

Abstract
Neurons in the cerebellar nuclei fire at accelerated rates for prolonged periods after trains of synaptic inhibition that interrupt spontaneous firing. Both in vitro and in vivo, however, this prolonged rebound firing is favored by strong stimulation of afferents, suggesting that neurotransmitters other than GABA may contribute to the increased firing rates. Here, we tested whether metabotropic glutamate receptors modulate excitability of nuclear cells in cerebellar slices from mouse. In current clamp, the prolonged rebound firing rate after high-frequency synaptic stimulation was reduced by a variety of group I mGluR antagonists, including CPCCOEt [7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester], JNJ16259685 (3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-(cis-4-methoxycyclohexyl)-methanone) plus MPEP, or 3-MATIDA (α-amino-5-carboxy-3-methyl-2-thiopheneacetic acid) plus MPEP, as long as both mGluR1 and mGluR5 were blocked. This mGluR-dependent acceleration of firing was reduced but still evident when IPSPs were prevented by GABAA receptor antagonists. In voltage clamp, voltage ramps revealed a non-inactivating, low-voltage-activated, nimodipine-sensitive current that was enhanced by the selective group I mGluR agonist s-DHPG [(S)-3,5-dihydroxyphenylglycine]. This putative L-type current also increased when mGluRs were activated by trains of evoked synaptic currents instead of direct application of agonist. In current clamp, blocking L-type Ca channels with the specific blocker nifedipine greatly reduced prolonged poststimulus firing and occluded the effect of adding group I mGluR antagonists. Thus, potentiation of a low-voltage-activated L-type current by synaptically released glutamate accounted nearly fully for the mGluR-dependent acceleration of firing. Together, these data suggest that prolonged rebound firing in the cerebellar nuclei in vivo is most likely to occur when GABAA and mGluRs are simultaneously activated by concurrent excitation and inhibition.
Introduction
Neurons in the cerebellar nuclei form the final stage of cerebellar processing. These spontaneously firing cells integrate synaptic inhibition from Purkinje cells of the cerebellar cortex with synaptic excitation from mossy fibers and inferior olivary fibers to generate the sole output of the cerebellum. After high-frequency trains of IPSPs evoked with fast excitatory transmission blocked, cerebellar nuclear cells in slices often display prolonged rebound firing, i.e., an elevation of firing rates above the prestimulus rate that persists for a few hundred milliseconds after stimulus offset (Aizenman and Linden, 1999; Molineux et al., 2006; Alviña et al., 2008; Zheng and Raman, 2009; Tadayonnejad et al., 2010). Such postinhibitory rebound increases in firing rate are thought to be one means by which Purkinje cells can regulate the output firing of the cerebellum. Despite the repeated documentation of this phenomenon, the ionic mechanisms and physiological conditions favoring prolonged rebound firing remain uncertain.
Prolonged rebound firing can be elicited reliably in vivo with strong stimulation to the inferior olive (Hoebeek et al., 2010). Surprisingly, however, most cerebellar nuclear cells show neither burst nor prolonged rebound firing after stimulation of exclusively Purkinje afferents in vivo (Alviña et al., 2008). Even in slice preparations, the likelihood of eliciting rebound firing decreases at lower stimulus strengths (Tadayonnejad et al., 2010). These findings suggest that specific types of stimuli may be necessary to facilitate rebound firing, possibly by engaging synaptic receptors other than GABAA receptors. Because high-frequency stimulation of glutamatergic afferents can activate group I mGluRs in cerebellar nuclear cells (Zhang and Linden, 2006), we tested whether these receptors can influence prolonged rebound firing in the cerebellar nuclei.
The results demonstrated that the firing rate after high-frequency synaptic stimulation was indeed reduced by blockade by group I mGluR antagonists. Conversely, an mGluR-dependent acceleration of firing was evident whether or not IPSPs were simultaneously evoked. In voltage clamp, hyperpolarizing ramps revealed a non-inactivating, voltage-gated, nimodipine-sensitive, likely L-type current, which was enhanced when group I mGluRs were activated by either agonist application or synaptically released glutamate. In current clamp, blocking L-type Ca channels with nifedipine reduced the mGluR-mediated increase in poststimulus firing. Thus, prolonged rebound firing in cerebellar nuclear cells depends primarily on the potentiation of L-type Ca current by activated group I mGluRs. Moreover, this acceleration of firing does not require inhibition. These data reveal an unanticipated relationship between group I mGluRs and L-type channels in the cerebellar nuclei. Additionally, the data offer an explanation for why inferior olivary activation but not simple stimulation of Purkinje cells more effectively induces prolonged rebound firing in vivo.
Materials and Methods
Preparation of cerebellar slices.
All animal studies were performed in accordance with institutional guidelines and were approved by the Institutional Animal Care and Use Committee of Northwestern University. Slices (300 μm) were made as in the study by Pugh and Raman (2006) from P13 to P15 C57BL/6 mouse pups or, as noted, from P22 to P28 weanling mice of either sex (Charles River). Mice were anesthetized with isoflurane and transcardially perfused with artificial CSF (ACSF) (4°C, pups; 35°C, weanling) containing the following (in mm): 123.5 NaCl, 3.5 KCl, 1.5 CaCl2, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, and 10 glucose. Mice were decapitated, and the cerebella were transferred into ice-cold (pups) or warmed (weanling) oxygenated ACSF. Parasagittal cerebellar slices were cut on a vibratome (VT 100S; Leica), incubated for 1 h at 35°C in oxygenated (95% O2/5% CO2) ACSF, and then maintained at room temperature.
Electrophysiological recording.
To minimize variance, recordings were made from large cells in the interpositus and the medial portion of the lateral nucleus, which most likely include the large glutamatergic projection neurons (Uusisaari et al., 2007). Most of these cells had somatic diameters of 15–20 μm. The selection criteria were identical to those in the study by Zheng and Raman (2009). Although bursting was not assessed in the present work, the majority of cells in that study produced rebound bursts in response to strong hyperpolarizing current injection to potentials less than −90 mV, but only prolonged rebound firing in response to IPSP trains. Patch pipettes (3–5 MΩ) were pulled from borosilicate glass on a Sutter Instruments P97 puller. Voltage-clamp recordings were made with an Axopatch 200B amplifier and current-clamp recordings with an Axoclamp 2B amplifier, and data were acquired with pClamp acquisition software (Molecular Devices). Pipettes were filled with an intracellular solution composed of the following (in mm): 120 K-gluconate, 4 Na-gluconate, 4 NaCl, 2 MgCl2, 14 Tris-creatine phosphate, 4 MgATP, 0.3 Tris-GTP, 10 HEPES, and 20 sucrose, pH 7.4 with KOH. In a subset of experiments (those in which 3-MATIDA (α-amino-5-carboxy-3-methyl-2-thiopheneacetic acid) was applied), 650 μm Ca was included in the intracellular solutions to give ∼100 nm free Ca. The range of firing rates and increase in rates (Δrate) measured in control solutions before application of antagonist overlapped with and without added free Ca, and the datasets were not distinguished further. For voltage-clamp recordings, 0.6 mm QX-314 was added to the intracellular solution to block Na current. As reported previously, Ih is also blocked by QX-314 (Aizenman and Linden, 1999; Zheng and Raman, 2009). During recordings, slices were superfused with warm (33–35°C) oxygenated ACSF solution. 6,7-Dinitroquinoxaline-2,3-dione (DNQX) at 5 μm, 10 μm CPP [(RS)-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid], and 10 μm SR95531 [6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid hydrobromide] were added to the bath as indicated to block AMPA, NMDA, and GABAA receptors. Blocking GABAA receptors had little effect on baseline firing rates, because most Purkinje axons are cut during slicing, yielding a low rate of spontaneous IPSPs (Telgkamp and Raman, 2002). In current clamp, nuclear cells were spontaneously active and fired at a range of rates. To minimize variance, hyperpolarizing current was applied to maintain the mean spontaneous rate near 20 spikes/s. Synaptic transmission was evoked by 45–100 μs voltage pulses delivered through a concentric bipolar electrode (FHC) to the white matter surrounding the cerebellar nuclei. For pharmacological studies, slices were perfused with ACSF containing 20 μm (S)-3,5-dihydroxyphenylglycine (s-DHPG), 10 μm nimodipine or nifedipine, 125 μm CPCCOEt [7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester], 40 μm MPEP, 0.2 μm JNJ16259685 (3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-(cis-4-methoxycyclohexyl)-methanone), and/or 150 μm 3-MATIDA. To ensure complete equilibration with the drug, recordings were made 10–15 min after perfusion was initiated. For perforated-patch recordings (Zheng and Raman, 2009), the pipette solution contained the following (in mm): 120 K-gluconate, 8 Na gluconate, 2 MgCl2, 10 HEPES, and 20 sucrose, pH 7.4 with KOH. Gramicidin in DMSO (20 mg/ml) was added to the pipette solution just before recording to give a final concentration of 50 μg/ml.
