

Abstract
Adenosine-induced coronary vasodilation is predominantly A2A adenosine receptor (AR)-mediated, whereas A1 AR is known to negatively modulate the coronary flow (CF). However, the coronary responses to adenosine in hyperlipidemia and atherosclerosis are not well understood. Using hyperlipidemic/atherosclerotic apolipoprotein E (APOE)–knockout mice, CF responses to nonspecific adenosine agonist (5′-N-ethylcarboxamide adenosine, NECA) and specific adenosine agonists (2-chloro-N6-cyclopentyl-adenosine [CCPA, A1 AR-specific] and CGS-21680, A2A AR-specific) were assessed using isolated Langendorff hearts. Western blot analysis was performed in the aorta from APOE and their wild-type (WT) control (C57BL/6J). Baseline CF (expressed as mL/min/g heart weight) was not different among WT (13.23 ± 3.58), APOE (13.22 ± 2.78), and APOE on high-fat diet (HFD) for 12 weeks (APOE-HFD, 12.37 ± 4.76). Concentration response curves induced by CGS-21680 were significantly shifted to the left in APOE and APOE-HFD when compared with WT. CCPA induced an increase in CF only at 10−6 M in all groups and the effect was reversed by the addition of a selective A2A AR antagonist, SCH-58261 (10−6 M), and a significant decrease in CF from baseline was observed. Western blot analysis showed a significant upregulation of A2A AR in the aorta from APOE and APOE-HFD. This study provides the first evidence that CF responses to A2A AR stimulation were upregulated in hyperlipidemic/atherosclerotic animals. The speculation is that the use of A2A AR-specific agonist for myocardial perfusion imaging (such as regadenoson) could overestimate the coronary reserve in coronary artery disease patients.
Keywords: hyperlipidemia, atherosclerosis, apolipoprotein E–knockout mice, coronary flow regulation, A2A adenosine receptor
Introduction
Coronary heart disease, caused mostly by atherosclerosis, is one of the leading causes of death in the USA and around the world. Elevated plasma cholesterol is one of the major independent risk factors for the development of atherosclerosis. More than 50% of heart disease cases are attributable to lipid abnormalities.1 The danger of atherosclerosis is thromboembolism, evolving from atherosclerotic plaques or by thrombotic vessel occlusion, leading to cardiac ischemia and infarction. The damage to tissue causes an increase in the release of adenosine, which is one of the major local regulators of coronary vessel tone and also plays a significant protective role in damage caused by ischemia-reperfusion injury. Pharmacological interventions using adenosine or its analogues are being investigated mostly toward adenosine-mediated effects in the cardiovascular system, such as the treatment of supraventricular arrhythmia, pharmacological stress myocardial perfusion imaging, congestive heart failure, controlling blood pressure, attenuating reperfusion injury following regional myocardial infarction, reducing infarct size, and improving postischemic cardiac function.2–5 Of the four adenosine receptor (AR) subtypes (A1, A2A, A2B, and A3), A1 and A2A ARs are predominantly expressed in coronary arteries. Adenosine-induced coronary vasodilation is predominantly A2A AR-mediated, whereas A1 AR is known to negatively modulate vasodilation.6–10 Recently, an A2A AR agonist, Lexiscan® (regadenoson, Astellas Pharma US, Inc, Deerfield, IL), has been approved by the US Food and Drug Administration (FDA) for myocardial perfusion imaging due to its selectivity (predominant coronary flow [CF] effect) and its avoidance of the A1 AR-mediated heart rate (HR) reducing side effects of adenosine, as well as A1 AR-mediated bronchoconstriction that was previously used.11
Arguably, the most critical advancement in the elucidation of factors affecting atherogenesis has been the development of mouse models of atherosclerosis. Among available models, the apolipoprotein E (APOE)–knockout mouse is particularly popular because of its tendency to spontaneously develop hyperlipidemia and atherosclerotic lesions on a standard chow diet, whereas a high-fat diet (HFD) exacerbates it.12 All known different stages of the atherosclerotic process are seen in the APOE, and are similar to the stages found in humans.13 Surprisingly, with the prevalence of coronary heart disease in Western countries and the importance of adenosine in CF regulation, few studies have been done to explore this effect of adenosine in hyperlipidemia/atherosclerosis.
Therefore, the objective of this study is to characterize the cardiovascular responses to adenosine analogues in a hyperlipidemic/atherosclerotic mouse model using isolated heart (Langendorff) experiments and isolated aorta experiments.
Materials and methods
Animals
All animals were cared for in accordance with protocol (No. 07-0801) approved by the Animal Care and Use Committee of the Health Science Center at West Virginia University, which conforms to the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). APOE mice and their wild-type (WT) control (C57BL/6J) were originally purchased from Jackson Laboratory (Bar Harbor, ME). All animals used in a given experiment originated from the same breeding pairs and were matched for age and weight. Standard laboratory food and water were available ad libitum. Animals were housed at a temperature that was held constant at 23 ± 2°C and humidity was 60% ± 10%. An inverted light–dark cycle of 12:12 h was used. All mice of either sex were 20–24 weeks old at the time of experiments.
One group of APOE mice (8 weeks) was placed on an HFD for 12 weeks to accelerate atherosclerosis (20 weeks old at the time of the experiments). The authors’ previous experiments have shown that after 12 weeks of HFD, lesions from APOE exhibited more extensive atherosclerotic lesions than APOE fed normal chow at the same age.14 The HFD was purchased from Harlan-Teklad (Madison, WI; catalog no. TD88137), consisting of 0.2% cholesterol, 21.2% fat, 0.75% methionine, 1.43 mg/g choline, 2.23 mg/kg folate, 32.5 µg/kg B12, and 22.25 mg/kg B6.
Langendorff preparation
Isolated heart experiments were performed in accordance with the methods previously described.9,15,16 Animals were anesthetized with intraperitoneal (ip) sodium pentobarbital (50 mg/kg); the depth of anesthesia was assessed by nonresponsiveness to toe and tail pinch. A thoracotomy was then performed, and the hearts were excised into an ice-cold heparinized (5 U/mL) modified Krebs-Henseleit buffer containing (in mM): NaCl 120, NaHCO3 25, KCl 4.7, KH2PO4 1.2, CaCl2 2.5, MgSO4 1.2, glucose 15, and ethylenediaminetetraacetic acid (EDTA) 0.05. After removal of lung and surrounding tissue, the aortas were rapidly cannulated with a 20-gauge blunt-ended needle, and retrograde coronary perfusion was initiated at constant pressure of 80 mm Hg with the Krebs-Henseleit buffer. The perfusate was equilibrated with 95% O2–5% CO2 at 37°C (pH of 7.4) and a pO2 of ~550 mm Hg. The left atrium was removed, and the left ventricle was vented with a small polyethylene apical drain. A fluid-filled balloon constructed of plastic film was inserted into the left ventricle across the mitral valve and connected to a pressure transducer permitting continuous measurement of left ventricular pressure. Balloon volume was modified through a stopcock attached to the ventricular pressure transducer using a 500-µL glass syringe (Hamilton Co, Reno, NV) to maintain a left ventricular diastolic pressure of 2–5 mm Hg. Hearts were immersed in a water-jacketed perfusate bath maintained at 37°C, and the bath was rinsed once at the onset of perfusion to clear blood from the bathing environment. CF was continuously monitored via a Doppler flow probe (Transonic Systems, Ithaca, NY) located in the aortic perfusion line. CF, aortic pressure, and left ventricular pressure were recorded in a PowerLab® multichannel data acquisition system (ADInstruments, Castle Hill, Australia) connected to a computer. Data were expressed as percentage of change from baseline for each parameter before the application of drugs.
