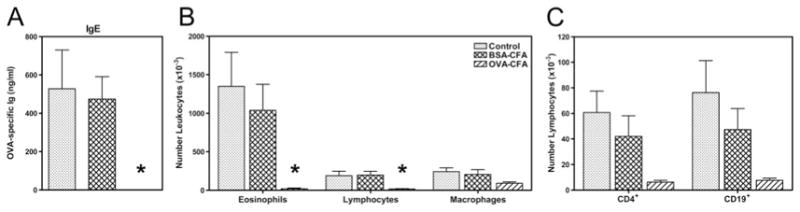
Maternal transmission of resistance to development of allergic airway disease in offspring is Ag specific. Pups were from mothers with Th1-type immunity to OVA or BSA (sensitized with OVA-CFA or BSA-CFA and OVA or BSA aerosol challenged 7– 8 wk prior) subjected to secondary challenge with the respective aerosolized OVA or BSA during pregnancy. Control pups were from naive mothers never exposed to OVA or BSA. Following weaning, allergic airway disease was elicited in 4- to 5-wk-old progeny by two immunizations with OVA-Al(OH)3 followed by challenge for 4 days with 1% aerosolized OVA (daily exposure time 60 min); all pups were immunized and challenged identically. The differences between groups of offspring were restricted solely to factors transmitted as a result of maternal sensitization and exposure to recall Ag during pregnancy. Parameters of disease severity measured in progeny of OVA- or BSA-immune or naive mothers were (A) OVA-specific IgE levels in serum determined by ELISA; (B) distribution of leukocyte populations in the airways determined from analysis of cytocentrifuged slide preparations stained with Wright-Giemsa; and (C) distribution of lymphocytes (CD4+ T cells and CD19+ B cells) determined by fluorescence flow cytometry. Bar labels refer to conditions of maternal sensitization. Results are expressed as mean ± SE and represent five to nine mice per group. *, p ≤ 0.05 when compared with between groups.
Discussion
The rationale for undertaking this study was to explore our hypothesis that transmitted maternal factors affect development of immune responsiveness in offspring. Numerous epidemiological studies suggest an inverse relationship between childhood exposure to Th1-inducing pathogens or nonviable microbial products and susceptibility to development of inflammatory diseases, such as asthma. The idea is that early life exposures to microbes or microbial products are important to direct maturation of the immune system in childhood toward a healthy balance of Th1- and Th2-type immune responsiveness. Based on this, we hypothesized that maternal factors transmitted pre- or postnatally would profoundly impact development or severity of allergic airway disease in progeny. Our findings supported this concept wherein pups from mothers with Th1-type immunity to OVA were more protected from developing severe allergic airway disease than pups of mothers with Th2-type immunity to OVA. Interestingly, progeny of mothers with Th1-type immunity to a heterologous albumin, BSA, were not protected from developing OVA-induced allergic airway disease. Together, these data suggest that protection of offspring from allergic airway disease in this model of maternal Th1-type immunity was Ag specific and not explained by maternal transmission of factors to affect development or maturation of the innate immune system in their progeny.
Other models have been developed that examine maternal transmission of asthma susceptibility. Interestingly, a variety of outcomes have been observed following immunization and aerosol challenge of progeny that are based on history of maternal allergen exposures or immunizations. The resultant data span the complete range from increased risk to increased protection. In addition, some transmitted maternal effects on susceptibility or severity of disease in offspring are Ag specific (61– 64), whereas others are independent of Ag (61, 65– 69). The explanation for the great diversity in disease outcome depends on the model of maternal transfer studied and is likely determined by the contribution of unique immunological mechanisms expressed in the context of specific genetic, developmental, and environmental factors. Taken together, the data illustrate profound maternal influences transferred to offspring pre- and/or postnatally capable to exacerbate or attenuate severity of allergic airway disease. It is likely that this diversity represents the human situation and could provide the basis for different outcomes reported in epidemiological studies, analyzed when stratification of the population was irrespective of maternal allergies or exposures before, or during, pregnancy and breastfeeding. As such, overall conclusions from these studies could potentially represent populations and their behavior within particular environments as has been reported for two genes encoding immune mediators known to be determinants of disease susceptibility (32, 70–72).
