

The complete genome sequences of Escherichia coli and Helicobacter pylori provide insights into the biology of these species
Recently, we biologists have been treated to a feast of the complete genome sequences of two gut bacteria: and Helicobacter pylori reported by Tomb et al. in Nature (Tomb et al, 1997) and Escherichia coli reported by Blattner et al. in Science. (Blattner et al. 1997). Complete sequences of eight microbes have now been published (Table 1), and there are over 30 additional projects underway and slated for completion in the next 12–18 months. The finished genome sequence of E. coli -- metabolic generalist, workhorse of biochemical genetics, molecular biology and biotechnology, and occasional pathogen -- has special, almost emotional, significance to today’s biologists, many of whom have grown up with its cultures in one form or another. By contrast, H. pylori -- metabolic specialist, gastric pathogen and causative agent of peptic ulcers -- is a relative newcomer to the scientific scene (Fig. 1).
Table 1.
Complete Genomes
| Species | Classification | Size (mb) | Orfs | Ref. | Web Site |
|---|---|---|---|---|---|
| Bacteria | |||||
| Mycoplasma genitalium G-37 | LowGC gram positive | 0.58 | 470 | Fraser et al. (1995) | http://www.tigr.org/tdb/mdb/mgdb/mgdb.html |
| Mycoplasma pneumoniae M129 | LowGC gram positive | 0.82 | 679 | Himmelreich et al. (1996) | http://www.zmbh.uni-heidelberg.de/M_pneumoniae/MP_Home.html |
| Escherichia coli K-12 | Proteobacteria (γ) | 4.60 | 4288 | Blattner et al. (1997) | http://www.genetics.wisc.edu:80/index.html |
| Haemophilus influenzae KW20 | Proteobacteria (γ) | 1.83 | 1743 | Fleischman et al. (1995) | http://www.tigr.org/tdb/mdb/hidb/hidb.html |
| Helicobacter pylori 26695 | Proteobacteria (ε) | 1.67 | 1590 | Tomb et al. (1997) | http://www.tigr.org/tdb/mdb/hpdb/hpdb.html |
| Synechocystis sp. PCC6803 | Cyanobacteria | 3.57 | 3168 | Kaneko et al. (1996) | http://www.kazusa.or.jp/cyano/cyano.html |
| Archaea | |||||
| Methanococcus jannascii | Euryarchaeota | 1.66 | 1738 | Bult et al. (1996) | http://www.tigr.org/tdb/mdb/mjdb/mjdb.html |
Eukaryote Saccharomyces cerevisiaeFungi13.05885Goffeau et al. (1997) http://genome-www.stanford.edu/Saccharomyces/
Figure 1.
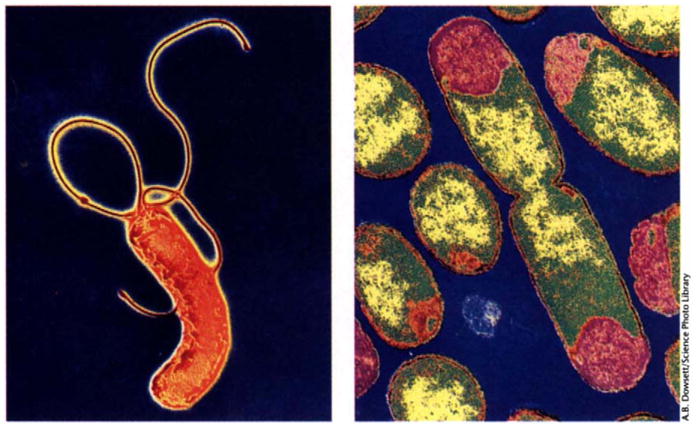
Transmission electron micrographs of H. pylori (left) and E. coli (right).
There are numerous reasons for going to the trouble of determining complete and accurate genome sequences of micro-organisms. In those microbes with pathogenic properties, the total set of instructions provides a potentially powerful basis for developing vaccines and other therapeutic agents. Genomic sequences offer insights into the range of functions an organism possesses, the relative importance natural selection attaches to each function, and the organism’s evolutionary history. In addition, the availability of complete genome sequences has spawned a enormous array of creative approaches for global functional analysis of genes and gene networks. There is particular virtue in having contiguous sequence of an entire genome; not only is it possible to predict all or almost all of the proteins that are present in the organism, but what is absent also becomes meaningful.