Data analysis and chemicals.
Data were analyzed with IGOR-Pro (Wavemetrics) and are presented as mean ± SEM. Statistical significance was assessed with two-tailed t tests, paired or unpaired as noted, with a α level of 0.05. A 6 mV junction potential was subtracted from all data, measured by the technique of Neher (1992). Stimulus artifacts have been digitally reduced in all figures. DNQX, CPP, SR95531, s-DHPG, CPCCOEt, MPEP, JNJ16259685, and 3-MATIDA were from Tocris Cookson, and TTX was from Alomone Labs. All other drugs were from Sigma-Aldrich.
Results
To explore whether mGluR activation modulates action potentials of cerebellar nuclear cells, we first tested the effect of the competitive antagonist CPCCOEt, on the acceleration of postinhibitory prolonged rebound firing. In the presence of 5 μm DNQX and 10 μm CPP to block AMPA and NMDA receptors, we evoked synaptic transmission with 500 ms, 100 Hz stimulus trains in slices from 2-week-old mouse pups (P13–P15) and compared firing rates of current-clamped cerebellar nuclear neurons before and after stimulation. During the stimulus train, spontaneous firing was interrupted. As reported previously (Zheng and Raman, 2009), the firing rate averaged over 300 ms after the train increased by 23.6 ± 2.6 Hz relative to the prestimulus rate (n = 9) (Fig. 1A,B). This increase, or Δrate, can be taken as a measure of the strength of prolonged rebound firing. A similar increase was seen when recordings were repeated in older animals, P22–P28 (25.9 ± 4.6 Hz). When CPCCOEt (125 μm) was added to the bath, the postinhibitory firing rate in the same cells was significantly reduced (pups, Δrate = 8.8 ± 2.8 Hz, n = 9, p = 0.0002 vs control, paired; weanlings, Δrate = 7.7 ± 2.3 Hz, n = 5, p = 0.005 vs control, paired). Subsequent recordings were made in mouse pups, except as noted. Because CPCCOEt applied at concentrations above 100 μm antagonizes both mGluR1 and mGluR5 (Annoura et al., 1996), these data suggest that glutamate released by stimulus trains designed to elicit only IPSPs can also activate group I mGluRs, which in turn facilitate prolonged rebound firing in cerebellar nuclear cells.
Figure 1.

Prolonged rebound firing is reduced by antagonizing group I mGluRs and does not require synaptic inhibition. A, Action potentials in a cerebellar nuclear cell, with a 500 ms, 100 Hz train of stimulation, before and after application of CPCCOEt. Fast excitatory input was blocked by DNQX and CPP. B, Summary of firing rate changes after stimulus trains with or without CPCCOEt. In this and all related figures, lines connect within-cell observations. Black symbols, Data from pups (P13–P15); gray symbols, data from weanlings (P22–P28). C, Action potentials recorded from another cell under the same conditions as in A, except inhibitory input was blocked by SR95531 (SR). D, Summary of the firing rate changes in SR95531 after stimulus trains with or without CPCCOEt. White symbols represent individual cells, and black symbols are mean ± SEM. In all figures, asterisks in all figures represent statistical significance (p < 0.05) for all comparisons by paired t tests; N.S., not significant; ctrl, control.
To test whether synaptic inhibition was necessary for this putatively mGluR-dependent acceleration of firing, we repeated the experiment with GABAA receptors blocked by 10 μm SR95531. Even without IPSPs, cells fired still significantly faster after stimulus trains, increasing their rates by 12.8 ± 3.0 Hz (n = 8, p = 0.004 vs baseline, paired) (Fig. 1C,D). This increase is smaller than with IPSPs, consistent with the idea that the hyperpolarization and interruption of spiking also contributes to prolonged rebound firing. Nevertheless, CPCCOEt completely prevented this increase (Δrate = 0.8 ± 0.8 Hz, n = 8, p = 0.003 vs control, no IPSPs, paired), indicating that the CPCCOEt-sensitive currents that accelerate firing do not need to be recruited by hyperpolarization. In the absence of CPCCOEt, the Δrate value varied across cells, with or without inhibition intact. This variance was not a function of age (Fig. 1B), and, because recordings were made from cells with similar soma sizes from a fairly restricted region of the nuclei, it did not seem to reflect responses of clearly distinguishable cell classes. Even nuclear cells with common morphological features, however, can differ quite widely in the density of their intrinsic currents, such as Na, K, and Ca currents (Raman et al., 2000; Zheng and Raman 2009). The variance in Δrate, therefore, suggests that the current(s) modulated by synaptic stimulation and responsible for persistent rebound firing also varies in density across cells.
The specificity of CPCCOEt has been debated, however. In addition to its antagonism of group I mGluRs, CPCCOEt apparently facilitates a net depolarizing current in Purkinje cells, which is thought to be independent of mGluRs, although the affected ion channels are unknown (Fukunaga et al., 2007). In contrast, a separate report suggests that CPCCOEt antagonizes a Ca-dependent slow inward current in Purkinje cells, which activates on a timescale of minutes (Shin et al., 2009). Therefore, to test whether the effects that we observed were indeed mediated by group I mGluRs, we repeated the experiments with different antagonists. In these experiments, GABAA receptors remained unblocked. Like CPCCOEt, application of both the mGluR1 antagonist JNJ16259685 (0.2 μm) and the mGluR5 inverse agonist MPEP (40 μm) consistently reduced Δrate (Fig. 2A,B) (from 33.0 ± 9.5 to 22.8 ± 3.9 Hz, n = 9, p = 0.03, paired). Likewise, when the competitive mGluR1 antagonist 3-MATIDA (150 μm) replaced JNJ16259685 and was applied with MPEP, Δrate decreased significantly (Fig. 2B) (from 41.1 ± 8.3 to 27.4 ± 9.1 Hz, n = 8, p = 0.0009, paired), consistent with a role of group I mGluRs. Application of either JNJ16259685 or MPEP alone, however, did not significantly affect the firing rate increase, although in MPEP, Δrate decreased in a majority of the cells (Fig. 2C–F) (JNJ16259685, from 26.0 ± 4.0 to 29.9 ± 4.3 Hz, n = 9, p = 0.4, paired; MPEP, from 34.0 ± 6.5 to 28.0 ± 6.7 Hz, n = 7, p = 0.07, paired). These data indicate that activation of either mGluR1 or mGluR5 alone is sufficient to accelerate firing rates of cerebellar nuclear cells.
Figure 2.