Isolated aorta experiments
While the mice were under deep anesthesia with pentobarbital sodium (100 mg/kg ip), a thoracotomy was performed and the aorta was gently removed. Fat and connective tissue were removed and the aorta was cut transversely into three or four rings 3–4 mm wide. The rings were mounted vertically between two stainless steel wire hooks with extreme care being taken to avoid damaging the endothelium. The rings were suspended in 10-mL organ baths that contained Krebs-Henseleit solution continuously gassed with 95% O2–5% CO2 (37°C, pH 7.4). Aortic rings were allowed to equilibrate for 60 min at an initial resting tension of 1 g. Composition of the Krebs-Henseleit solution was (in mM): 118 NaCl, 4.8 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 2.5 CaCl2, and 11 glucose. Changes in the isometric tension were measured with a fixed-range precision-force transducer (model TSD 125C; BIOPAC Systems, Goleta, CA) connected to a differential amplifier (model DA 100B; BIOPAC Systems). Data were recorded on a Dell (Round Rock, TX) computer using an MP 100 WSW digital acquisition system (BIOPAC Systems) and were analyzed using AcqKnowledge 3.5.7 software (BIOPAC Systems).
After rings were equilibrated, the responsiveness of each individual ring was checked by successive administration of a submaximally effective concentration of KCl (50 mM). The integrity of the vascular endothelium was tested pharmacologically by acetylcholine (1 µM)-induced relaxation of 0.1 µM phenylephrine-precontracted rings. Tissues that did not elicit a reproducible and stable contraction with phenylephrine and relaxed with acetylcholine were discarded from the study. Preparations were then washed several times with Krebs-Henseleit solution and allowed to relax fully for 30 min before the start of experiment.
Western blot
Mouse aorta proteins were isolated by using an ice-cold tissue lysis buffer consisting of 0.05 M Tris-buffered saline (TBS), pH 7.4, 1% Triton X-100, 0.25% sodium deoxycholate, 150 mM sodium chloride, 1 mM EDTA, 1 mM phenylmethylsulfonyl, and Halt® Protease Inhibitor Cocktail (Thermo Scientific, Waltham, MA) by way of glass mortar and pestle. Samples were then centrifuged for 15 min at 13,000 revolutions per minute (rpm) and the supernatant was stored at −80°C. Protein extracts (30 µg protein per well) were separated on NuPAGE® 4%–12% Bis-Tris Gels (Invitrogen, Carlsbad, CA) along with Novex® Sharp Protein Standard, 3.5–260 kDa (Invitrogen), run in parallel. Proteins were then transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA), blocked by 5% milk for 1 hour and then probed with anti-A2A AR rabbit polyclonal immunoglobulin G (IgG) antibody that was produced in the authors’ lab17 with a dilution of 1:1000 in TBS with Tween® (TBST; Promega, Madison, WI) +0.5% milk overnight at 4°C or with anti-β-actin (Santa Cruz Biotechnology, Inc, Santa Cruz, CA) at a dilution of 1:5000 at room temperature for 1 hour. This was followed by incubation with a secondary horseradish peroxidase-conjugated antibody (antimouse and antirabbit immunoglobulins, respectively; Santa Cruz Biotechnology, Inc) for 1 hour at room temperature. For detection of bands, the membranes were treated with an enhanced chemiluminescence reagent (GE Healthcare, Waukesha, WI) for 1 min and subsequently exposed to ECL Hyperfilm® (GE Healthcare). Relative band intensities were quantified by densitometry, and each sample was normalized to the β-actin values.
Experimental protocol
Isolated heart experiments
After a 30-min equilibration period, CF, HR, and developed pressure (systolic pressure minus diastolic pressure) were examined in WT, APOE, and APOE fed HFD (APOE-HFD), and baseline data for these parameters were collected at the end of equilibration period. A total of 18 WT, 15 APOE, and 23 APOE-HFD of both sexes were used in the experiments. The number of mice used in each group of the following experiments is listed in the figures and table.
Dose–response relationships were constructed for 5′-N-ethylcarboxamide adenosine (NECA, 10−10–10−6 M), a nonspecific adenosine agonist, by infusing the drug into the coronary perfusate through an injection port directly proximal to the aortic cannula. The infusion rate was controlled to a maximum of 1% of CF with a microinjection infusion pump (Harvard Apparatus, Holliston, MA). After baseline data were acquired, each heart was exposed to progressively increasing concentrations of NECA to develop a dose–response relationship. Each concentration of NECA was infused for 5 min, and data were sampled at the end of the 5-min infusion period. After the infusion of each concentration of NECA, a minimum of 5 min of perfusion was allowed for complete drug washout (with recovery to baseline parameters occurring uniformly between 5 and 10 min). Data were again collected at the end of each washout period and used as references for normalizing response to subsequent agonist concentration.
A2A AR-specific agonist, CGS-21680 (10−11–10−6 M), and A1 AR-specific agonist, 2-chloro-N6-cyclopentyl-adenosine (CCPA, 10−11–10−6 M), were also used to construct concentration–response curves.
In addition, CCPA was found to induce vasodilation at high concentration (10−6 M) in both WT and APOE-HFD. Therefore, an A2A AR antagonist, 2-(2-Furanyl)-7-(2-phenylethyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c] pyrimidin-5-amine (SCH-58261), was used to block the possible A2A AR-mediated vasodilation at a high concentration of CCPA. The antagonist, SCH-58261, was infused at final concentration of 10−6 M for 10 min before the start of CCPA and throughout the rest of the experiments.
Isolated aorta experiments
After equilibration and verification of arterial ring integrity, the concentration–response curves for NECA were obtained by cumulative addition of the agonists in phenylephrine (0.1 µM)-contracted tissues. The agonists were added to yield the next higher concentration only when the response to the earlier dose reached a steady state (usually about 5–10 min). At the end of the highest concentration, baths were washed four times. After baseline was established, 10 µM of sodium nitroprusside (SNP) was added to ensure that the maximal relaxation was achieved. The relaxation responses were expressed as percent decrease of total tension induced by 0.1 µM phenylephrine.