Some conditions of exposure to maternal-derived allergen or immunity that determine susceptibility or severity of disease in offspring have been identified in animal models. However to date, limited information on specific cells or molecules transferred from mothers to progeny and the mechanisms by which they instigate changes in asthma susceptibility or severity have been identified. This is a challenge inherent in these types of studies, including the one presented in this manuscript, where the readout is exacerbated or attenuated symptoms of disease. This is due to the fact that identical outcomes can be achieved via interventions that impact any one of a number of immunological pathways. A BALB/c model developed by L. Kobzik and colleagues (66, 68, 69) clearly reveals transmission of increased risk for development of allergic airway disease to offspring from mothers with Th2-biased OVA-specific immunity. In addition, suboptimal immunization of progeny facilitated their determination that both pre- and/or postnatal periods of exposure to allergic mothers are capable to enhance risk for development of asthma (66, 68, 69). In our study, C57BL/6 progeny from mothers with Th2-type immunity to OVA demonstrated no increase in susceptibility or severity of allergic airway disease when compared with progeny of naive mothers. However, in addition to the strain difference, it was determined in the BALB/c model that exposure to aerosolized Ag, i.e., restoration of allergic airway disease, immediately before mating (but not during pregnancy) was absolutely required for maternal transmission of risk. In our studies using a C57BL/6 model, secondary allergic airway disease in mothers was not reinstated until during pregnancy. Thus, lack of maternal transmission of increased risk in our model is consistent with the similar studies performed in BALB/c mice.
In contrast to the previous work, one goal of our study was to directly compare development of allergic airway disease in offspring, with the only difference between groups being the predominant character (Th1 vs Th2 type) of immunological memory and airway inflammation in their mothers. In this way, immune mothers subjected to challenge with recall aerosolized Ag during pregnancy would expose their offspring systemically to those immunological mediators and Ags transferred via the placenta. We suspect that further transfer of maternal factors generated as a result of the recalled immune response during pregnancy also occurred in early postnatal life via breast milk during nursing. The presence of maternal Ig in offspring nursed on immune, but not naive, mothers was supportive of this idea (see Fig. 6). Dramatic differences between the response to immunization with OVA-Al(OH)3 vs OVA-CFA were observed in C57BL/6 mice following primary challenge with aerosolized OVA and following secondary challenge with aerosolized OVA during pregnancy (see Figs. 3–5). Most unique to OVA-CFA-primed mice was the predominance of OVA-specific IFN-γ production by T cells following sensitization, the virtual absence of eosinophil recruitment to the airways following aerosol challenge and high levels of OVA-specific IgG2a in serum. It has been shown that treatment of pregnant CD1 mice with rIFN-γ (100 IU at E6.5) protects offspring from developing severe allergic airway disease upon sensitization and aerosol challenge (73). An analogous correlate in humans is the demonstration that maternal stable work (i.e., exposure to environments rich in microbial products) during pregnancy is more protective against development of atopy in children than maternal stable work during lactation (32). These data imply that in certain conditions prenatal transfer of cytokines or TLR agonists from mothers to offspring occurs, presumably via the placenta. We suspect similar prenatal transfer of maternal Th1-type soluble factors occurred in OVA-CFA and BSA-CFA immune mothers during the secondary challenge during pregnancy. Despite this, offspring of BSA-CFA immune mothers were not protected from OVA-induced allergic airway disease.
One of the most interesting findings in our model was the demonstration that maternal OVA-specific IgE was present in serum of 2-wk-old offspring nursing on Th1- or Th2-immune mothers subjected to recall aerosol challenge during pregnancy. Because offspring of naive mothers were devoid of OVA-specific IgE and the fact that no mechanism to facilitate transplacental transport of IgE is thought to exist in mice or humans, the presence of OVA-specific IgE in serum of nursing offspring suggested that acquisition of maternal IgE from immune mothers was a result of intestinal absorption from breast milk. The low-affinity IgE receptor, CD23, is expressed by intestinal epithelial cells and specific isoforms are responsible for absorption of IgE or IgE immune complexes from the intestinal lumen (74, 75). The potential for repercussions from maternally transmitted IgE or IgE immune complexes from breast milk are likely significant in offspring. Interestingly, maternal IgE to cat allergen (76) or maternal history of asthma in conjunction with exposure to cat allergen is associated with increased risk of wheezing in childhood (77).