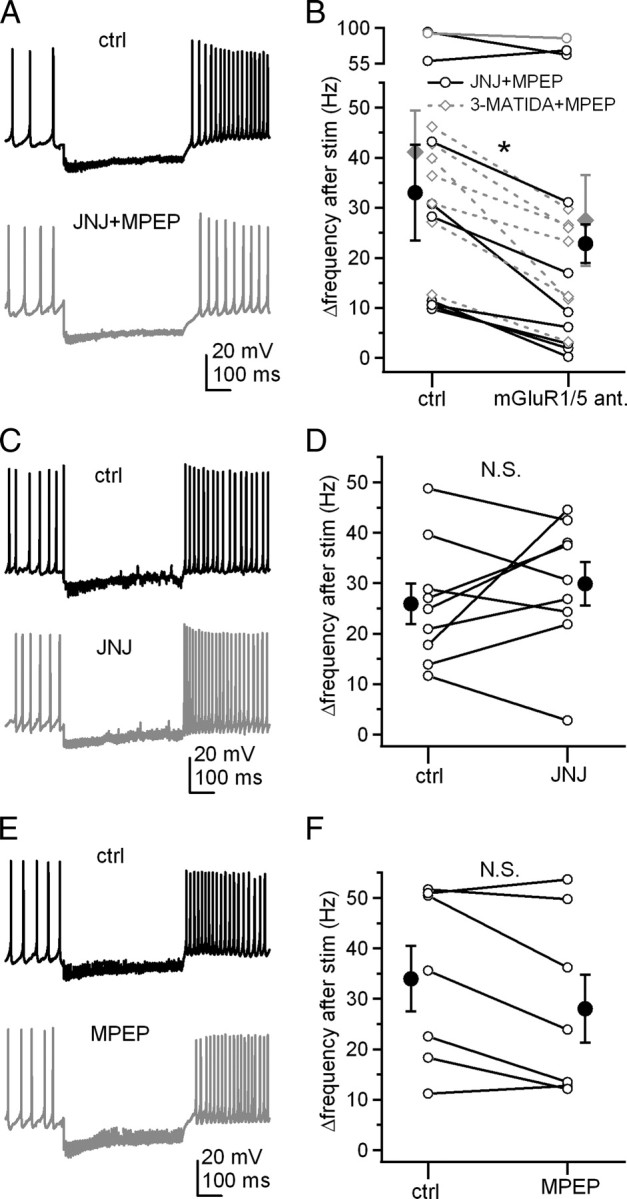
Either mGluR1 or mGluR5 is sufficient to facilitate prolonged rebound firing. A, Action potentials in a cerebellar nuclear cell, with a 500 ms, 100 Hz stimulus train, before and after application of JNJ16259685 (JNJ) and MPEP together. Fast excitatory input was blocked by DNQX and CPP. B, Summary of firing rate changes after stimulus trains before and after application of JNJ16259685 and MPEP together as in A or 3-MATIDA and MPEP together. ant., Antagonists. C, Same protocol in another cell, before and after application of JNJ16259685 alone. D, Summary of firing rate changes after stimulus trains as in C. E, Same protocol in another cell, before and after application of MPEP alone. F, Summary of firing rate changes after stimulus trains as in E. White symbols represent individual cells, and black symbols indicate mean ± SEM.
To identify the currents that are modified by group I mGluR activation, we recorded from voltage-clamped cerebellar nuclear cells before and after bath application of s-DHPG, a selective group I mGluR agonist. Fast synaptic receptors were blocked with DNQX, CPP, and SR95531, and Na current was blocked with intracellular QX-314 (60 μm). At −66 mV, 20 μm s-DHPG increased the holding current by −126 ± 29 pA (n = 7, from −288 ± 43 pA in control to −414 ± 55 pA in s-DHPG, p = 0.005), suggesting that mGluR activation might modulate one or more currents that were tonically active at this voltage. To characterize these putative leak currents, the voltage was stepped from −66 to −46 mV for 150 ms, to let inactivation of voltage-gated currents occur, and then ramped down to −96 mV over 500 ms. After 100 ms at −96 mV, the voltage was ramped back up to −56 mV over 250 ms. If the holding current at −66 mV is dominated by a voltage-independent leak, these ramps should evoke currents that vary fairly linearly with voltage. Slow ramps at subthreshold potentials with Na channels blocked were selected to optimize voltage control. Remarkably, in addition to a linear leak, this protocol revealed a non-monotonic current. As shown in Figure 3A, during the hyperpolarizing ramp in control solution, the inward current first increased in amplitude (from approximately −46 to −51 mV), then decreased (from approximately −51 to −61 mV), and then increased linearly until the end of the downward ramp. We reasoned that the step to −46 mV evoked a voltage-gated inward current that remained on as the voltage was ramped downward, so that the increasing driving force resulted in a larger current. With additional hyperpolarization, this voltage-gated current began to deactivate, reducing the total current amplitude. Ultimately, leak currents dominated, increasing as the voltage became still more negative. During the depolarizing ramp, the process was reversed, following the trajectory predicted for a non-inactivating component of voltage-gated current. Thus, this protocol reveals the presence of a long-lasting, voltage-gated current in cerebellar nuclear cells that activates at voltages as negative as −60 mV.
Figure 3.
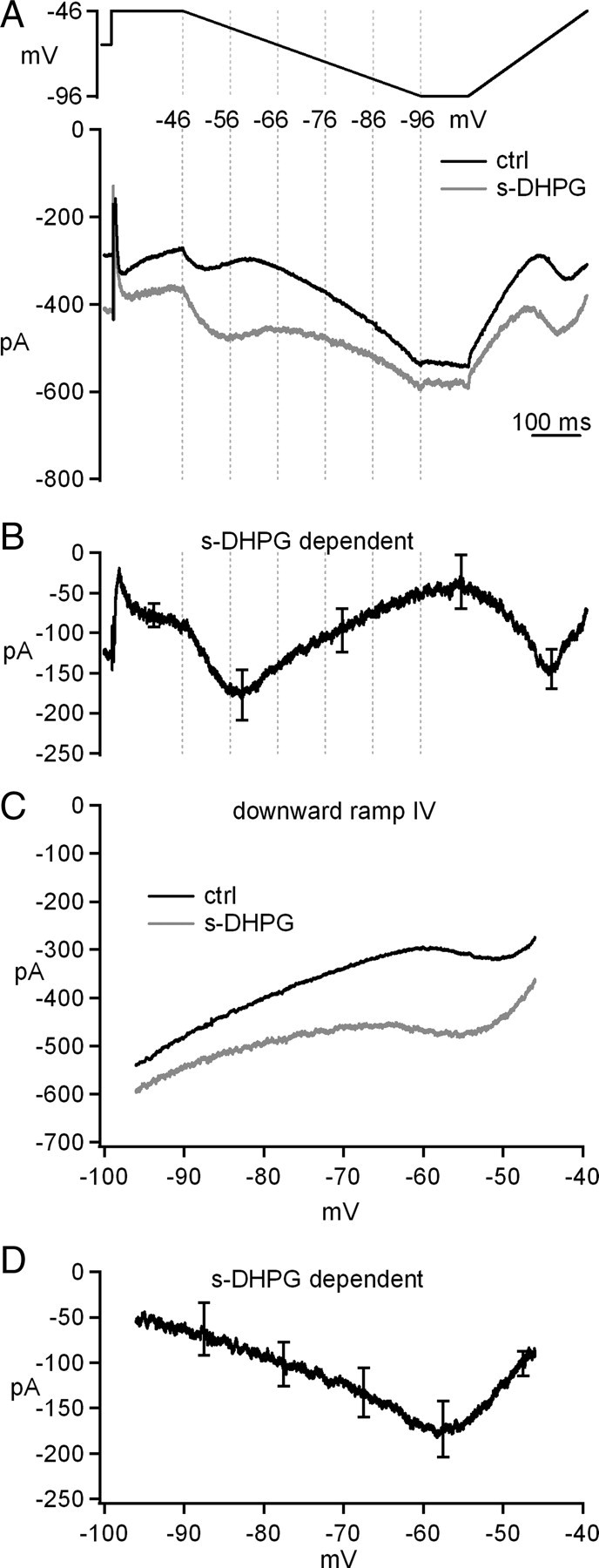
Voltage-gated as well as voltage-independent currents are modulated by group I mGluR agonists. A, Voltage protocol and evoked currents before and after application of s-DHPG. B, s-DHPG-dependent current obtained by subtraction. C, Current–voltage curves for currents during the downward ramp before and after application of s-DHPG. D, Current–voltage curve for s-DHPG-dependent current during the downward ramp. Synaptic transmission was blocked by SR95531, DNQX, and CPP. Na current was blocked by QX-314.