Materials
NECA, CCPA, CGS-21680, phenylephrine, SNP, and all the chemicals were purchased from Sigma-Aldrich (St Louis, MO). SCH-58261 was purchased from Tocris Bioscience (Ellisville, MO).
Statistical analysis
Unless stated otherwise, all data were expressed as mean ± standard error of the mean (SEM). All concentration–response curves were analyzed using nonlinear regression using GraphPad Prism3® (GraphPad Software, Inc, San Diego, CA). EC50 were derived from these curves. For statistical comparison between curves, pEC50 (negative logarithm of the molar concentration of agonist producing 50% of the maximum response) of the curve was used.18–20 Maximum effect (Emax) elicited by the agonist was also used to compare between groups. When significant differences between curves were found, the data were further analyzed between groups at the same concentrations, then analyzed using Student’s t-test or one-way analysis of variance (ANOVA) followed by Bonferroni correction for multiple comparisons. P < 0.05 was considered statistically significant.
Results
Isolated heart experiments
Table 1 summarizes baseline functional parameters (CF, HR, and developed pressure) after 30 min of equilibration in isolated mouse hearts perfused at constant pressure. Body weight, heart weight, heart-to-body weight ratio, baseline CF, HR, and left ventricular developed pressure (LVDP) were not significantly different among groups.
Table 1.
Baseline functional data of C57BL/6 J (WT), APOE and APOE-HFD
| WT (n = 16) | APOE (n = 15) | APOE-HFD (n = 14) | |
|---|---|---|---|
| Body weight (g) | 28.40 ± 4.57 | 26.01 ± 3.38 | 28.31 ± 5.35 |
| Heart weight (mg) | 157.40 ± 22.70 | 150.60 ± 21.85 | 171.59 ± 24.38 |
| Heart-to-body weight ratio (%) | 0.53 ± 0.12 | 0.53 ± 0.04 | 0.58 ± 0.08 |
| Coronary flow (mL/min/g) | 13.23 ± 3.58 | 13.22 ± 2.78 | 12.37 ± 4.76 |
| Heart rate (beat/min) | 381.51 ± 34.25 | 372.39 ± 22.50 | 369.06 ± 34.18 |
| Developed pressure (mm Hg) | 141.61 ± 22.00 | 118.46 ± 30.88 | 123.62 ± 16.66 |
Notes: All values are means ± SEM; functional parameters were measured after 30 min of normothermic aerobic perfusion using a standard Langendorff preparation; P < 0.05.
Abbreviations: APOE, apolipoprotein E knockout; APOE-HFD, APOE fed high-fat diet; n, number of hearts; SEM, standard error of the mean; WT, wild type.
The CF pEC50s for the nonspecific agonist, NECA, were not significantly different among groups (Figure 1A: 8.43 ± 0.15 in WT, 8.49 ± 0.15 in APOE, and 8.54 ± 0.21 in APOE-HFD), although the graph shows a trend to leftward shift (Figure 1A). The maximal CF responses (Emax) to NECA were not different among different groups (Figure 1A: 211.26% ± 12.14% in WT, 249.02% ± 29.53% in APOE, and 253.05% ± 4.95% in APOE-HFD). The NECA-induced negative chronotropic responses (pEC50s) were significantly shifted to the right in APOE-HFD groups in comparison to WT and APOE groups (Figure 1B: HR pEC50s for WT, APOE, and APOE-HFD were 7.26 ± 0.12, 7.04 ± 0.19, and 6.24 ± 0.13). The maximal HR responses (Emax) to NECA were not different between groups (Figure 1B: 32.69% ± 2.70%, 47.43% ± 4.93%, and 43.01% ± 5.69%). There is a significant leftward shift in LVDP concentration–response curves (pEC50s) in APOE and APOE-HFD when compared with WT (Figure 1C: pEC50 for WT, APOE, and APOE-HFD were 6.86 ± 0.41, 8.33 ± 0.11, and 8.84 ± 0.45).
Figure 1.
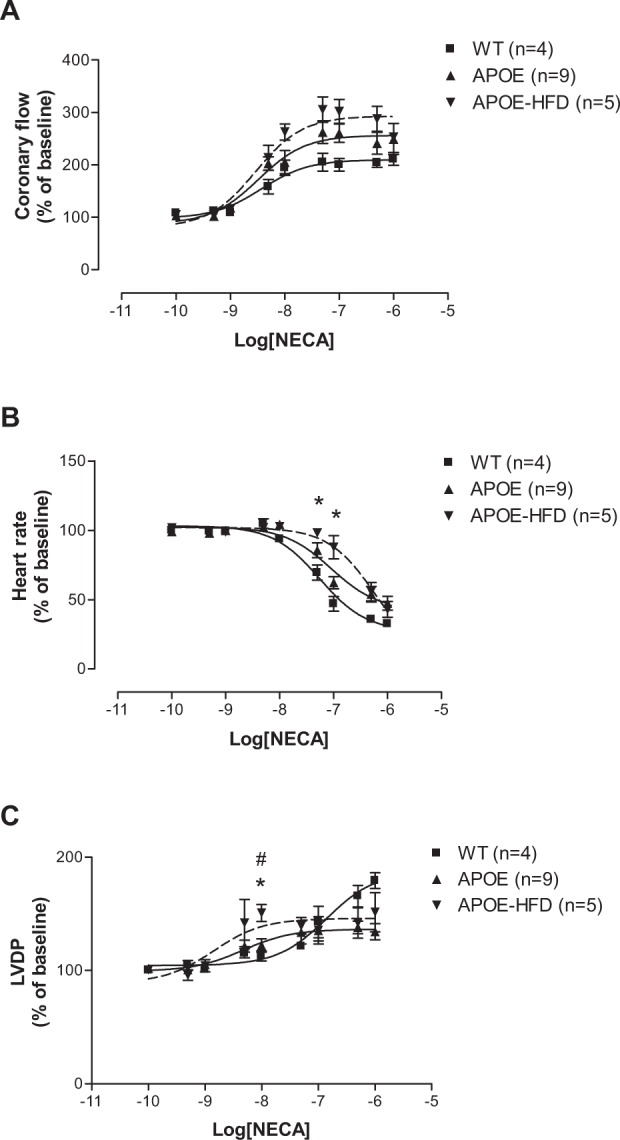
Concentration–response curves for coronary flow (A), heart rate (B), and left ventricular developed pressure (C) for NECA in isolated perfused hearts from WT apolipoprotein E–knockout mice, and APOE-HFD.
Notes: *Significant difference from WT; #significant difference from APOE.
Abbreviations: APOE, apolipoprotein E; APOE-HFD, APOE fed high-fat diet; HR, heart rate; LVDP, left ventricular developed pressure; NECA, 5′-N-ethylcarboxamide adenosine; WT, wild type.