Importantly, our data demonstrated that transferred protection from severe allergic airway disease in offspring of Th1-immune mothers was Ag specific. The most logical mediators of allergen specific asthma resistance in this model could be allergen-specific IgGs or IgA transferred postnatally from mothers to progeny in breast milk. Although our preliminary experiments suggest a significant role for maternal Ig in transmission of asthma resistance, further analyses are required to prove this hypothesis. This is consistent with the observations of Jarrett et al. (34) that demonstrate transfer of maternal OVA-specific IgG is sufficient to protect offspring from development of OVA-specific IgE responses following immunization, although non-IgG mediated protective effects are also noted (34). Furthermore, if allergen-specific maternal Ig is involved in Ag-specific resistance to development of allergic airway disease, it will be important to determine the contributions of IgG, IgA, Ig receptors (e.g., FcRn, FcγR, CD23, or FcαR) and complement to the protective mechanism. In addition, the origin of cells transducing these effects (i.e., maternal or offspring) remain to be identified. Together, these data suggest that allergen immunization before pregnancy could provide protection from allergic sensitization and potentially from development of atopic disease in early life. Much further investigation of animal models and application of their findings to longitudinal studies in humans are required to determine the feasibility of such an approach.
Acknowledgments
We are indebted to John B. Morris (University of Connecticut School of Pharmacy, Storrs, CT) for setting up the aerosol generators and exposure chambers for uniform, reproducible delivery of aerosolized Ags. We thank Amanda L. Marzo (University of Connecticut Health Center) and Karen M. Laky (National Institute of Allergy and Infectious Diseases, National Institutes of Health) for their help with development and analysis of T cell cytokine production by ELISPOT and Matthew E. Poynter (University of Vermont) for assistance in accurate presentation of Penh data. We also thank Michelle M. Cloutier and the other members of the Pulmonary Research Consortium for their helpful advice as we undertook this project. We are grateful to Leo Lefrançois and Carol A. Wu for their critical evaluation of the manuscript.
Footnotes
This work was supported by National Institutes of Health Grants HL069083, HL08058, and HL066963 (to L.P.) and American Lung Association of Connecticut Grants (to L.P. and to A.P.M.). D.M.S. was supported in part by National Institutes of Health Training Grant AI007080.
Abbreviations used in this paper: E, embryonic day; BAL, bronchoalveolar lavage; Penh, enhanced pause.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Walker C, Kaegi MK, Braun P, Blaser K. Activated T cells and eosinophilia in bronchoalveolar lavages from subjects with asthma correlated with disease severity. J Allergy Clin Immunol. 1991;88:935–942. doi: 10.1016/0091-6749(91)90251-i. [DOI] [PubMed] [Google Scholar]
- 2.McFadden ER, I, Gilbert A. Asthma. N Engl J Med. 1992;327:1928–1924. doi: 10.1056/NEJM199212313272708. [DOI] [PubMed] [Google Scholar]
- 3.Corrigan CJ, Kay AB. Asthma: role of T-lymphocytes and lymphokines. Br Med Bull. 1992;48:72– 84. doi: 10.1093/oxfordjournals.bmb.a072543. [DOI] [PubMed] [Google Scholar]
- 4.Corrigan CJ, Kay AB. T cells and eosinophils in the pathogenesis of asthma. Immunol Today. 1992;13:501–507. doi: 10.1016/0167-5699(92)90026-4. [DOI] [PubMed] [Google Scholar]