The genome sequence of an organism is like the Rosetta stone: it is impressive to see but it must be translated to have value. The most important initial steps in translating a genome are identifying all of the genes and assigning functions to them. Genes can be identified by genetic and biochemical experiments or predicted by computational analysis of the genome sequence. Functions of genes can be assigned also by experimental and computational methods, but accurate prediction of function based solely on sequence information is not so straightforward. In the case of E. coli, computational prediction of gene function is less important because of the vast wealth of genetic and biochemical data collected from this organism over the last fifty years (Riley, 1993). However, for H. pylori and for most of the species for which complete genome sequences are published, far less experimentally derived functional information is available. Thus, analysis of these genomes, and most of the ones that will be sequenced in the future, depends heavily on computational methods.
Tomb, et al. use the BLAZE program (Brutlag et al., 1993) to assign function to each predicted H. pylori gene based on the function of the previously characterized gene in the sequence database that is most similar in sequence to the predicted gene, but only if the likelihood of the match is much higher than that expected by chance. Blattner’s group go one step further. They identify multiple similar sequences in existing databases, and if most of these genes appear to have the same physiological role, this function is assigned to the new gene. If the top scoring sequences have different physiological roles, attempts are made to identify a common denominator, such as transport activity, and this general activity, with unknown specificity, is then assigned to the new gene. Although both approaches are likely to result in correct functional assignments for most genes, there are many cases where either approach will lead to incorrect predictions.
One example where caution seems warranted is in the prediction that H. pylori is capable of mismatch repair, based on the assignment of methyl transferase, MutS, and UvrD functions to several of its genes. (Tomb, et al., 1997). However, it is unlikely that this DNA repair process is present in H. pylori because its genome sequence does not contain a homolog of MutL, a protein required for mismatch repair in all organisms studied from bacteria to humans (Modrich and Lahue, 1996). Furthermore, phylogenetic analysis suggests that there has been an ancient duplication in the mutS gene family, and that the “mutS” gene (HP0621) in H. pylori is not an orthologue (a gene whose origininating from a speciation event), but is rather a paralogue (a gene originating from a gene duplication event) of the E. coli mutS gene (Fig. 2). Genes that are orthologs of the E. coli mutS gene, (Fig. 2, blue), are absolutely required for mismatch repair in many bacterial species. By contrast, the mutS paralogs, (Fig. 2, red), have no known function. Why was the HP0621 gene called mutS, and not identified as a mutS paralog? Analysis of the database search used by Tomb, et al. (see their web site, Table 1) indicates that the gene was given this designation because its highest sequence similarity hit was with the gene sll1772 from Synechocystis sp. (strain PCC6803), a cyanobacterium. The researchers annotating the Synechocystis sp. genome sequence earlier gave the name mutS to gene sll1772, again because it scored highly similar to mutS in a similar type of analysis. However, gene sll1772 is only one of two mutS-like genes in Synechocystis sp.; a second gene (gene sll1165) predicted from its genome sequence is much more similar to mutS from E. coli, and is the likely mutS orthologue in Synechocystis sp. H. pylori, for unknown reasons, does not encode an orthologue of the mutS genes known to be involved in mismatch repair. As this example shows, database errors are often self-propagating.
Figure 2.

Reconstruction of the evolution of MutS-like proteins in bacteria using molecular phylogenetics. MutS-like protein sequences were aligned and a tree of these sequences was generated using molecular phylogenetic methods (details are available from the authors on request). left, The tree of MutS-like proteins (thin lines) is shown embedded within the species tree (thick grey lines). The gene duplication event (marked by an asterisk) occurred prior to the divergence of these bacterial species and led to the presence of two paralogous MutS-like subgroups (distinguished by different colors and gene subscripts a or b). Gene loss in some lineages is indicated when the MutS tree stops within the species tree. right, The MutS tree is extracted from the species tree and untwisted to better show the relaitonships among the different MutS forms. Only one lineage (labeled in blue) includes genes with established roles in mismatch repair. The genes in the second lineage (in red) have no known function. Because the H. pylori gene is a member of this second lineage, it should not be assigned the MutS function.
This difficulty in assigning function on the basis of sequence data is likely to be widespread, particularly because so many microbial genome sequences are forthcoming. Some simple precautions may help to alleviate the problem. Perhaps the most obvious rule is to avoid assuming that a function assigned to a sequence is correct just because it already appears in a database. The method used by Blattner et al. of examining many high scoring sequences at once may reduce the likelihood of being misled by a single database misannotation, because it assigns a function only if many of the top scoring genes have the same function. A second simple precaution is to recognize that sequence similarity indicates only the potential for a biochemical activity. Close similarity does not readily identify the physiological role for a protein and is not definitive evidence that two proteins have the same biochemical activity. Likewise, the absence of a homologous gene in a whole genome sequence does not necessarily mean that the activity is absent in the organism.