The CF pEC50s for the A2A AR-specific agonist, CGS-21680, showed a significant difference between various groups (Figure 2A: 8.14 ± 0.06 in WT, 9.02 ± 0.11 in APOE, and 8.75 ± 0.13 in APOE-HFD). The curves for CF shifted significantly to the left in APOE and APOE-HFD groups from their WT control (Figure 2A). The maximal CF response to CGS-21680 was not significantly different between groups (204.37% ± 19.30% in WT, 240.34% ± 37.86% in APOE, and 246.68% ± 29.16% in APOE-HFD). CGS-21680 had no effect on HR in all three groups (Figure 2B). Also, CGS-21680 induced a concentration-dependent increase in LVDP in all three groups (Figure 2C), but they were not significantly different from each other (Figure 2C).
Figure 2.
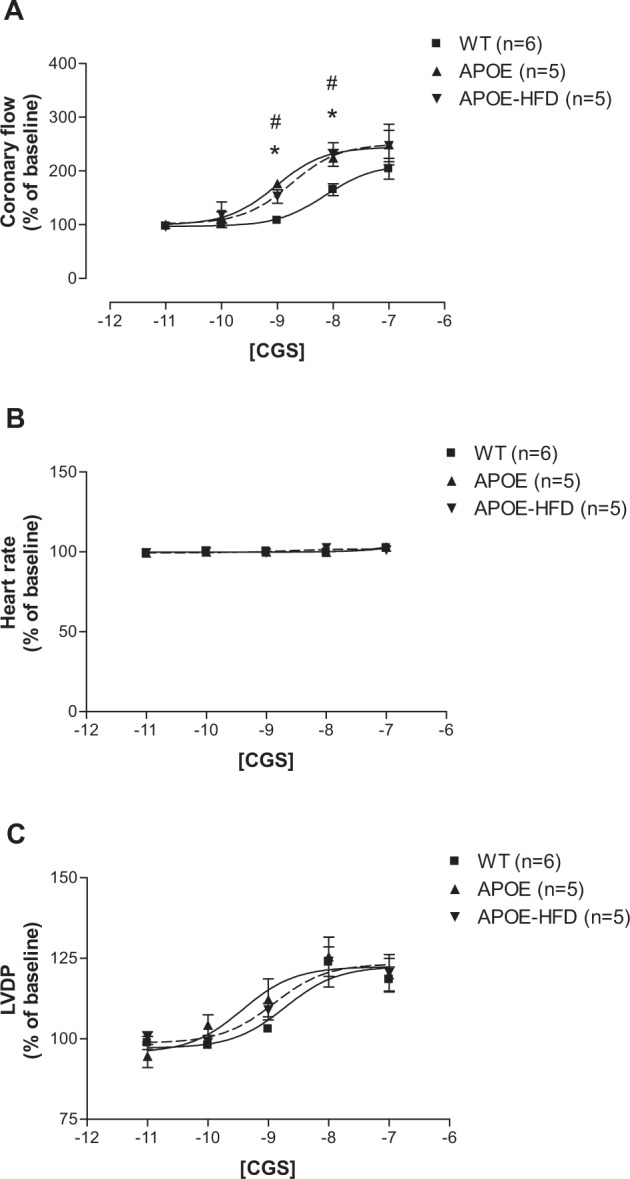
Concentration–response curves for coronary flow (A), heart rate (B), and LVDP (C) for CGS-21680 (CGS) in isolated perfused hearts from WT, APOE, and APOE-HFD.
Notes: *Significant difference from WT; #significant difference from APOE.
Abbreviations: APOE, apolipoprotein E; APOE-HFD, APOE fed high-fat diet; LVDP, left ventricular developed pressure; WT, wild type.
Since the most significant changes in NECA-induced negative chronotropic effect was in the APOE-HFD group, the concentration–response curves for CCPA, the A1 AR-specific agonist, were only constructed in WT and APOE fed HFD (APOE-HFD) groups (Figure 3). Due to observed CF increase at a concentration of 10−6 M CCPA, 1 µM SCH-58261 (A2A selective antagonist) was used throughout to confirm the possible nonspecific A2A effect of CCPA. Therefore, four groups were divided in these experiments: CCPA in WT, CCPA in the presence of SCH-58621 in WT (WT + SCH), CCPA in APOE-HFD (APOE-HFD), and CCPA in the presence of SCH-58621 in APOE-HFD (APOE-HFD + SCH).
Figure 3.
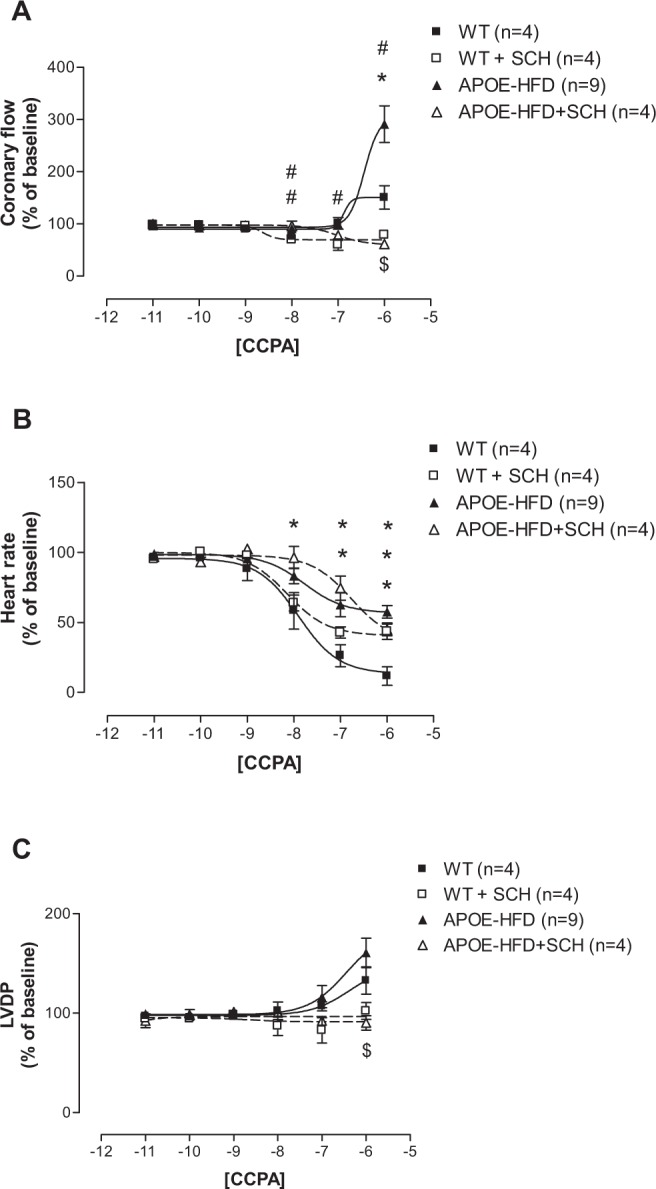
Concentration–response curves for coronary flow (A), Heart rate (B), and LVDP (C) for CCPA with or without 1 µM SCH-58261 (SCH) in isolated perfused hearts from WT and APOE-HFD.