- 5.Wilson JW, Djukanovic R, Howarth PH, Holgate ST. Lymphocyte activation in bronchoalveolar lavage and peripheral blood in atopic asthma. Am Rev Respir Dis. 1992;145:958–960. doi: 10.1164/ajrccm/145.4_Pt_1.958. [DOI] [PubMed] [Google Scholar]
- 6.Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant Th2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 7.Nakajima H, Iwamoto S, Tomoe S, Matsumura R, Tomioka H, Takatse K, Yoshida S. CD4+ T-lymphocytes and interleukin-5 mediate antigen-induced eosinophil infiltration into the mouse trachea. Am Rev Respir Dis. 1992;146:374–377. doi: 10.1164/ajrccm/146.2.374. [DOI] [PubMed] [Google Scholar]
- 8.Gavett SH, Chen X, Finkelman F, Wills-Karp M. Depletion of murine CD4+ T lymphocytes prevents antigen-induced airway hyperreactivity and pulmonary eosinophilia. Am J Respir Cell Mol Biol. 1994;10:587–593. doi: 10.1165/ajrcmb.10.6.8003337. [DOI] [PubMed] [Google Scholar]
- 9.Martin TR, Takeishi T, Katz HR, Austen KF, Drazen JM, Galli SJ. Mast cell activation enhances airway responsiveness to methacholine in the mouse. J Clin Invest. 1993;91:1176–1182. doi: 10.1172/JCI116277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oshiba A, Hamelmann E, Takeda K, Bradley KL, Loader JE, Larsen GL, Gelfand EW. Passive transfer of immediate hypersensitivity and airway hyperresponsiveness by allergen-specific immunoglobulin (Ig) E and IgG1 in mice. J Clin Invest. 1996;97:1398–1408. doi: 10.1172/JCI118560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korsgren M, Erjefält JS, Korsgren O, Sundler F, Persson CGA. Allergic eosinophil-rich inflammation develops in lungs and airways of B cell-deficient mice. J Exp Med. 1997;185:885– 892. doi: 10.1084/jem.185.5.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hogan SP, Mould A, Kikutani H, Ramsay AJ, Foster PS. Aeroallergen-induced eosinophilic inflammation, lung damage, and airways hyperreactivity in mice can occur independently of IL-4 and allergen-specific immunoglobulins. J Clin Invest. 1997;99:1329–1339. doi: 10.1172/JCI119292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamelmann E, Takeda K, Schwarze J, Vella AT, Irvin CG, Gelfand EW. Development of eosinophilic airway inflammation and airway hyperresponsiveness requires interleukin-5 but not immunoglobulin E or B lymphocytes. Am J Respir Cell Mol Biol. 1999;21:480– 489. doi: 10.1165/ajrcmb.21.4.3659. [DOI] [PubMed] [Google Scholar]
- 14.Maclean JS, Sauty A, Luster AD, Drazen JM, De Sanctis GT. Antigen-induced airway hyperresponsiveness, pulmonary eosinophilia and chemokine expression in B-cell deficient mice. Am J Respir Cell Mol Biol. 1999;20:379–387. doi: 10.1165/ajrcmb.20.3.3291. [DOI] [PubMed] [Google Scholar]
- 15.Forsthuber T, Yip HC, Lehmann PV. Induction of Th1 and Th2 immunity in neonatal mice. Science. 1996;271:1728–1730. doi: 10.1126/science.271.5256.1728. [DOI] [PubMed] [Google Scholar]
- 16.Culley FJ, Pollott J, Openshaw PJ. Age at first viral infection determines the pattern of T cell-mediated disease during reinfection in adulthood. J Exp Med. 2002;196:1381–1386. doi: 10.1084/jem.20020943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Upham JW, Rate A, Rowe J, Kusel M, Sly PD, Holt PG. Dendritic cell immaturity during infancy restricts the capacity to express vaccine-specific T-cell memory. Infect Immun. 2006;74:1106–1112. doi: 10.1128/IAI.74.2.1106-1112.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ridge JP, Fuchs EJ, Matzinger P. Neonatal tolerance revisited: turning on newborn T cells with dendritic cells. Science. 1996;271:1723–1726. doi: 10.1126/science.271.5256.1723. [DOI] [PubMed] [Google Scholar]