When s-DHPG was added to the bath, the inward current was increased at all points in the double-ramp protocol, consistent with an augmentation of both the linear leak component and the voltage-gated current. Moreover, the voltage of the maximal current during the downward ramp shifted to a more negative potential (Fig. 3A). Subtracting the control from s-DHPG records gave the total s-DHPG-dependent current (Fig. 3B). Plotting the data as current versus voltage during the downward ramp illustrated that s-DHPG both increased the current and negatively shifted its activation voltage (Fig. 3C). The maximal increase in current was −173 ± 32 pA at −57.5 ± 1.2 mV (n = 6) (Fig. 3D). During the upward ramp, the maximal s-DHPG-dependent current was similar to that evoked the downward ramp (−169 ± 29 pA), supporting the idea that the s-DHPG-dependent voltage-gated current inactivates only slightly. Together, these data indicate that group 1 mGluRs potentiate the long-lasting, low-voltage-activated current in cerebellar nuclear cells.
To verify that the modulation by s-DHPG did not simply result from disrupting the intracellular contents during whole-cell recording, we repeated the experiments with gramicidin perforated-patch recordings. Although voltage-clamp recordings are compromised by the high access resistance in perforated-patch mode, we reasoned that we could still test whether s-DHPG could augment the current evoked by slow voltage changes at subthreshold potentials. Not surprisingly, on depolarization to −46 mV, escaping currents were evident, which subsided as the voltage was hyperpolarized to subthreshold values. In the presence of s-DHPG, the non-monotonic current clearly increased (Fig. 4A,B). In fact, the amplitude and voltage of the maximal s-DHPG-activated current with perforated-patch and whole-cell recordings were indistinguishable (Fig. 4C) (gramicidin, −178 ± 24 pA at −56.5 ± 1.7 mV, n = 7; whole cell, −173 ± 32 pA at −57.5 ± 1.2 mV, n = 6; amplitude, p = 0.8; voltage, p = 0.7, unpaired). These data validated the initial observations and justified additional experiments with whole-cell recording, in which series resistance is lower, enabling voltage control at subthreshold potentials, even in dendrites (Pugh and Raman, 2008).
Figure 4.
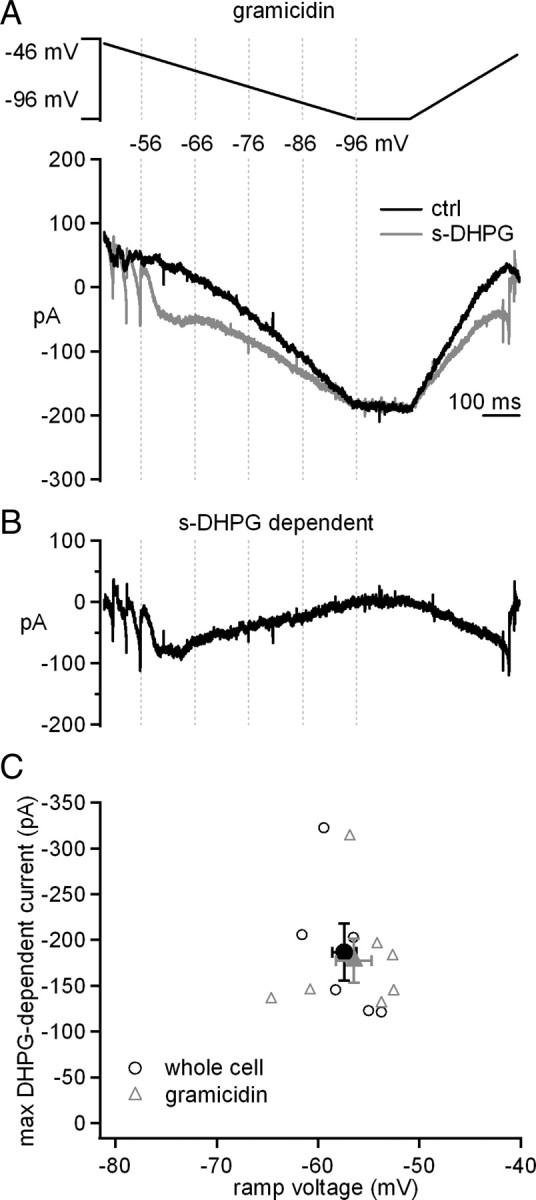
Modulation of voltage-gated currents is present in perforated-patch mode. A, Voltage protocol and evoked currents before and after application of s-DHPG in gramicidin perforated patch recordings. B, s-DHPG-dependent current obtained by subtraction. C, The maximal s-DHPG-dependent current versus voltage at which it occurred during the downward ramp in whole-cell (black open circles) and gramicidin recordings (gray open triangles). Black filled symbols, mean ± SEM. Synaptic transmission was blocked by SR95531, DNQX, and CPP.
We next considered candidate voltage-gated currents that might be modulated by group I mGluRs. The most likely possibility for a relatively low-voltage-activated but essentially non-inactivating, QX-314-insensitive current was L-type Ca current. We therefore repeated the experiment in the presence of 10 μm nimodipine, an L-type Ca current antagonist. Without s-DHPG, the current evoked during the downward ramp was almost linear, as expected for a leak current with no voltage sensitivity (Fig. 5A). Like in control solutions, application of s-DHPG increased the holding current at −66 mV, but only by −87.1 ± 22.3 pA, indicating that approximately one-third of the s-DHPG-dependent holding current increase was nimodipine sensitive. During the ramps, s-DHPG in nimodipine increased the current at all potentials but to a lesser extent than in the absence of the blocker. Subtractions indicated that the s-DHPG-dependent current at −57.5 mV during the downward ramp was only −84 ± 20 pA (n = 8), approximately one-half that in control conditions (Fig. 5B). The current–voltage curves for the raw and s-DHPG-dependent currents with and without nimodipine demonstrate that s-DHPG indeed increases an inward leak current but that a substantial fraction of the current potentiated by s-DHPG at physiological, subthreshold potentials (−70 to −50 mV) is a nimodipine-sensitive, likely L-type current (Fig. 5C,D).
Figure 5.
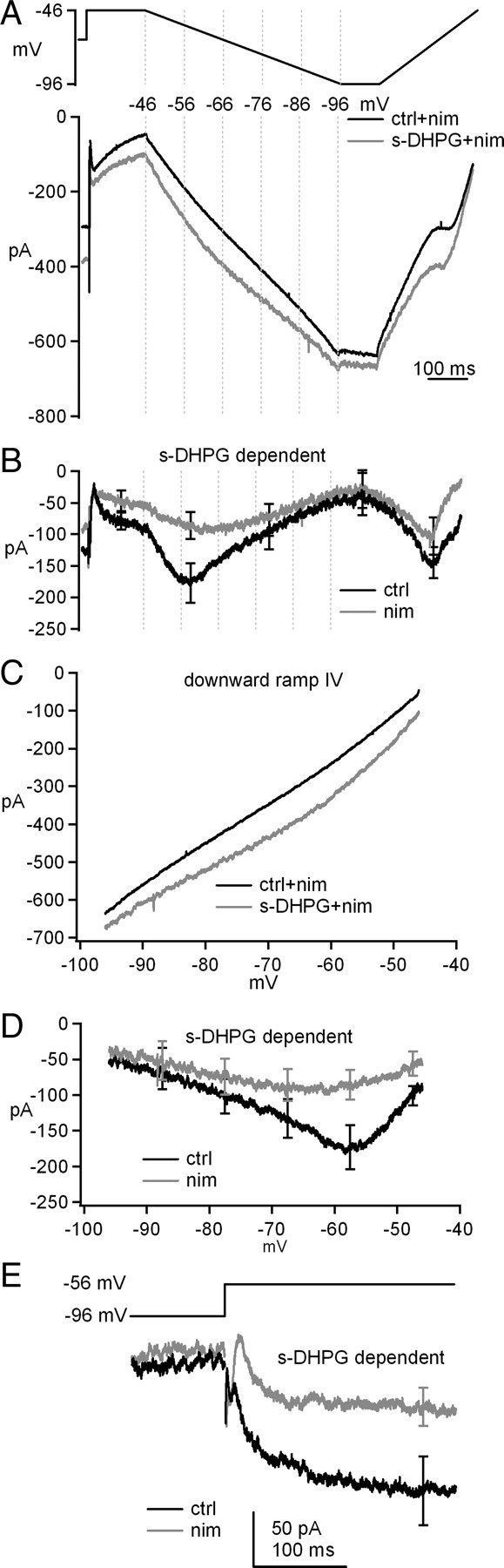
Group I mGluRs potentiate a nimodipine-sensitive L-type current. A, Voltage protocol and evoked currents in nimodipine (nim) before and after application of s-DHPG. B, s-DHPG-dependent currents in the presence of nimodipine. The record without nimodipine from Figure 3C is overlaid for comparison. C, Current–voltage curves for currents during downward ramps in nimodipine before and after application of s-DHPG. D, Overlay of current–voltage curves for s-DHPG-dependent currents during downward ramps with nimodipine and without nimodipine (from Fig. 3E, for comparison). E, s-DHPG-dependent current evoked by step depolarizations, with or without nimodipine. Synaptic transmission was blocked by SR95531, DNQX, and CPP. Na current was blocked by intracellular QX-314.