Notes: *Significant difference from WT; #significant difference from WT+SCH; $significant difference from APOE-HFD.
Abbreviations: APOE, apolipoprotein E; APOE-HFD, APOE fed high-fat diet; CCPA, 2-chloro-N6-cyclopentyl-adenosine; LVDP, left ventricular developed pressure; WT, wild type.
CCPA by itself did not change CF until it reached 10−6 M, where it increased CF in both WT and APOE-HFD (Figure 3A solid squares and solid triangles). The maximal CF increase induced by CCPA at 10−6 M in APOE-HFD was also significantly greater than in WT (Emax were: 150.64% ± 22.44% in WT vs 291% ± 35.15% in APOE-HFD). After the addition of SCH-58261, the CCPA-induced increase in CF at 10−6 M was completely blocked in both WT and APOE-HFD groups (Figure 3A open squares and triangles, Emax were: 78.99% ± 6.69% in WT with SCH-58261 and 61.35% ± 13.99% in APOE-HFD with SCH-58261). In addition, SCH-58261 also “unmasked” the CF-reducing effect of CCPA at a concentrations ranging from 10–8–10–6 M in WT and APOE-HFD groups (Figure 3A open square and triangle), with Emax at 60%–80% range.
As in the case with NECA, a negative chronotropic (HR) response to CCPA in APOE-HFD was significantly less than in WT (curve shifted to the right, Figure 3B). Interestingly, SCH-58261 seems to have an effect in shifting the CCPA concentration–response curve to the right when compared with CCPA without SCH-58261 in both WT and APOE-HFD (Figure 3B, pEC50s were: 7.9 ± 0.07 in WT, 8.14 ± 0.16 in WT+SCH-58621, 7.77 ± 0.03 in APOE-HFD, and 6.77 ± 0.18 in APOE-HFD+SCH-58621). In addition, SCH-58621 also blocked the maximal negative chronotropic effect of CCPA in WT, but not in APOE-HFD, when compared with CCPA alone (Figure 3B, Emax were: 11.71% ± 6.58% in WT, 43.34% ± 5.51% in WT+SCH-58621, 57.58% ± 4.42% in APOE, and 44.97% ± 4.50% in APOE-HFD+SCH-58621).
Similar to CF concentration–response curves, CCPA did not increase LVDP until the highest concentration (10−6 M) in WT and APOE-HFD groups (Figure 3C). There was no significant difference between WT and APOE-HFD at 10−6 M concentration without SCH-58261 (Figure 3C). However, the concentration–response curve was significantly blocked by the addition of 1 µM SCH-58261 in both WT and APOE-HFD (Figure 3C).
Organ bath experiments
NECA induced a concentration-dependent contraction until 10−7 M and then relaxation at 10−6 and 10−5 M in WT and APOE (Figure 4A). There was no significant difference between these two groups. In the APOE-HFD group, however, the concentration–response curve was significantly different from WT at 10−7 and 10−6 M of NECA. In effect, no significant vasoconstriction was found in the APOE-HFD group. Further analysis of the aortic responses to acetylcholine at maximal tension induced by phenylephrine showed that the response in the APOE-HFD group was significantly less when compared with the other two groups (Figure 4B, 31.24% ± 18.89% reduction in APOE-HFD vs 63.01% ± 15.35% and 67.16% ± 18.91% reduction in WT and APOE groups, respectively).
Figure 4.
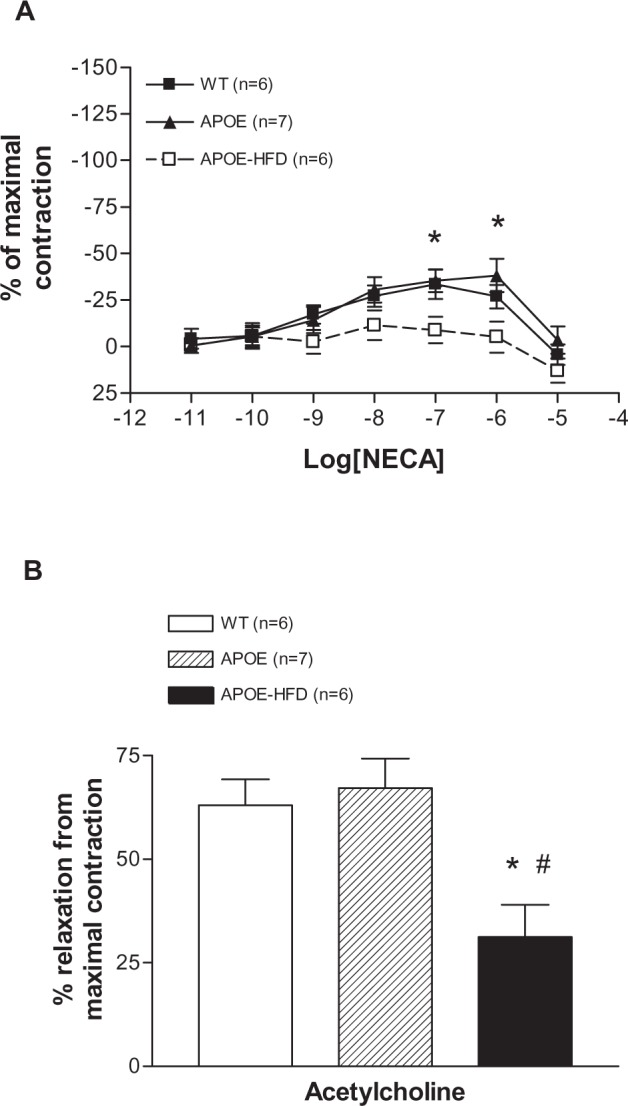
Concentration–response curves for changes in isometric tension produced by NECA in isolated aorta from WT, APOE, and APOE-HFD (A). Responses to 1 µM acetylcholine after preconstriction with 0.1 µM phenylnephrine in aortas from WT, APOE, and APOE-HFD (B).
Notes: *Significant difference from WT; #significant difference from APOE.
Abbreviations: APOE, apolipoprotein E; APOE-HFD, APOE fed high-fat diet; NECA, 5′-N-ethylcarboxamide adenosine; WT, wild type.
Western blot experiments
A2A AR expression was found to be significantly higher in the aortas from APOE and APOE-HFD when compared with the aorta from WT as demonstrated in Figure 5.
Figure 5.