- 19.Adkins B, Bu Y, Guevara P. The generation of Th memory in neonates versus adults: prolonged primary Th2 effector function and impaired development of Th1 memory effector function in murine neonates. J Immunol. 2001;166:918–925. doi: 10.4049/jimmunol.166.2.918. [DOI] [PubMed] [Google Scholar]
- 20.Strachan DP. Hay fever, hygiene, and household size. Br Med J. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347:911–920. doi: 10.1056/NEJMra020100. [DOI] [PubMed] [Google Scholar]
- 22.Wills-Karp M, Santeliz J, Karp CL. The germless theory of allergic disease: revisiting the hygiene hypothesis. Nat Rev Immunol. 2001;1:69–75. doi: 10.1038/35095579. [DOI] [PubMed] [Google Scholar]
- 23.Gern JE, Busse WW. Relationship of viral infections to wheezing illnesses and asthma. Nat Rev Immunol. 2002;2:132–138. doi: 10.1038/nri725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedlander SL, Jackson DJ, Gangnon RE, Evans MD, Li Z, Roberg KA, Anderson EL, Carlson-Dakes KT, Adler KJ, Gilbertson-White S, et al. Viral infections, cytokine dysregulation and the origins of childhood asthma and allergic diseases. Pediatr Infect Dis J. 2005;24(Suppl):S170–S176. doi: 10.1097/01.inf.0000187273.47390.01. [DOI] [PubMed] [Google Scholar]
- 25.Schaub B, Lauener R, von Mutius E. The many faces of the hygiene hypothesis. J Allergy Clin Immunol. 2006;117:969–977. doi: 10.1016/j.jaci.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Celedon JC, Wright RJ, Litonjua AA, Sredl D, Ryan L, Weiss ST, Gold DR. Day care attendance in early life, maternal history of asthma, and asthma at the age of 6 years. Am J Respir Crit Care Med. 2003;167:1239–1243. doi: 10.1164/rccm.200209-1063OC. [DOI] [PubMed] [Google Scholar]
- 27.Ramsey CD, Celedon JC. The hygiene hypothesis and asthma. Curr Opin Pulm Med. 2005;11:14–20. doi: 10.1097/01.mcp.0000145791.13714.ae. [DOI] [PubMed] [Google Scholar]
- 28.Moffatt MF, Cookson WO. The genetics of asthma: maternal effects in atopic disease. Clin Exp Allergy. 1998;28(Suppl 1):56–66. doi: 10.1046/j.1365-2222.1998.0280s1056.x. [DOI] [PubMed] [Google Scholar]
- 29.Holt PG, Macaubas C, Stumbles PA, Sly PD. The role of allergy in the development of asthma. Nature. 1999;402(Suppl 6760):B12–B17. doi: 10.1038/35037009. [DOI] [PubMed] [Google Scholar]
- 30.Holloway JA, Warner JO, Vance GH, Diaper ND, Warner JA, Jones CA. Detection of house-dust-mite allergen in amniotic fluid and umbilical-cord blood. Lancet. 2000;356:1900–1902. doi: 10.1016/S0140-6736(00)03265-7. [DOI] [PubMed] [Google Scholar]
- 31.Wills-Karp M, Brandt D, Morrow AL. Understanding the origin of asthma and its relationship to breastfeeding. Adv Exp Med Biol. 2004;554:171–191. doi: 10.1007/978-1-4757-4242-8_16. [DOI] [PubMed] [Google Scholar]
- 32.Ege MJ, Bieli C, Frei R, van Strien RT, Riedler J, Ublagger E, Schram-Bijkerk D, Brunekreef B, van Hage M, Scheynius A, et al. Prenatal farm exposure is related to the expression of receptors of the innate immunity and to atopic sensitization in school-age children. J Allergy Clin Immunol. 2006;117:817– 823. doi: 10.1016/j.jaci.2005.12.1307. [DOI] [PubMed] [Google Scholar]
- 33.Zinkernagel RM. Maternal antibodies, childhood infections, and autoimmune diseases. N Engl J Med. 2001;345:1331–1335. doi: 10.1056/NEJMra012493. [DOI] [PubMed] [Google Scholar]
- 34.Jarrett EEE, Hall E. IgE suppression by maternal IgG. Immunology. 1983;48:49–58. [PMC free article] [PubMed] [Google Scholar]
- 35.Schramm CM, Puddington L, Wu CA, Guernsey L, Gharaee-Kermani M, Phan SH, Thrall RS. Chronic inhaled ovalbumin exposure induces antigen-dependent but not antigen-specific inhalational tolerance in a murine model of allergic airway disease. Am J Pathol. 2004;164:295–304. doi: 10.1016/S0002-9440(10)63119-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karulin AY, Hesse M, Tary-Lehmann M, Lehmann PV. Single-cytokine-producing CD4 memory cells predominate in type 1 and type 2 immunity. J Immunol. 2000;164:1862–1872. doi: 10.4049/jimmunol.164.4.1862. [DOI] [PubMed] [Google Scholar]