The downward ramp is an informative biophysical measure, because it allows us to isolate non-inactivating current components and distinguish voltage-gated from leak currents, but a current that participates in rebound firing must be activated by depolarizations. To test whether step depolarizations would reveal a similar increase in L-type current, we next measured the nimodipine sensitivity of the s-DHPG-dependent current evoked by a step from −96 to −56 mV. This current was −154 ± 32 pA without nimodipine and fell to −77 ± 19 pA in the presence of nimodipine, confirming that L-type current accounts for approximately one-half the current increased by s-DHPG (Fig. 5E). The increase in current was long lasting, persisting at least 250 ms after depolarization. Together, these data support the idea that group I mGluR activation enhances multiple non-inactivating currents, of which the primary voltage-gated component comes from L-type Ca channels.
Next, we tested whether L-type Ca current can be amplified by stimulated glutamate release rather than bath application of agonist. In these experiments, 100 Hz, 500 ms stimulus trains were applied at −66 mV with only GABAA and NMDA receptors blocked, so that the AMPA receptor-mediated fast EPSCs would serve as an indicator of glutamate release. Fifty ms after the stimulus train, the voltage was stepped to −46 mV for 100 ms, followed by a 250 ms ramp down to −96 mV. The difference between these currents and ramp-evoked currents without previous synaptic stimulation gave a measure of the stimulation-dependent increase in current (Fig. 6A). Like the data obtained with s-DHPG, the subtracted records revealed an increase in two components of current. First, the train increased the non-monotonic voltage-gated current evoked during the ramp. Importantly, this increase disappeared when the experiment was repeated in nifedipine (10 μm), an L-type Ca channel blocker that, unlike nimodipine at this concentration, should have no effect on T-type channels (Stengel et al., 1998). Second, immediately after the stimulus train, a long-lasting inward current was evident. This component of current was only partly reduced by nifedipine. Because nifedipine at the applied concentration fully blocks the voltage-dependent (L-type) current, the residual, nifedipine-insensitive current is likely to represent the slow mGluR1-dependent EPSC described in cerebellar nuclear cells by Zhang and Linden (2006). Across cells, the ramp-evoked currents varied, as evident in raw records from three sample cells in Figure 6B; this variation in the mGluR response is likely to contribute to the range of Δrate values observed in current clamp. In all cases, however, the stimulus train increased current amplitudes. Moreover, nifedipine linearized ramp-evoked currents and reduced the magnitude of the stimulus-dependent increase in inward current. The current–voltage relation for the ramp-evoked current averaged across all cells (n = 7) confirmed that trains of EPSCs potentiate nifedipine-sensitive L-type Ca currents, as well as nifedipine-insensitive voltage-independent putative slow EPSCs (Fig. 6C). The increase in L-type current was maximal at −56.7 mV (control, −100.1 ± 15.2 pA; nifedipine, −47.3 ± 12.0 pA), nearly the same voltage at which the s-DHPG-dependent current was maximal (−57.5 mV). These data suggest that synaptic glutamate release evoked by trains of stimuli is sufficient to activate mGluRs and enhance L-type Ca current in cerebellar nuclear cells.
Figure 6.
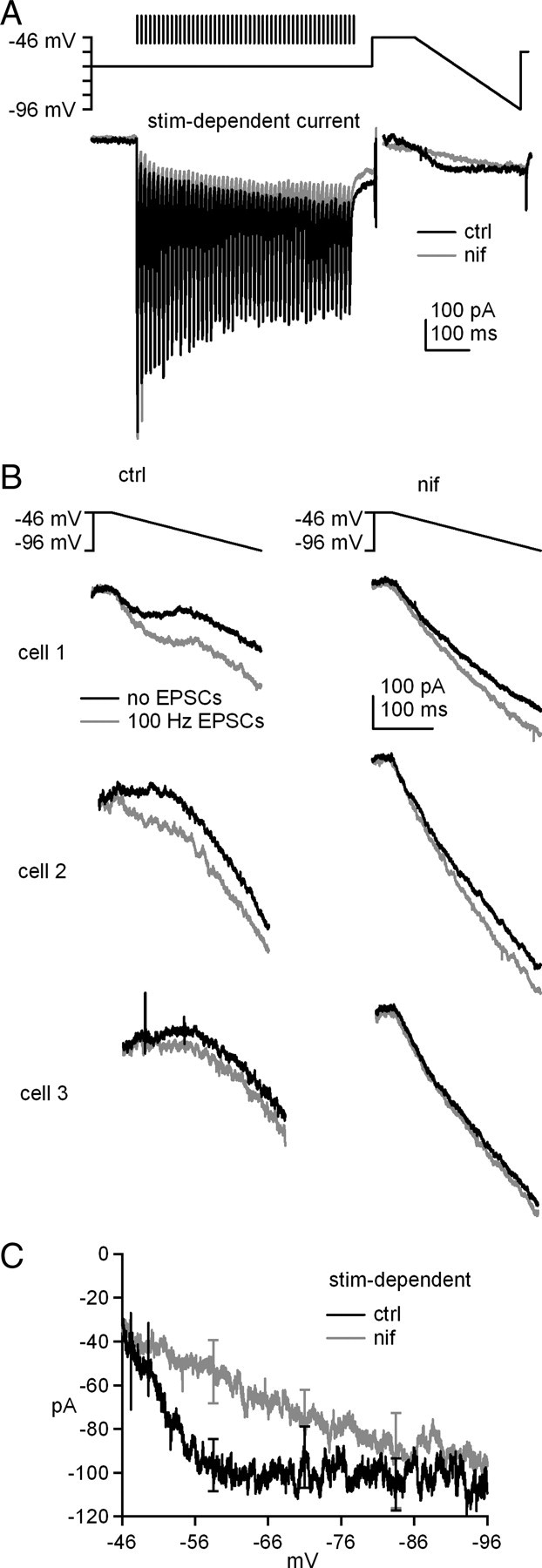
Synaptically released glutamate can potentiate L-type Ca current. A, Voltage protocol with conditioning train of EPSCs evoked at 100 Hz for 500 ms and mean stimulation-dependent currents with or without nifedipine (nif). B, Voltage protocol and evoked currents in three representative cells with or without conditioning stimulation with or without nifedipine. GABAA and NMDA receptors were blocked by SR95531 and CPP. C, Current–voltage curves for stimulation-dependent currents during the downward ramp.