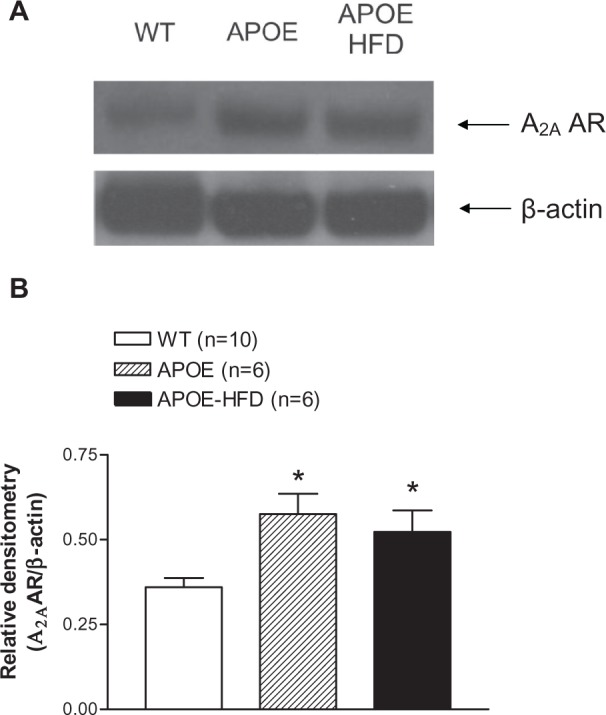
A representative A2A AR Western blot (A) and the densitometric analysis (B) of aortas from WT, APOE, and APOE-HFD. β-actin was used as a loading control.
Note: *Significant difference from WT.
Abbreviations: APOE, apolipoprotein E; APOE-HFD, APOE fed high-fat diet; A2A AR, A2A adenosine receptor; WT, wild type.
Discussion
The major finding of this study is that A2A AR function is significantly upregulated in APOE, both with and without HFD. The CF concentration–response curves for A2A AR agonist (CGS-21680, Figure 2) shifted to the left in APOE and APOE-HFD. Also observed was an increase in response to A1 AR agonist (CCPA, Figure 3) in APOE-HFD at the highest concentration (10−6 M) when compared with WT and the effect was completely blocked by A2A AR-specific antagonist, SCH-58261. In addition, similar responses were also observed in isolated mouse aorta (Figure 4A), which demonstrated less NECA-induced vasoconstriction in APOE-HFD group. These data support the enhanced response to A2A AR stimulation in hyperlipidemic/atherosclerotic mice. Finally, the Western blot experiments in the aorta from WT, APOE, and APOE-HFD provided the molecular evidence for A2A AR upregulation in the authors’ hyperlipidemia/atherosclerosis model (Figure 5).
The mechanism of the A2A AR upregulation is not clear. Previous studies in another atherosclerosis mouse model (LDL/ApoB48 double knockout mice) showed a decrease in coronary reserve,21 which is opposite to the authors’ findings. However, these mice were older and in a more severe state of the disease (57–70 weeks old after 2–3 months of HFD feeding) than in this study (20–24 weeks old after 3 months of HFD feeding). It is possible that the younger mice in this study were in the early stage of the disease and the upregulation of A2A AR is part of the compensatory mechanism to cope with the narrowing of the coronary arteries, which can be severe in different segments of the coronary artery. This assumption can be supported by a previous study that the vasodilatory response to adenosine is enhanced in hypoxic porcine coronary arteries and the effect can be modulated by A2A antagonist, ZM-241385.22 Studies in PC-12 cells demonstrated that A2A AR expression is upregulated to enhance cell viability during hypoxia.23 Thus, the A2A AR upregulation found in this study may be due to a similar response to hypoxia leading to a compensatory decrease in CF by upregulating A2A AR.
Adenosine and dipyridamole (inhibits the uptake of adenosine) are routinely used in detecting CF reserve, especially in patients suspected of coronary artery disease. It is mainly due to the activation of A2A AR. Regadenoson (Lexiscan), a low affinity selective A2A AR agonist that has pronounce coronary vasodilatory effect and minimal systemic effect, is marketed specifically for this application.24 However, if A2A AR response is upregulated in human coronary circulation as in this study, there may be a phase in patients who will have mild to severe coronary blockade due to atherosclerosis but show little change in coronary reserve. The change will only be exhibited when the compensatory response is exhausted, such as in the older LDL-ApoB48 double-knockout mice in Saraste’s study.21 Another study, using transthoracic echocardiography measuring CF reserve between WT and APOE mouse groups, showed diminished CF reserve (by adenosine) in APOE.25 However, APOE mice were much older than WT (40 vs 18 weeks) in this study. Since CF reserve is known to decrease with age, it is not clear if that claim is valid. Further investigations are needed to clarify these issues.
These data demonstrate that CCPA-induced vasoconstriction is unmasked after the application of SCH-58261. Previous studies from the authors’ lab and others have demonstrated that A1 AR plays an opposing role to A2A AR vasodilation in coronary circulation.7,26 In this study, the blockade of A2A AR by SCH-58261 in CCPA-induced CF changes reveals that A1 AR did actively constrict CF in both groups of mice (Figure 3). With the use of SCH-58261, CCPA induced a significant vasoconstriction at a concentration of 10−8 M or higher in WT but only at 10−7 M or higher in APOE-HFD, clearly supporting the authors’ findings. These data also suggest that A1 AR function in coronary circulation may also be downregulated in APOE-HFD. Further understanding of these observations is needed.
Furthermore, the response to endothelial-dependent acetylcholine was lower only in APOE-HFD (Figure 4B), suggesting endothelial dysfunction in atherosclerosis in this group. This is also in agreement with previous findings that the endothelial dysfunction was only found in atherosclerotic section of the vessel.27,28 Previous studies from the authors’ lab and others demonstrate that A2A AR-mediated coronary vasodilation is both endothelium dependent and independent and nitric oxide (NO) release is part of the A2A AR-mediated vasodilatory response.15,29–32 In the isolated aorta experiments, the contractile response to NECA was significantly lower only in APOE-HFD group (Figure 4A), which suggests an upregulation of A2A AR and this is further supported by the Western blot experiments (Figure 5). Although this may also suggest a downregulation of A1 AR, it is not supported by Western blot analysis in the authors’ previous study.14 In addition, the endothelial dysfunction (less responses to acetylcholine) in APOE-HFD also suggests that the A2A AR upregulation may be on smooth muscle cells only.