- 37.Seymour BWP, Gershwin LJ, Coffman RL. Aerosol-induced immunoglobulin (Ig)-E unresponsiveness to ovalbumin does not require CD8+ or T cell receptor (TCR)-γ/δ+ T cells or interferon (IFN)-γ in a murine model of allergen sensitization. J Exp Med. 1998;187:721–731. doi: 10.1084/jem.187.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacky JP. A plethysmograph for long-term measurements of ventilation in unrestrained animals. J Appl Physiol. 1978;45:644– 647. doi: 10.1152/jappl.1978.45.4.644. [DOI] [PubMed] [Google Scholar]
- 39.Bates JH, Irvin CG. Measuring lung function in mice: the phenotyping uncertainty principle. J Appl Physiol. 2003;94:1297–1306. doi: 10.1152/japplphysiol.00706.2002. [DOI] [PubMed] [Google Scholar]
- 40.Mojtabavi N, Dekan G, Stingl G, Epstein MM. Long-lived Th2 memory in experimental allergic asthma. J Immunol. 2002;169:4788– 4796. doi: 10.4049/jimmunol.169.9.4788. [DOI] [PubMed] [Google Scholar]
- 41.Jotereau F, Heuze F, Salomon-Vie V, Gascan H. Cell kinetics in the fetal mouse thymus: precursor cell input, proliferation, and emigration. J Immunol. 1987;138:1026–1030. [PubMed] [Google Scholar]
- 42.Rennert PD, James D, Mackay F, Browning JL, Hochman PS. Lymph node genesis is induced by signaling through the lymphotoxin β receptor. Immunity. 1998;9:71–79. doi: 10.1016/s1074-7613(00)80589-0. [DOI] [PubMed] [Google Scholar]
- 43.Yoshida H, Honda K, Shinkura R, Adachi S, Nishikawa S, Maki K, Ikuta K, Nishikawa SI. IL-7 receptor α+ CD3− cells in the embryonic intestine induces the organizing center of Peyer’s patches. Int Immunol. 1999;11:643– 655. doi: 10.1093/intimm/11.5.643. [DOI] [PubMed] [Google Scholar]
- 44.Kincade PW, Owen JJ, Igarashi H, Kouro T, Yokota T, Rossi MI. Nature or nurture: steady-state lymphocyte formation in adults does not recapitulate ontogeny. Immunol Rev. 2002;187:116–125. doi: 10.1034/j.1600-065x.2002.18710.x. [DOI] [PubMed] [Google Scholar]
- 45.Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol. 2006;7:344–353. doi: 10.1038/ni1330. [DOI] [PubMed] [Google Scholar]
- 46.Allison JP, Havran WL. The immunobiology of T cells with invariant γδ antigen receptors. Annu Rev Immunol. 1991;9:679–705. doi: 10.1146/annurev.iy.09.040191.003335. [DOI] [PubMed] [Google Scholar]
- 47.Anderson G, Jenkinson EJ, Moore NC, Owen JJT. MHC class II-positive epithelium and mesenchyme cells are both required for T-cell development in the thymus. Nature. 1993;362:70–73. doi: 10.1038/362070a0. [DOI] [PubMed] [Google Scholar]
- 48.Kung TT, Jones H, Adams GK, III, Umland SP, Kreutner W, Egan RW, Chapman RW, Watnick AS. Characterization of a murine model of allergic pulmonary inflammation. Int Arch Allergy Immunol. 1994;105:83–90. doi: 10.1159/000236807. [DOI] [PubMed] [Google Scholar]
- 49.Ohkawara Y, Lei XF, Stämpfli MR, Marshall JS, Xing Z, Jordana M. Cytokine and eosinophil responses in the lung, peripheral blood, and bone marrow compartments in a murine model of allergen-induced airways inflammation. Am J Respir Cell Mol Biol. 1997;16:510–520. doi: 10.1165/ajrcmb.16.5.9160833. [DOI] [PubMed] [Google Scholar]
- 50.Schramm CM, Puddington L, Yiamouyiannis CA, Lingenheld EG, Whiteley HE, Wolyniec WW, Noonan TC, Thrall RS. Proinflammatory roles of TCRγδ and TCRαβ lymphocytes in a murine model of asthma. Am J Respir Cell Mol Biol. 2000;22:218–225. doi: 10.1165/ajrcmb.22.2.3620. [DOI] [PubMed] [Google Scholar]