If prolonged rebound firing depends on an increase in L-type Ca current by group I mGluRs, then blocking these channels should reduce the poststimulus acceleration of firing. We therefore repeated the current-clamp experiments of Figure 1 in the presence of 10 μm nifedipine. With AMPA and NMDA receptors blocked, the firing rate increased by only 8.7 ± 2.6 Hz in nifedipine (n = 14), considerably less than the 31.3 ± 3.0 Hz obtained by pooling data from all control records (n = 42, p < 1e-6, unpaired). These data are consistent with a major role for L-type current in prolonged rebound firing. Addition of CPCCOEt did not further reduce the firing rate increase (Δrate = 14.0 ± 3.0 Hz before and 11.5 ± 3.6 Hz after antagonist, n = 8, p = 0.12, paired) (Fig. 7A,C,G). The results were similar when recordings were repeated in cells from weanling mice (Δrate = 18.6 ± 5.8 Hz before and 17.9 ± 3.8 Hz after antagonist, n = 5, p = 0.82, paired). Likewise, when a combination of MPEP and 3-MATIDA was applied instead of CPCCOEt, Δrate did not change significantly (1.8 ± 2.7 Hz before and −1.4 ± 1.9 Hz after antagonists, n = 6, p = 0.26, paired) (Fig. 7C). Together, these data support the hypothesis that the mGluR-dependent component of prolonged rebound firing is primarily dependent on potentiation of L-type current.
Figure 7.
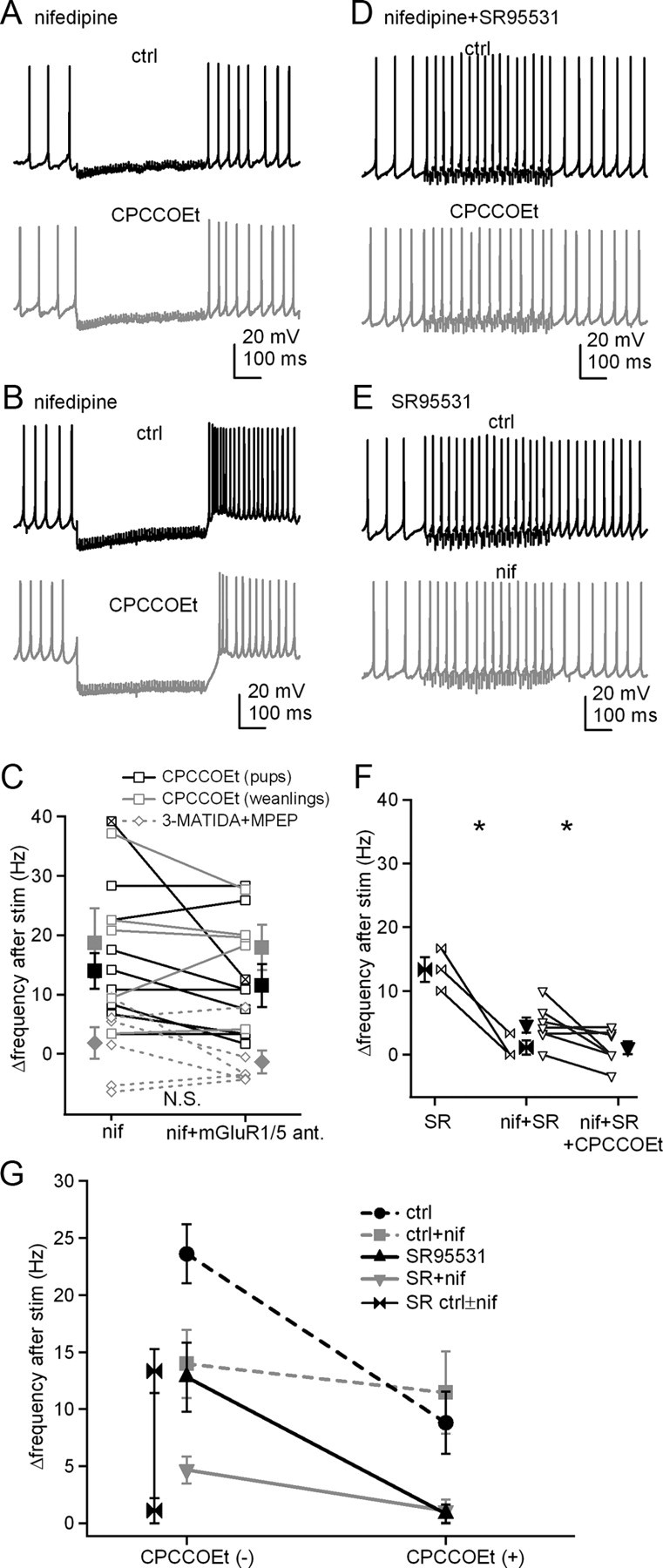
Prolonged rebound firing is reduced by blockade of L-type Ca channels. A, Action potentials in a cerebellar nuclear cell, with a 500 ms, 100 Hz stimulus train before and after application of CPCCOEt in nifedipine (nif). Fast excitatory transmission was blocked by DNQX and CPP. B, Action potentials recorded from the one cell showing rebound burst firing under the same conditions as in A, with or without CPCCOEt. C, Summary of firing rate changes after stimulus trains in nifedipine with or without CPCCOEt or nifedipine with or without 3-MATIDA and MPEP. Open symbols represent individual cells, and filled symbols indicate mean ± SEM. The x-box symbol indicates the bursting cell in B and was excluded from the statistical analysis. Black squares, Data from pups (P13–P15) with CPCCOEt; gray squares, data from weanlings (P22–P28) with CPCCOEt; gray diamonds, data from pups with 3-MATIDA and MPEP. ant., Antagonists. D, Action potentials from another cell under the same conditions as in A, except with inhibitory input blocked by SR95531 (SR). E, Action potentials from a cell with stimulus trains applied before and after exposure to nifedipine without CPCCOEt, with inhibitory and fast excitatory transmission blocked. F, Summary of firing rate increases with or without nifedipine and/or CPCCOEt, with inhibitory input blocked. G, Summary of firing rate changes for all conditions tested.
In this dataset, one neuron generated a clear burst of spikes riding on a depolarization immediately after the offset of inhibition; this cell was the only one in the full study to exhibit such a burst. The Δrate value in this neuron was 39.2 Hz in nifedipine. Interestingly, however, after addition of CPCCOEt, the firing rate increase fell to 12.5 Hz and the burst had fewer spikes (Fig. 7B,C). Because L-type channels could not have contributed to rebound firing in this cell, it is most likely that T-type current drove the bursting (Molineux et al., 2006). If so, the decreased bursting in CPCCOEt suggests that T-type current in nuclear cells is also potentiated by mGluR activation, consistent with recent work demonstrating that group I mGluRs can upregulate these currents in Purkinje cells (Hildebrand et al., 2009).
Last, to test whether the potentiation of L-type current is responsible for the acceleration of firing after stimulation even with synaptic inhibition blocked, we repeated the experiment in SR95531 as well as nifedipine, CPP, and DNQX. Under these conditions, the poststimulation firing rate increase was only 4.7 ± 1.2 Hz (n = 7, p = 0.03 vs no nifedipine, unpaired) (Fig. 7D,F), demonstrating that potentiation of L-type current also dominates the poststimulus firing rate acceleration in the absence of inhibition. Application of CPCCOEt abolished the residual rate increase (Δrate = 1.1 ± 1.0 Hz, n = 7, p = 0.04, paired) (Fig. 7D,F,G), suggesting that the leak-like slow EPSC can make a small but measurable contribution to poststimulation firing. To verify that an influence of L-type current on firing was evident in within-cell comparisons, we also recorded without CPCCOEt before and after perfusion of nifedipine. The results confirmed that nifedipine decreased the firing rate change after stimulation from 13.3 ± 1.9 to 1.1 ± 1 Hz (n = 3, p = 0.03, paired) (Fig. 7E–G). Overall, these data demonstrate that activation of group I mGluRs accelerates firing by potentiating L-type Ca channels and does not require inhibition.