These data also demonstrate that NECA and CCPA-induced negative chronotropic responses were affected by hyperlipidemia/atherosclerosis. Previous studies from the authors’ group and others suggest that adenosine-induced negative chronotropic effect is predominantly mediated by A1 AR mainly due to its blockade of atrioventricular node (AV) node conduction.6,33 Therefore, it was reasonable to presume that A1 AR may be downregulated in hyperlipidemia/atherosclerosis. However, real-time reverse-transcription polymerase chain reaction (RT PCR) analysis of the heart did not show changes in A1 AR expression between WT and APOE on HFD (data not shown). These data imply that the modulated responses to NECA and CCPA in APOE may not be due to changes in A1 AR expression. Although hyperlipidemia was known to interfere with various ion channels in isolated cardiac cells,34 the baseline HR from both groups of mice did not differ from each other. This implies that the baseline electrical conduction of the heart may not be affected by the hyperlipidemic condition. Therefore, the modulating effect of hyperlipidemia/atherosclerosis on A1 AR-mediated negative chronotropic effect may lie at the second messenger level, such as a pertussis toxin-sensitive G protein, which is a downstream effector for A1 AR-mediated effects on HR.35
Interestingly, the use of SCH-58261seems to have a modulating effect on CCPA-induced decrease in HR in both WT and APOE-HFD. Previous studies have suggested that A2A AR agonists-induced tachycardia is mediated by a baroreceptor reflex trigger by a decrease in arterial pressure and/or direct stimulation of arterial chemoreceptor by A2A AR.24 Since the Langendorff system is an isolated heart preparation; this may not be the case. A recent study demonstrated that A2A ARs are also located in the atrium and may be able to activate ryanodine receptor.36 Another study also demonstrated that β-adrenergic stimulation of HR may be due to synchronization of ryanodine receptor related Ca2+ release.37 In addition, A2A AR is known to inhibit A1 AR-mediated β-adrenergic inhibition on cardiac contractility (ie, A2A AR will enhance β-adrenergic positive inotropic effect).38,39 Therefore, it is possible that A2A AR may also play a role in HR through the ryanodine receptor that is a part of the β-adrenergic mediated chronotropic responses.
In conclusion, this study demonstrates that hyperlipidemia and/or atherosclerosis in APOE mice have an effect on adenosine-induced CF and HR regulation. The increase in response to A2A AR agonist, CGS-21680, may be due to an upregulation of A2A AR in coronary vasculature, possibly on smooth muscle. Due to the use of adenosine and its analogues as a tool for cardiac imaging in assessing coronary reserve in atherosclerosis-suspected patients, the possible A2A AR upregulation in hyperlipidemic/atherosclerotic condition should be taken into consideration.
Acknowledgments
The authors would like to thank Kevin P Roush for his technical assistance. Supported by Research Development Grant from West Virginia University and NIH HL 027339 and HL 094447.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Sorrentino MJ. Cholesterol reduction to prevent CAD. What do the data show? Postgrad Med. 2000;108(7):40–42. 45–46, 49–52. doi: 10.3810/pgm.2000.12.1310. [DOI] [PubMed] [Google Scholar]
- 2.Geraets D, Kienzle M. Clinical use of adenosine. Iowa Med. 1992;82(1):25–28. [PubMed] [Google Scholar]
- 3.Smits GJ, McVey M, Cox BF, Perrone MH, Clark KL. Cardioprotective effects of the novel adenosine A1/A2 receptor agonist AMP 579 in a porcine model of myocardial infarction. J Pharmacol Exp Ther. 1998;286(2):611–618. [PubMed] [Google Scholar]
- 4.Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. 2007;356(11):1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 5.Peart JN, Headrick JP. Adenosinergic cardioprotection: multiple receptors, multiple pathways. Pharmacol Ther. 2007;114(2):208–221. doi: 10.1016/j.pharmthera.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Mustafa SJ, Morrison RR, Teng B, Pelleg A. Adenosine receptors and the heart: role in regulation of coronary blood flow and cardiac electrophysiology. Handb Exp Pharmacol. 2009;(193):161–188. doi: 10.1007/978-3-540-89615-9_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tawfik HE, Teng B, Morrison RR, Schnermann J, Mustafa SJ. Role of A1 adenosine receptor in the regulation of coronary flow. Am J Physiol Heart Circ Physiol. 2006;291(1):H467–H472. doi: 10.1152/ajpheart.01319.2005. [DOI] [PubMed] [Google Scholar]
- 8.Tawfik HE, Schnermann J, Oldenburg PJ, Mustafa SJ. Role of A1 adenosine receptors in regulation of vascular tone. Am J Physiol Heart Circ Physiol. 2005;288(3):H1411–H1416. doi: 10.1152/ajpheart.00684.2004. [DOI] [PubMed] [Google Scholar]
- 9.Morrison RR, Teng B, Oldenburg PJ, Katwa LC, Schnermann JB, Mustafa SJ. Effects of targeted deletion of A1 adenosine receptors on postischemic cardiac function and expression of adenosine receptor subtypes. Am J Physiol Heart Circ Physiol. 2006;291(4):H1875–H1882. doi: 10.1152/ajpheart.00158.2005. [DOI] [PubMed] [Google Scholar]
- 10.Belardinelli L, Shryock JC, Snowdy S, et al. The A2A adenosine receptor mediates coronary vasodilation. J Pharmacol Exp Ther. 1998;284(3):1066–1073. [PubMed] [Google Scholar]
- 11.Wilson CN, Nadeem A, Spina D, Brown R, Page CP, Mustafa SJ. Adenosine receptors and asthma. Handb Exp Pharmacol. 2009;193:329–362. doi: 10.1007/978-3-540-89615-9_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meir KS, Leitersdorf E. Atherosclerosis in the apolipoprotein- E-deficient mouse: a decade of progress. Arterioscler Thromb Vasc Biol. 2004;24(6):1006–1014. doi: 10.1161/01.ATV.0000128849.12617.f4. [DOI] [PubMed] [Google Scholar]
- 13.Hofker MH, van Vlijmen BJ, Havekes LM. Transgenic mouse models to study the role of APOE in hyperlipidemia and atherosclerosis. Atherosclerosis. 1998;137(1):1–11. doi: 10.1016/s0021-9150(97)00266-9. [DOI] [PubMed] [Google Scholar]
- 14.Teng B, Roush KP, Nadeem A, Morrison RR, Mustafa SJ. Reduced atherosclerotic lesions in A1 adenosine receptor (AR)/apolipoprotein E (APOE) double knockout mice: role for A1 AR in the development of atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29(7):e73–e74. [Google Scholar]