- 51.Whitacre CC, Reingold SC, O’Looney PA. A gender gap in autoimmunity. Science. 1999;283:1277–1278. doi: 10.1126/science.283.5406.1277. [DOI] [PubMed] [Google Scholar]
- 52.Palaszynski KM, Smith DL, Kamrava S, Burgoyne PS, Arnold AP, Voskuhl RR. A yin-yang effect between sex chromosome complement and sex hormones on the immune response. Endocrinology. 2005;146:3280–3285. doi: 10.1210/en.2005-0284. [DOI] [PubMed] [Google Scholar]
- 53.Lambert KC, Curran EM, Judy BM, Milligan GN, Lubahn DB, Estes DM. Estrogen receptor α (ERα) deficiency in macrophages results in increased stimulation of CD4+ T cells while 17 β-estradiol acts through ERα to increase IL-4 and GATA-3 expression in CD4+ T cells independent of antigen presentation. J Immunol. 2005;175:5716–5723. doi: 10.4049/jimmunol.175.9.5716. [DOI] [PubMed] [Google Scholar]
- 54.Marzo AL, Vezys V, Williams K, Tough DF, Lefrançois L. Tissue-level regulation of Th1 and Th2 primary and memory CD4 T cells in response to Listeria infection. J Immunol. 2002;168:4504– 4510. doi: 10.4049/jimmunol.168.9.4504. [DOI] [PubMed] [Google Scholar]
- 55.Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, Ghiran S, Gerard NP, Yu C, Orkin SH, Gerard C. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305:1776–1779. doi: 10.1126/science.1100283. [DOI] [PubMed] [Google Scholar]
- 56.Yiamouyiannis CA, Schramm CM, Puddington L, Stengel P, Baradaran-Hosseini E, Whiteley HE, Thrall RS. Shifts in lung lymphocyte profiles correlate with the sequential development of acute allergic and chronic tolerant stages in a murine asthma model. Am J Pathol. 1999;154:1911–1921. doi: 10.1016/S0002-9440(10)65449-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cohn L, Tepper JS, Bottomly K. IL-4-independent induction of airway hyperresponsiveness by Th2, but not Th1, cells. J Immunol. 1998;161:3813–3816. [PubMed] [Google Scholar]
- 58.Hansen G, Berry G, DeKruyff RH, Umetsu DT. Allergen-specific Th1 cells fail to counterbalance Th2 cell-induced airway hyperreactivity but cause severe airway inflammation. J Clin Invest. 1999;103:175–183. doi: 10.1172/JCI5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 60.Smith KM, Pottage L, Thomas ER, Leishman AJ, Doig TN, Xu D, Liew FY, Garside P. Th1 and Th2 CD4+ T cells provide help for B cell clonal expansion and antibody synthesis in a similar manner in vivo. J Immunol. 2000;165:3136–3144. doi: 10.4049/jimmunol.165.6.3136. [DOI] [PubMed] [Google Scholar]
- 61.Uthoff H, Spenner A, Reckelkamm W, Ahrens B, Wolk G, Hackler R, Hardung F, Schaefer J, Scheffold A, Renz H, Herz U. Critical role of preconceptional immunization for protective and nonpathological specific immunity in murine neonates. J Immunol. 2003;171:3485–3492. doi: 10.4049/jimmunol.171.7.3485. [DOI] [PubMed] [Google Scholar]
- 62.Bednar-Tantscher E, Mudde GC, Rot A. Maternal antigen stimulation downregulates via mother’s milk the specific immune responses in young mice. Int Arch Allergy Immunol. 2001;126:300–308. doi: 10.1159/000049527. [DOI] [PubMed] [Google Scholar]
- 63.Melkild I, Groeng EC, Leikvold RB, Granum B, Lovik M. Maternal allergen immunization during pregnancy in a mouse model reduces adult allergy-related antibody responses in the offspring. Clin Exp Allergy. 2002;32:1370–1376. doi: 10.1046/j.1365-2745.2002.01458.x. [DOI] [PubMed] [Google Scholar]
- 64.Victor JR, Jr, Fusaro AE, Duarte AJ, Sato MN. Preconception maternal immunization to dust mite inhibits the type I hypersensitivity response of offspring. J Allergy Clin Immunol. 2003;111:269–277. doi: 10.1067/mai.2003.39. [DOI] [PubMed] [Google Scholar]