The MutS story above and many other examples provide evidence that classifying members of multigene families is one of the most difficult parts of assigning function. Molecular phylogenetics is probably a better method for dividing multigene families into groups of orthologous genes than simply relying on database searches. As orthologues frequently have functions distinct from paralogues, a “phylogenomic” methodology is likely to improve the accuracy of function assignment to members of multigene families identified in complete genome sequences. In addition, assignment of function on the basis of DNA sequence data will likely become more accurate as we learn how to integrate knowledge about biochemical pathways and regulatory networks into the computational methods.
In addition to stimulating predictions of the functions of individual genes, the complete genome sequences of H. pylori and E. coli provide clues about their global metabolic capabilities. One striking difference between these two organisms is that H. pylori has many fewer genes than E. coli. Three of the other bacteria whose complete sequences are published also have reduced genome sizes. How can this phenomenon be explained? One argument is that organisms with broad ecological niches need more genes (Hinegardner, 1976). For example, E. coli, with a genome of 4.6 million base pairs, can be thought of as a metabolic generalist because it is capable of growing under a variety of conditions. It is equipped to grow in the lower gut of animals where it meets a variety of sugars that have not been absorbed by its host’s digestive tract. Absorption being an efficient process, the residual sugars and amino acids are dilute. The lower gut is also anaerobic; E. coli is a facultative anaerobe, capable of fermentative metabolism. E. coli survives when it is released to the environment where it can be disseminated to new hosts. It grows faster in air than in the gut, metabolizing carbon to CO2. Its metabolic generalism shows in its genome; there are many different transport proteins to accumulate dilute substrates from the gut contents. There are 700 known gene products for central intermediary metabolism, degradation of small molecules, and energy metabolism. Helping E. coli adjust to a variety of growth conditions are the 400 regulatory genes (some known on the basis of experiments and some attributed for reasons of sequence similarity), or 4.5% of the total genome. By contrast, H. pylori, with a genome of only 1.66 million base pairs, is an ecological specialist, apparently living nowhere but in the mucosa of the stomach. To survive in this highly acidic environment, H. pylori encodes genes that allow it to develop a positive inside membrane potential and has double the number of basic amino acids in most of its proteins compared to other microbes. Consistent with this restricted ecological niche, the genome sequence of H. pylori indicates that it is much more limited in its metabolic capabilities and its regulatory networks (Tomb, et al., 1997). The genome sequence also provides clues as to how H. pylori survives in the highly acidic environment of the stomach. The proteins encoded by the H. pylori genome have twice the number of basic amino acids compared to proteins of other microbes; this may help in establishing a positive inside membrane potential. These comparisons provide but one of the many valuable insights that can be learned from sequences of complete genomes.
We have every reason to be delighted by the feast that has just been served to us. These complete genomic sequences have a major impact on the study of these two gut bacteria, and will likely speed up our understanding of the mechanisms by which they cause disease. Because these and the other available bacterial sequences are from widely divergent microbes, we are already getting an idea of which genes are universal and perhaps form the core of a micro-organism (Mushegian and Koonin, 1996). By contrast, as complete genome sequences from closely related pairs of microbes become available, we will learn more about mutation and recombination processes, as well as features such as codon usage, genome structure, and horizontal gene transfer, that change on a shorter evolutionary time scale (for example, Lawrence and Ochman, 1997). Today’s feast will likely seem meager in comparison to the lavish smorgasbord expected in the future.
References
- Blattner FR, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Brutlag DL, Dautricourt JP, Diaz R, Fier J, Moxon B, Stamm R. BLAZE: An implementation of the Smith-Waterman comparison algorithm on a massively parallel computer. Computers and Chemistry. 1993;17:203–207. [Google Scholar]
- Bult CJ, et al. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- Fleischmann RD, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Fraser CM, et al. The minimal gene complement of Mycoplasma genitalium. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- Goffeau A, et al. The yeast genome directory. Nature. 1997;387(Suppl):5–105. [PubMed] [Google Scholar]
- Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li BC, Herrmann R. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996;24:4420–4449. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinegardner R. Evolution of genome size. In: Ayala FJ, editor. Molecular Evolution. Sinauer; Sunderland, MA: 1976. pp. 179–199. [Google Scholar]
- Kaneko T, et al. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803 II Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 1996;3:109–136. doi: 10.1093/dnares/3.3.109. [DOI] [PubMed] [Google Scholar]
- Lawrence JG, Ochman H. Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol. 1997;44:383–397. doi: 10.1007/pl00006158. [DOI] [PubMed] [Google Scholar]
- Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Ann Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- Mushegian AR, Koonin EV. A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc Natl Acad Sci U S A. 1996;93:10268–10273. doi: 10.1073/pnas.93.19.10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley M. Functions of the gene products of Escherichia coli. Micro Rev. 1993;57:862–952. doi: 10.1128/mr.57.4.862-952.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomb JF, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]