

Abstract
A series of synthetic aporphine derivatives structurally related to domesticine and nantenine (ring A, N6 and ring C truncated analogs), was evaluated in MTS cytotoxicity assays against the human colon cancer cell lines, HCT-116 and Caco-2. In general, the C1 position of ring A is tolerant of alkoxy substituents as well as a benzoyl ester functionality. Other modifications evaluated resulted in a decrease in cytotoxic activity. The most potent compounds identified had IC50 values in the range 23μM-38μM, comparable to the known cytotoxic agent, etoposide.
Keywords: aporphine, nantenine, cytotoxic, HCT-116, Caco-2
Cancer is the second leading cause of death in developed countries and colon cancer is the third most common cancer in the world, with prevalence mainly in Western countries. Despite the fact that many new chemotherapeutic drugs have been developed and many aggressive treatments are available, cancer death rates continue to rise. 1 Many chemotherapeutic agents act through cytotoxicity which leads to inhibition of carcinogenesis. Development of resistance to approved chemotherapy medications as well as a myriad of side-effects of these compounds in cancer patients, has contributed to the continued challenge in treating various forms of cancer. Thus, there is a constant search for new cytotoxic agents that can serve as leads for the development of chemotherapeutics. Furthermore, the discovery of new cytotoxic agents may afford the opportunity to obtain a more detailed understanding of the mechanistic underpinnings of this deadly scourge.
Aporphine alkaloids are endowed with a range of biological activities and may well be considered to be privileged drug discovery scaffolds. For example, naturally occurring and synthetic aporphines have been investigated as acetylcholinesterase inhibitors, 2-4 CNS receptor ligands, 5-8 and as antimicrobial 9, antimalarial 10, 11 and antiviral 12, 13 agents. In addition, there are a number of reports on the cytotoxic activity of some members of this alkaloid class. 14-16 Prior studies on aporphines as cytotoxic agents have focused exclusively on naturally-occurring aporphines. The biological targets involved in the cytotoxic activity of aporphines are yet to be fully elucidated although DNA-mediated and topoisomerase-related mechanisms appear to play a role in some instances. 17-20 Interpretation of currently available information with regards to the SAR of these molecules as cytotoxic agents is unreliable because of the diversity of assay systems and cell lines employed by various investigators. To date, no systematic study of the structure-activity relationships (SARs) of aporphines as cytotoxic agents has been conducted. An understanding of structure-activity effects is a necessary aspect of any future undertaking to further understand the mechanism of action of these molecules as well as to capitalize on their therapeutic potential.
Herein, we present results from an SAR evaluation of the cytotoxic activity of a set of synthetic aporphine derivatives (some of which we have acquired in the process of other investigations) in the human colon cancer cell lines HCT-116 and Caco-2.
Based on the structural similarity of domesticine (1) and nantenine (2) (Table 1) to other known cytotoxic aporphines, we initially screened these compounds in HCT-116 and Caco-2 colon cancer cell lines. In these assays, 1 had moderate activity, (approximately 3 and 4-fold less potent respectively than the benchmark etoposide). However, we were enthused to find that 2 had potency comparable to that of etoposide in both cell lines. This suggested that a C1 phenolic group on the aporphine scaffold was detrimental to activity and we therefore decided to investigate the effect of replacement of this phenolic group with other moieties. Thus compounds 3-5 were targeted for cytotoxicity studies to aid in understanding the SAR at this position. In addition, to probe the effects of other structural changes in ring A and N6 of the aporphine nucleus, compounds 6-12 were evaluated. An assessment of the requirement for an intact aporphine skeleton was obtained via evaluation of the seco-ring C analogs 13 and 14.
Table 1.
Ring A, N6 and seco-ring C analogs evaluated
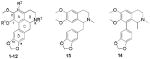 | |||
|---|---|---|---|
| Compound # | R1 | R2 | R3 |
| 1 | H | H | CH3 |
| 2 | CH3 | H | CH3 |
| 3 | (CH3CH2)2CHCH2 | H | CH3 |
| 4 | CH3(CH2)3CH2 | H | CH3 |
| 5 | PhCO | H | CH3 |
| 6 | CH3 | Br | CH3 |
| 7 | CH3 | H | CH3CO |
| 8 | CH3 | H | CH3CH2OCO |
| 9 | CH3 | H | CH3SO2 |
| 10 | H | H | CH3CH2OCO |
| 11 | CH3CH2 | H | CH3CH2OCO |
| 12 | PhCH2O | H | CH3CH2OCO |
Compounds 1, 2, 4 and 7-13 were prepared as described by us previously. 2, 7, 8, 21, 22. Compound 3 was prepared in three steps by alkylation of readily available phenol 108 followed by treatment of the carbamate product with lithium aluminium hydride (Scheme 1) and alkylation of the secondary amine product thus formed (due to cleavage of the N-carbamate group). Compound 5 (Scheme 1) was synthesized via lithium aluminium hydride reduction of 10. This reduction gave predominantly the secondary amine; the crude mixture was subsequently subjected to reductive amination to yield domesticine (1). Acylation of 1 with benzoic anhydride afforded 5.