- 65.Herz U, Ahrens B, Scheffold A, Joachim R, Radbruch A, Renz H. Impact of in utero Th2 immunity on T cell deviation and subsequent immediate-type hypersensitivity in the neonate. Eur J Immunol. 2000;30:714–718. doi: 10.1002/1521-4141(200002)30:2<714::AID-IMMU714>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 66.Hamada K, Suzaki Y, Goldman A, Ning YY, Goldsmith C, Palecanda A, Coull B, Hubeau C, Kobzik L. Allergen-independent maternal transmission of asthma susceptibility. J Immunol. 2003;170:1683–1689. doi: 10.4049/jimmunol.170.4.1683. [DOI] [PubMed] [Google Scholar]
- 67.Fedulov A, Silverman E, Xiang Y, Leme A, Kobzik L. Immuno-stimulatory CpG oligonucleotides abrogate allergic susceptibility in a murine model of maternal asthma transmission. J Immunol. 2005;175:4292– 4300. doi: 10.4049/jimmunol.175.7.4292. [DOI] [PubMed] [Google Scholar]
- 68.Leme AS, Hubeau C, Xiang Y, Goldman A, Hamada K, Suzaki Y, Kobzik L. Role of breast milk in a mouse model of maternal transmission of asthma susceptibility. J Immunol. 2006;176:762–769. doi: 10.4049/jimmunol.176.2.762. [DOI] [PubMed] [Google Scholar]
- 69.Hubeau C, Apostolou I, Kobzik L. Adoptively transferred allergen-specific T cells cause maternal transmission of asthma risk. Am J Pathol. 2006;168:1931–1939. doi: 10.2353/ajpath.2006.051231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eder W, Klimecki W, Yu L, von Mutius E, Riedler J, Braun-Fahrlander C, Nowak D, Martinez FD. Opposite effects of CD 14/−260 on serum IgE levels in children raised in different environments. J Allergy Clin Immunol. 2005;116:601– 607. doi: 10.1016/j.jaci.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 71.Maier LM, Howson JM, Walker N, Spickett GP, Jones RW, Ring SM, McArdle WL, Lowe CE, Bailey R, Payne F, et al. Association of IL13 with total IgE: evidence against an inverse association of atopy and diabetes. J Allergy Clin Immunol. 2006;117:1306–1313. doi: 10.1016/j.jaci.2005.12.1354. [DOI] [PubMed] [Google Scholar]
- 72.Vercelli D, Martinez FD. The Faustian bargain of genetic association studies: bigger might not be better, or at least it might not be good enough. J Allergy Clin Immunol. 2006;117:1303–1305. doi: 10.1016/j.jaci.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 73.Lima C, Souza VM, Faquim-Mauro EL, Hoshida MS, Bevilacqua E, Macedo MS, Tavares-de-Lima W, Vargaftig BB. Modulation of the induction of lung and airway allergy in the offspring of IFNγ-treated mother mice. J Immunol. 2005;175:3554–3559. doi: 10.4049/jimmunol.175.6.3554. [DOI] [PubMed] [Google Scholar]
- 74.Bevilacqua C, Montagnac G, Benmerah A, Candalh C, Brousse N, Cerf-Bensussan N, Perdue MH, Heyman M. Food allergens are protected from degradation during CD23-mediated transepithelial transport. Int Arch Allergy Immunol. 2004;135:108–116. doi: 10.1159/000080653. [DOI] [PubMed] [Google Scholar]
- 75.Montagnac G, Molla-Herman A, Bouchet J, Yu LC, Conrad DH, Perdue MH, Benmerah A. Intracellular trafficking of CD23: differential regulation in humans and mice by both extracellular and intracellular exons. J Immunol. 2005;174:5562–5572. doi: 10.4049/jimmunol.174.9.5562. [DOI] [PubMed] [Google Scholar]
- 76.Platts-Mills TA, Erwin EA, Allison AB, Blumenthal K, Barr M, Sredl D, Burge H, Gold D. The relevance of maternal immune responses to inhalant allergens to maternal symptoms, passive transfer to the infant, and development of antibodies in the first 2 years of life. J Allergy Clin Immunol. 2003;111:123–130. doi: 10.1067/mai.2003.10. [DOI] [PubMed] [Google Scholar]
- 77.Celedon JC, Litonjua AA, Ryan L, Platts-Mills T, Weiss ST, Gold DR. Exposure to cat allergen, maternal history of asthma, and wheezing in first 5 years of life. Lancet. 2002;360:781–782. doi: 10.1016/S0140-6736(02)09906-3. [DOI] [PubMed] [Google Scholar]