- 15.Teng B, Ledent C, Mustafa SJ. Up-regulation of A(2B) adenosine receptor in A(2A) adenosine receptor knockout mouse coronary artery. J Mol Cell Cardiol. 2008;44:905–914. doi: 10.1016/j.yjmcc.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morrison RR, Jones R, Byford AM, et al. Transgenic overexpression of cardiac A(1) adenosine receptors mimics ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2000;279(3):H1071–H1078. doi: 10.1152/ajpheart.2000.279.3.H1071. [DOI] [PubMed] [Google Scholar]
- 17.Marala RB, Mustafa SJ. Immunological characterization of adenosine A2A receptors in human and porcine cardiovascular tissues. J Pharmacol Exp Ther. 1998;286(2):1051–1057. [PubMed] [Google Scholar]
- 18.Irat AM, Aslamaci S, Karasu C, Ari N. Alteration of vascular reactivity in diabetic human mammary artery and the effects of thiazolidinediones. J Pharm Pharmacol. 2006;58(12):1647–1653. doi: 10.1211/jpp.58.12.0012. [DOI] [PubMed] [Google Scholar]
- 19.Wihlborg AK, Wang L, Braun OO, et al. ADP receptor P2Y12 is expressed in vascular smooth muscle cells and stimulates contraction in human blood vessels. Arterioscler Thromb Vasc Biol. 2004;24(10):1810–1815. doi: 10.1161/01.ATV.0000142376.30582.ed. [DOI] [PubMed] [Google Scholar]
- 20.Zatta AJ, Matherne GP, Headrick JP. Adenosine receptor-mediated coronary vascular protection in post-ischemic mouse heart. Life Sci. 2006;78(21):2426–2437. doi: 10.1016/j.lfs.2005.09.035. [DOI] [PubMed] [Google Scholar]
- 21.Saraste A, Kytö V, Laitinen I, et al. Severe coronary artery stenoses and reduced coronary flow velocity reserve in atherosclerotic mouse model: Doppler echocardiography validation study. Atherosclerosis. 2008;200(1):89–94. doi: 10.1016/j.atherosclerosis.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 22.Frobert O, Haink G, Simonsen U, Gravholt CH, Levin M, Deussen A. Adenosine concentration in the porcine coronary artery wall and A2A receptor involvement in hypoxia-induced vasodilatation. J Physiol. 2006;570(Pt 2):375–384. doi: 10.1113/jphysiol.2005.100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi S, Millhorn DE. Stimulation of expression for the adenosine A2A receptor gene by hypoxia in PC12 cells. A potential role in cell protection. J Biol Chem. 1999;274(29):20358–20365. doi: 10.1074/jbc.274.29.20358. [DOI] [PubMed] [Google Scholar]
- 24.Dhalla AK, Wong MY, Wang WQ, Biaggioni I, Belardinelli L. Tachycardia caused by A2A adenosine receptor agonists is mediated by direct sympathoexcitation in awake rats. J Pharmacol Exp Ther. 2006;316(2):695–702. doi: 10.1124/jpet.105.095323. [DOI] [PubMed] [Google Scholar]
- 25.Wikstrom J, Gronros J, Gan LM. Adenosine induces dilation of epicardial coronary arteries in mice: relationship between coronary flow velocity reserve and coronary flow reserve in vivo using transthoracic echocardiography. Ultrasound Med Biol. 2008;34(7):1053–1062. doi: 10.1016/j.ultrasmedbio.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Sato A, Terata K, Miura H, et al. Mechanism of vasodilation to adenosine in coronary arterioles from patients with heart disease. Am J Physiol Heart Circ Physiol. 2005;288(4):H1633–H1640. doi: 10.1152/ajpheart.00575.2004. [DOI] [PubMed] [Google Scholar]
- 27.Bonthu S, Heistad DD, Chappell DA, Lamping KG, Faraci FM. Atherosclerosis, vascular remodeling, and impairment of endothelium-dependent relaxation in genetically altered hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 1997;17(11):2333–2340. doi: 10.1161/01.atv.17.11.2333. [DOI] [PubMed] [Google Scholar]
- 28.Crauwels HM, Van Hove CE, Holvoet P, Herman AG, Bult H. Plaque-associated endothelial dysfunction in apolipoprotein E-deficient mice on a regular diet. Effect of human apolipoprotein AI. Cardiovasc Res. 2003;59(1):189–199. doi: 10.1016/s0008-6363(03)00353-5. [DOI] [PubMed] [Google Scholar]
- 29.Abebe W, Hussain T, Olanrewaju H, Mustafa SJ. Role of nitric oxide in adenosine receptor-mediated relaxation of porcine coronary artery. Am J Physiol. 1995;269(5 Pt 2):H1672–H1678. doi: 10.1152/ajpheart.1995.269.5.H1672. [DOI] [PubMed] [Google Scholar]
- 30.Flood AJ, Willems L, Headrick JP. Coronary function and adenosine receptor-mediated responses in ischemic-reperfused mouse heart. Cardiovasc Res. 2002;55(1):161–170. doi: 10.1016/s0008-6363(02)00329-2. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Fenton RA, Wheeler HB, et al. Adenosine A2a receptors increase arterial endothelial cell nitric oxide. J Surg Res. 1998;80(2):357–364. doi: 10.1006/jsre.1998.5439. [DOI] [PubMed] [Google Scholar]
- 32.Teng B, Qin W, Ansari HR, Mustafa SJ. Involvement of p38-mitogen-activated protein kinase in adenosine receptor-mediated relaxation of coronary artery. Am J Physiol Heart Circ Physiol. 2005;288(6):H2574–H2580. doi: 10.1152/ajpheart.00912.2004. [DOI] [PubMed] [Google Scholar]
- 33.Belardinelli L, West GA, Clemo SHF. Regulation of atrioventricular node function by adenosine. In: Gerlach E, Becker B, editors. Topics and Perspectives of Adenosine Research. Berlin: Springer-Verlag; 1987. pp. 344–355. [Google Scholar]
- 34.Levitan I, Fang Y, Rosenhouse-Dantsker A, Romanenko V. Cholesterol and ion channels. Subcell Biochem. 2010;51:509–549. doi: 10.1007/978-90-481-8622-8_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu J, Tong H, Wang L, Hurt CM, Pelleg A. Endogenous adenosine, A1 adenosine receptor, and pertussis toxin sensitive guanine nucleotide binding protein mediate hypoxia induced AV nodal conduction block in guinea pig heart in vivo. Cardiovasc Res. 1993;27(1):134–140. doi: 10.1093/cvr/27.1.134. [DOI] [PubMed] [Google Scholar]
- 36.Hove-Madsen L, Prat-Vidal C, Llach A, et al. Adenosine A2A receptors are expressed in human atrial myocytes and modulate spontaneous sarcoplasmic reticulum calcium release. Cardiovasc Res. 2006;72(2):292–302. doi: 10.1016/j.cardiores.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 37.Lakatta EG, Vinogradova TM, Bogdanov KY. Beta-adrenergic stimulation modulation of heart rate via synchronization of ryanodine receptor Ca2+ release. J Card Surg. 2002;17(5):451–461. [PubMed] [Google Scholar]
- 38.Monahan TS, Sawmiller DR, Fenton RA, Dobson JG., Jr Adenosine A(2a)-receptor activation increases contractility in isolated perfused hearts. Am J Physiol Heart Circ Physiol. 2000;279(4):H1472–H1481. doi: 10.1152/ajpheart.2000.279.4.H1472. [DOI] [PubMed] [Google Scholar]
- 39.Tikh EI, Fenton RA, Dobson JG., Jr Contractile effects of adenosine A1 and A2A receptors in isolated murine hearts. Am J Physiol Heart Circ Physiol. 2006;290(1):H348–H356. doi: 10.1152/ajpheart.00740.2005. [DOI] [PubMed] [Google Scholar]