Scheme 1.
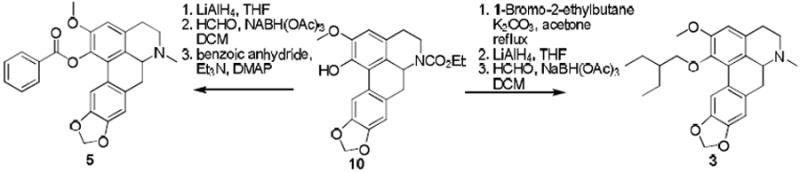
Synthesis of compounds 3 and 5
Phenol 15, was prepared in a manner analogous to the synthesis of 10, 8 and was brominated with NBS to give 16 (Scheme 2). In a sequential manner, compound 16 was O-methylated, the boc group removed by treatment with ZnBr2, and the amine alkylated via reductive amination to give the bromo-nantenine analog, compound 6. (We also attempted to synthesize 6 via direct NBS bromination of 2 but found that this method gave poly-halogenation of the aromatic rings).
Scheme 2.
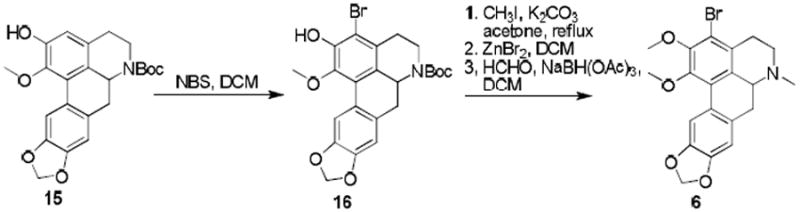
Synthesis of compound 6
Compounds 1-14 were evaluated in MTS assays as described by us 23 with modifications [24]. Table 2 summarizes the data from this evaluation. Replacement of the C1 hydroxyl group of domesticine (1) with alkyloxy groups (ie compounds 2-4) resulted in improved cytotoxicity vs the cell lines tested. The branched alkyl analog 3 was the most potent against the cell lines evaluated and was approximately 2-fold selective for colon cancer cells vs normal colon fibroblasts (CCD-18Co).
Table 2.
Cytotoxicity of compounds 1-14 against human colon cancer (HCT-116, Caco-2) and normal colon (CCD-18Co) cell lines
| Compound # | IC50 (μM)a | ||
|---|---|---|---|
| HCT-116 | Caco-2 | CCD-18Co | |
| 1 | 73.3±4.6 | 73.5 ±3.2 | 133.5±3.1 |
| 2 | 38.3±1.6 | 36.2±2.4 | 56.5±1.9 |
| 3 | 26.6±1.1 | 22.8±3.5 | 50.7±2.0 |
| 4 | 46.3±2.6 | 35.3±2.4 | 73.4±2.1 |
| 5 | 25.2±2.2 | 25.9±1.8 | 45.1±0.8 |
| 6 | 100.3±2.1 | 100.7±1.7 | 180.6±3.5 |
| 7 | 185.7±3.4 | 159.6±6.4 | 188.5±2.2 |
| 8 | 81.4±1.2 | 83.2±3.8 | 111.6±1.6 |
| 9 | 196.1±3.4 | 195.8±4.3 | n.d. |
| 10 | 72.7±2.4 | 71.7±2.8 | 127.9±5.4 |
| 11 | 73.3±4.6 | 73.5±3.2 | 133.5±3.1 |
| 12 | 93.3±4.7 | 99.8±2.3 | 109.6±0.6 |
| 13 | 190.9±4.0 | 191.3±2.5 | n.d. |
| 14 | 195.4±5.7 | 189.8±5.7 | n.d. |
| etoposide | 25.5±2.1 | 18.6±1.8 | 44.4±1.8 |
determined at 72 h
n.d.= not determined
The n-pentyl analog 4 was slightly less potent than nantenine (2) in the HCT-116 cell line but was equipotent in the Caco-2 cell line.