Discussion
These data demonstrate that prolonged rebound firing in the cerebellar nuclei in slices does not result solely or even primarily from the relief of inhibition from Purkinje cells. Instead, trains of stimulation that elicit IPSPs also activate group I mGluRs, which potentiate L-type Ca currents, which drive an increase in firing rate. Activation of either mGluR1 or mGluR5 alone appears sufficient to induce this effect. These results suggest that rebound firing is readily observed in slices primarily because stimulus trains release glutamate as well as GABA (Aizenman and Linden, 1999; Molineux et al., 2006; Alviña et al., 2008; Zheng and Raman, 2009; Tadayonnejad et al., 2010). More importantly, they provide an explanation for the observations in vivo that prolonged rebound firing is rarely elicited by exclusively GABAergic transmission evoked by stimulating Purkinje afferents in the cortex (Alviña et al., 2008) but reliably evoked by stimulating the inferior olive, which not only activates Purkinje cells but also sends a direct glutamatergic projection to the nuclei (Hoebeek et al., 2010). Thus, in intact cerebella, prolonged rebound firing may be generated most robustly when synaptic inhibition is strong enough to suppress firing but coincides with excitation that activates mGluRs.
Group I mGluR modulation of multiple inward currents
Activation of mGluRs is known to modulate Ca currents, but the most common effect is to reduce, not increase, the amplitudes of L-type and other high-voltage-activated Ca currents (Swartz and Bean, 1992; Sahara and Westbrook, 1993; Cheong et al., 2008). Group I mGluRs often also activate an inward current that produces a slow EPSC at negative potentials (Guatteo et al., 1999; Dzubay and Otis, 2002; Karakossian and Otis, 2004; Rae and Irving, 2004); this current is also present in the cerebellar nuclei (Zhang and Linden, 2006). The slow EPSC is nonselective, however, and thus not carried by voltage-gated Ca channels. Instead, it has the hallmarks of a transient receptor potential (TRP) channel (Gee et al., 2003) and, in Purkinje cells, is mediated by TRPC3 channels (Hartmann et al., 2008).
In cerebellar interneurons, mGluR-mediated TRP current can depolarize cells and indirectly activate L-type currents (Collin et al., 2009). In contrast, the present work indicates that potentiation of L-type currents in cerebellar nuclear cells occurs independently of activation of TRP and other leak-like conductances, because it is evident under voltage clamp. L-type currents are similarly potentiated in both cerebellar granule cells, by group I mGluR agonists, which mobilize Ca from intracellular stores (Chavis et al., 1995, 1996), and medullary inspiratory neurons, after hypoxia-induced activation of group I mGluRs (Mironov and Richter, 1998, 2000). The potentiation of L-type currents in cerebellar nuclear cells also occurs independently of synaptic inhibition: stimulating mGluRs in the presence of GABAA receptor antagonists still significantly accelerates firing, although to a lesser extent than with inhibition intact. Importantly from a physiological perspective, the potentiation of L-type channels alone by synaptically released glutamate is sufficient to increase the firing rates of cerebellar nuclear neurons for a few hundred milliseconds.
A rather striking attribute of the L-type current potentiated by group I mGluRs is its voltage dependence and kinetics. The current undergoes only minor inactivation over several hundred milliseconds, and the nifedipine-sensitive current flows at potentials as negative as −55 mV. After modulation by mGluRs, channels open at still more hyperpolarized voltages. These low activation voltages are unlikely to reflect voltage-clamp error, given the use of subthreshold voltage ramps and the primarily somatic localization of L-type channels, as evident from whole-cell recordings from isolated somata and Ca imaging from dendrites (Zheng and Raman, 2009). Instead, these currents may result from CaV1.3 channels, which are known to activate in the same low-voltage range as T-type currents (Lipscombe et al., 2004). These low activation voltages and the lack of inactivation make this L-type current well adapted to provide a consistent depolarizing drive at subthreshold potentials and thereby control firing rates for several hundred milliseconds.
Direct influence of inhibition on rebound firing
In the absence of group I mGluR activation, inhibition alone elicited weaker but significant prolonged rebound firing, with Δrate values ∼40% as great as with mGluRs unblocked. These data support the idea that recovery of intrinsic depolarizing currents contributes to prolonged rebound firing, consistent with studies in which inhibition is induced by dynamically clamped IPSPs (Sangrey and Jaeger, 2010) or photorelease of GABA (Alviña et al., 2008), i.e., without electrical stimulation. In fact, dynamic clamp experiments suggest that a long-lasting inward current, possibly persistent Na current, drives prolonged rebound firing. Indeed, Na currents of nuclear cells undergo slow inactivation during spontaneous spiking and recover partially during hyperpolarizations to −70 mV (Aman and Raman, 2007). Thus, they are likely to contribute at least to short-term accelerations of firing after the relief of inhibition; however, they inactivate before the prolonged rebound period is complete, suggesting that other currents must participate as well. Another long-lasting current that may be recruited by inhibition in nuclear cells is low-voltage-activated R-type Ca current. The R-type channel peptide blocker SNX-482 reduces the firing rate acceleration and decreases dendritic Ca signals after hyperpolarizing current injections (Zheng and Raman, 2009). Consistent with a role for this current, a nimodipine-insensitive, non-inactivating, R-type-like current in nuclear cells is also blocked by mibefradil (Zheng and Raman, 2009), and this drug prevents prolonged as well as burst rebound firing (Alviña et al., 2009). Another possibility is that inhibition may deactivate K currents, such as SK current, which in turn reduce a hyperpolarizing drive after relief of inhibition (Nelson et al., 2003). Thus, prolonged rebound firing likely results from multiple currents modulated by excitatory and inhibitory synaptic transmission. The variable density of these currents (Raman et al., 2000; Zheng and Raman, 2009) is expected to affect the extent of prolonged rebound firing, thereby contributing to the range of Δrates that we observed.
The most extensively studied candidate current for driving postinhibitory rebounds, and particularly burst firing, is T-type Ca current (Llinás and Mühlethaler, 1988). Although T-type current is present in these cells, it recovers by <10% at voltages reached by IPSP trains (Zheng and Raman, 2009), consistent with the observation that bursts occur only occasionally after trains of IPSPs (Alviña et al., 2008; Tadayonnejad et al., 2010), which hyperpolarize neurons no farther than ECl, approximately −75 mV in these neurons. The small fraction of recovered T-type current is indeed expected to activate on relief of inhibition, but, because of its rapid decay kinetics, it is mostly likely to contribute only to the earliest (burst) phase of rebound firing (Molineux et al., 2006). Interestingly, in the one bursting cell recording in this study, the burst frequency and number of spikes were both reduced by group I mGluR blockade. This observation, along with recent results showing that group I mGluRs can increase T-type currents (Hildebrand et al., 2009; Johnston and Delaney, 2010), offers an explanation for why rebound bursts can be elicited more readily with higher stimulus intensity (Tadayonnejad et al., 2010): Higher stimulus strengths are more likely to elicit sufficient glutamate release to activate mGluRs. Likewise, rebound bursts were evoked less frequently with GABA photorelease than with synaptic stimulation (Alviña et al., 2009), supporting the idea that simultaneous activation of glutamate and GABA receptors maximizes both burst and prolonged rebound firing in the cerebellar nuclei.
Implications for long-term plasticity in the cerebellar nuclei
The relationship between group I mGluRs and L-type Ca channels may be particularly relevant in the cerebellar nuclei because both have been implicated in complementary aspects of synaptic plasticity. Group I mGluR activation with depolarization is required to induce LTD of EPSCs in cerebellar nuclear cells (Zhang and Linden, 2006), whereas activation of L-type Ca current without hyperpolarization antagonizes LTP at the same synapses (Person and Raman, 2010). It remains unknown whether these forms of LTD and LTP operate on the same mechanism or are separate processes. In either case, the present results raise the possibility that, when activation of glutamatergic afferents is sufficient to activate group I mGluRs, the augmentation of L-type current may reduce the probability that a net potentiation of EPSCs is induced. During periods of reduced inhibition, L-type channels and group I mGluRs may therefore act synergistically to increase either the extent or the likelihood of LTD of EPSCs.