Together, these results suggest that C1 alkyl branching results in higher cytotoxicity but also indicates that increasing n-alkyl chain length is detrimental to activity vs HCT-116 cells. The benzoyl ester 5 had slightly improved activity as compared to 2 in HCT-116 and Caco-2 cells and was moderately more selective with regards to toxicity in non-tumorigenic human colon CCD-18Co cells. Addition of a bromine atom to position 3 of nantenine (ie compound 6) decreased cytotoxic activity in all cell lines. Replacement of the N-methyl group of nantenine (2) with acetyl, ethyl carbamate or methanesulfonyl groups was detrimental to cytotoxic activity (compounds 7-9 respectively). However, comparison of domesticine (1) to its N-ethyl carbamate analog 10 revealed no significant difference in cytotoxicity. Although C1 alkylation gave improved activity in the N-methyl series, this structural change was not associated with improved activity in the N-ethyl carbamate series of analogs, as comparison of 10 with 11 and 12 reveals. Cumulatively, the evaluation of 1-12 is indicative of a greater preference for the N-methyl group of the aporphine skeleton as compared to other N-protected functionalities in relation to cytotoxic activity. Additionally, the presence of an N-ethyl carbamate functionality appears to be better tolerated with a C1 hydroxy group than with a C1 methoxy group on the aporphine skeleton. The ring truncated analogs 13 and 14 had significantly lower activity as compared to 2. This points to a requirement for ring C of the aporphine nucleus to be intact for cytotoxic activity.
In conclusion, our study has uncovered potent cytotoxic activity of the aporphine alkaloid nantenine (2), the C1 alkoxy derivative (3) and the C1 benzoate derivative (5). Of these, 3 and 5 exhibit marginally higher selective cytotoxicity vs normal cells. These compounds have potencies comparable to that of the clinically available anticancer therapeutic drug, etoposide in vitro. This is the first account of the cytotoxic activity of aporphines in HCT-116 and Caco-2 human colon cancer cell lines. Our SAR evaluation indicates that the N-methyl group and the intact aporphine nucleus of 2 are important for its cytotoxic activity. Interestingly, this study also indicates that 2 may be manipulated via substitution of the C1 methyl group with other alkyl groups without severely compromising cytotoxicity. The findings herein are relevant to the study of aporphines as potential anticancer drugs and are also significant in other therapeutic areas (where cytotoxicity of the molecules described may not be desirable).
The importance of substituents on other portions of the nantenine aporphine core (eg ring D) remains to be determined. Furthermore, the selectivity vs other cancer cell lines and mechanism(s) involved in the cytotoxic actions of 2, 3 and 5 are unknown at present. These are dimensions that we intend to explore in future.
Acknowledgments
This publication was made possible by Grant Number RR03037 from the National Center for Research Resources (NCRR) of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH.
References and Notes
- 1.Jemal A, Siegel R, Xu J, Ward E. CA Cancer J Clin. 2010;60:277. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Pecic S, McAnuff MA, Harding WW. J Enzyme Inhib Med Chem. 2011;26:46. doi: 10.3109/14756361003671078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang H, Wei YB, Zhang C, Ning FX, Qiao W, Huang SL, Ma L, Huang ZS, Gu LQ. Eur J Med Chem. 2009;44:2523. doi: 10.1016/j.ejmech.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 4.Adsersen A, Kjolbye A, Dall O, Jager AK. J Ethnopharmacol. 2007;113:179. doi: 10.1016/j.jep.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Cabedo N, Berenguer I, Figadere B, Cortes D. Curr Med Chem. 2009;16:2441. doi: 10.2174/092986709788682100. [DOI] [PubMed] [Google Scholar]
- 6.Zhang A, Zhang Y, Branfman AR, Baldessarini RJ, Neumeyer JL. J Med Chem. 2007;50:171. doi: 10.1021/jm060959i. [DOI] [PubMed] [Google Scholar]
- 7.Pecic S, Makkar P, Chaudhary S, Reddy BV, Navarro HA, Harding WW. Bioorg Med Chem. 2010;18:5562. doi: 10.1016/j.bmc.2010.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaudhary S, Pecic S, Legendre O, Navarro HA, Harding WW. Bioorg Med Chem Lett. 2009;19:2530. doi: 10.1016/j.bmcl.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen JH, Du ZZ, Shen YM, Yang YP. Arch Pharm Res. 2009;32:3. doi: 10.1007/s12272-009-1111-7. [DOI] [PubMed] [Google Scholar]
- 10.Ayers S, Zink DL, Mohn K, Powell JS, Brown CM, Murphy T, Brand R, Pretorius S, Stevenson D, Thompson D, Singh SB. Planta Med. 2007;73:296. doi: 10.1055/s-2007-967124. [DOI] [PubMed] [Google Scholar]
- 11.Waechter AI, Cave A, Hocquemiller R, Bories C, Munoz V, Fournet A. Phytother Res. 1999;13:175. doi: 10.1002/(SICI)1099-1573(199903)13:2<175::AID-PTR395>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 12.Boustie J, Stigliani JL, Montanha J, Amoros M, Payard M, Girre L. J Nat Prod. 1998;61:480. doi: 10.1021/np970382v. [DOI] [PubMed] [Google Scholar]
- 13.Montanha JA, Amoros M, Boustie J, Girre L. Planta Med. 1995;61:419. doi: 10.1055/s-2006-958128. [DOI] [PubMed] [Google Scholar]
- 14.Makarasen A, Sirithana W, Mogkhuntod S, Khunnawutmanotham N, Chimnoi N, Techasakul S. Planta Med. 2011 doi: 10.1055/s-0030-1270743. [DOI] [PubMed] [Google Scholar]
- 15.Mohamed SM, Hassan EM, Ibrahim NA. Nat Prod Res. 2010;24:1395. doi: 10.1080/14786410902906959. [DOI] [PubMed] [Google Scholar]
- 16.Stevigny C, Block S, De Pauw-Gillet MC, de Hoffmann E, Llabres G, Adjakidje V, Quetin-Leclercq J. Planta Med. 2002;68:1042. doi: 10.1055/s-2002-35651. [DOI] [PubMed] [Google Scholar]
- 17.Hoet S, Stevigny C, Block S, Opperdoes F, Colson P, Baldeyrou B, Lansiaux A, Bailly C, Quetin-Leclercq J. Planta Med. 2004;70:407. doi: 10.1055/s-2004-818967. [DOI] [PubMed] [Google Scholar]
- 18.Goren AC, Zhou BN, Kingston DG. Planta Med. 2003;69:867. doi: 10.1055/s-2003-43224. [DOI] [PubMed] [Google Scholar]
- 19.Woo SH, Reynolds MC, Sun NJ, Cassady JM, Snapka RM. Biochem Pharmacol. 1997;54:467. doi: 10.1016/s0006-2952(97)00198-6. [DOI] [PubMed] [Google Scholar]
- 20.Stevigny C, Bailly C, Quetin-Leclercq J. Curr Med Chem Anticancer Agents. 2005;5:173. doi: 10.2174/1568011053174864. [DOI] [PubMed] [Google Scholar]
- 21.Chaudhary S, Pecic S, Legendre O, Harding WW. Tetrahedron Lett. 2009;50:2437. doi: 10.1016/j.tetlet.2009.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Legendre O, Pecic S, Chaudhary S, Zimmerman SM, Fantegrossi WE, Harding WW. Bioorg Med Chem Lett. 2010;20:628. doi: 10.1016/j.bmcl.2009.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, Henry GE, Seeram NP. J Agric Food Chem. 2009;57:7282. doi: 10.1021/jf901716j. [DOI] [PubMed] [Google Scholar]
- 24.Cell lines and culture conditions:Human colon cancer cell lines Caco-2 (adenocarcinoma) and HCT-116 (carcinoma) were obtained from American Type Culture Collection (Rockville, USA). Caco-2 cells were grown in EMEM medium supplemented with 10% v/v fetal bovine serum, 1% v/v nonessential amino acids, 1% v/v L-glutamine and 1% v/v antibiotic solution (Sigma). HCT-116 cells were grown in McCoy’s 5a medium supplemented with 10% v/v fetal bovine serum, 1% v/v nonessential amino acids, 2% v/v HEPES and 1% v/v antibiotic solution. Cells were maintained at 37 °C in an incubator under a 5% CO2/95% air atmosphere at constant humidity. Cells were counted using a hemacytometer and were plated at 5000-3,000 cells per well, in a 96-well format for 24 or 48 h prior to MS extract or pure compounds addition depending on the cell line. All of the test samples were solubilized in DMSO (<0.5 % in the culture medium) and were filter sterilised (0.2 μm) prior to addition to the culture media. Control cells were also run in parallel and subjected to the same changes in medium with a 0.5 % DMSO. In addition, cells were treated as indicated above for 24, 48 or 72 h to unravel the potential cytotoxicity of compounds against all cell lines.Cell cytotoxicty (MTS assay):At the end of 2 or 3 days of treatment with serially diluted test samples, 20 μL of the MTS reagent, in combination with the electron coupling agent, phenazine methosulfate, was added to the wells and cells were incubated at 37°C in a humidified incubator for 3 h. Absorbance at 490 nm (OD490) was monitored with a spectrophotometer (SpectraMax M2, Molecular Devices Corp., operated by SoftmaxPro v.4.6 software, Sunnyvale, CA, USA), to obtain the number of cells relative to control populations. 20 μL of etoposide 4 mg/mL (Sigma) was assayed as a negative control of proliferation. The results are expressed as the concentration that inhibit growth of cell by 50% vs. control cells (control medium used as negative control), IC50. Data are presented as the mean ± S.D. of three separated experiments on each cell line. Etoposide provided consistent IC50 values of 15-25 μM for the HCT-116 and Caco-2 cells.