Interestingly, L-type currents also regulate long-term plasticity of Purkinje-mediated inhibition in the cerebellar nuclei: a relatively low Ca influx, produced by L-type channels alone, favors LTD of IPSPs, but if Ca influx is augmented by activation of other channels, LTP of IPSPs is induced (Morishita and Sastry, 1996; Aizenman et al., 1998; Ouardouz and Sastry, 2000). The present results suggest that group I mGluR activation may also influence the plasticity of synaptic inhibition; in the extreme, if L-type-mediated Ca influx is sufficiently increased, the depression of IPSPs may even convert to potentiation. If so, it would be another means by which group I mGluRs work with L-type channels to reduce long-term net synaptic excitation onto cerebellar nuclear cells (over minutes to hours), even while increasing short-term intrinsic excitability (over hundreds of milliseconds).
Footnotes
This work was supported by NIH Grant NS39395 (I.M.R.). We thank members of the Raman laboratory, J. Pugh, T. Aman, A. Person, J. Bant, M. Benton, and A. Lewis for helpful discussion.
References
- Aizenman CD, Linden DJ. Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J Neurophysiol. 1999;82:1697–1709. doi: 10.1152/jn.1999.82.4.1697. [DOI] [PubMed] [Google Scholar]
- Aizenman CD, Manis PB, Linden DJ. Polarity of long-term synaptic gain change is related to postsynaptic spike firing at a cerebellar inhibitory synapse. Neuron. 1998;21:827–835. doi: 10.1016/s0896-6273(00)80598-x. [DOI] [PubMed] [Google Scholar]
- Alviña K, Walter JT, Kohn A, Ellis-Davies G, Khodakhah K. Questioning the role of rebound firing in the cerebellum. Nat Neurosci. 2008;11:1256–1258. doi: 10.1038/nn.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alviña K, Ellis-Davies G, Khodakhah K. T-type calcium channels mediate rebound firing in intact deep cerebellar neurons. Neuroscience. 2009;158:635–641. doi: 10.1016/j.neuroscience.2008.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman TK, Raman IM. Subunit dependence of Na channel slow inactivation and open channel block in cerebellar neurons. Biophys J. 2007;92:1938–1951. doi: 10.1529/biophysj.106.093500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annoura H, Fukunaga A, Uesugi M, Tatsuoka T, Horikawa Y. A novel class of antagonists for metabotropic glutamate receptors, 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylates. Bioorg Med Chem Lett. 1996;6:763–766. [Google Scholar]
- Chavis P, Fagni L, Bockaert J, Lansman JB. Modulation of calcium channels by metabotropic glutamate receptors in cerebellar granule cells. Neuropharmacology. 1995;34:929–937. doi: 10.1016/0028-3908(95)00082-h. [DOI] [PubMed] [Google Scholar]
- Chavis P, Fagni L, Lansman JB, Bockaert J. Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature. 1996;382:719–722. doi: 10.1038/382719a0. [DOI] [PubMed] [Google Scholar]
- Cheong E, Lee S, Choi BJ, Sun M, Lee CJ, Shin HS. Tuning thalamic firing modes via simultaneous modulation of T- and L-type Ca2+ channels controls pain sensory gating in the thalamus. J Neurosci. 2008;28:13331–13340. doi: 10.1523/JNEUROSCI.3013-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin T, Franconville R, Ehrlich BE, Llano I. Activation of metabotropic glutamate receptors induces periodic burst firing and concomitant cytosolic Ca2+ oscillations in cerebellar interneurons. J Neurosci. 2009;29:9281–9291. doi: 10.1523/JNEUROSCI.1865-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzubay JA, Otis TS. Climbing fiber activation of metabotropic glutamate receptors on cerebellar Purkinje neurons. Neuron. 2002;36:1159–1167. doi: 10.1016/s0896-6273(02)01052-8. [DOI] [PubMed] [Google Scholar]
- Fukunaga I, Yeo CH, Batchelor AM. The mGlu1 antagonist CPCCOEt enhances the climbing fibre response in Purkinje neurones independently of glutamate receptors. Neuropharmacology. 2007;52:450–458. doi: 10.1016/j.neuropharm.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Gee CE, Benquet P, Gerber U. Group I metabotropic glutamate receptors activate a calcium-sensitive transient receptor potential-like conductance in rat hippocampus. J Physiol. 2003;546:655–664. doi: 10.1113/jphysiol.2002.032961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guatteo E, Mercuri NB, Bernardi G, Knöpfel T. Group I metabotropic glutamate receptors mediate an inward current in rat substantia nigra dopamine neurons that is independent from calcium mobilization. J Neurophysiol. 1999;82:1974–1981. doi: 10.1152/jn.1999.82.4.1974. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008;59:392–398. doi: 10.1016/j.neuron.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand ME, Isope P, Miyazaki T, Nakaya T, Garcia E, Feltz A, Schneider T, Hescheler J, Kano M, Sakimura K, Watanabe M, Dieudonné S, Snutch TP. Functional coupling between mGluR1 and Cav3.1 T-type calcium channels contributes to parallel fiber-induced fast calcium signaling within Purkinje cell dendritic spines. J Neurosci. 2009;29:9668–9682. doi: 10.1523/JNEUROSCI.0362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoebeek FE, Witter L, Ruigrok TJ, De Zeeuw CI. Differential olivo-cerebellar cortical control of rebound activity in the cerebellar nuclei. Proc Natl Acad Sci U S A. 2010;107:8410–8415. doi: 10.1073/pnas.0907118107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J, Delaney KR. Synaptic activation of T-type Ca2+ channels via mGluR activation in the primary dendrite of mitral cells. J Neurophysiol. 2010;103:2557–2569. doi: 10.1152/jn.00796.2009. [DOI] [PubMed] [Google Scholar]
- Karakossian MH, Otis TS. Excitation of cerebellar interneurons by group I metabotropic glutamate receptors. J Neurophysiol. 2004;92:1558–1565. doi: 10.1152/jn.00300.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe D, Helton TD, Xu W. L-type calcium channels: the low down. J Neurophysiol. 2004;92:2633–2641. doi: 10.1152/jn.00486.2004. [DOI] [PubMed] [Google Scholar]
- Llinás R, Mühlethaler M. Electrophysiology of guinea-pig cerebellar nuclear cells in the in vitro brain stem-cerebellar preparation. J Physiol. 1988;404:241–258. doi: 10.1113/jphysiol.1988.sp017288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov SL, Richter DW. L-type Ca2+ channels in inspiratory neurones of mice and their modulation by hypoxia. J Physiol. 1998;512:75–87. doi: 10.1111/j.1469-7793.1998.075bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov SL, Richter DW. Hypoxic modulation of L-type Ca2+ channels in inspiratory brainstem neurones: intracellular signalling pathways and metabotropic glutamate receptors. Brain Res. 2000;869:166–177. doi: 10.1016/s0006-8993(00)02396-9. [DOI] [PubMed] [Google Scholar]
- Molineux ML, McRory JE, McKay BE, Hamid J, Mehaffey WH, Rehak R, Snutch TP, Zamponi GW, Turner RW. Specific T-type calcium channel isoforms are associated with distinct burst phenotypes in deep cerebellar nuclear neurons. Proc Natl Acad Sci U S A. 2006;103:5555–5560. doi: 10.1073/pnas.0601261103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita W, Sastry BR. Postsynaptic mechanisms underlying long-term depression of GABAergic transmission in neurons of the deep cerebellar nuclei. J Neurophysiol. 1996;76:59–68. doi: 10.1152/jn.1996.76.1.59. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Nelson AB, Krispel CM, Sekirnjak C, du Lac S. Long-lasting increases in intrinsic excitability triggered by inhibition. Neuron. 2003;40:609–620. doi: 10.1016/s0896-6273(03)00641-x. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Sastry BR. Mechanisms underlying LTP of inhibitory synaptic transmission in the deep cerebellar nuclei. J Neurophysiol. 2000;84:1414–1421. doi: 10.1152/jn.2000.84.3.1414. [DOI] [PubMed] [Google Scholar]
- Person AL, Raman IM. Deactivation of L-type Ca current by inhibition controls LTP at excitatory synapses in the cerebellar nuclei. Neuron. 2010;66:550–559. doi: 10.1016/j.neuron.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh JR, Raman IM. Potentiation of mossy fiber EPSCs in the cerebellar nuclei by NMDA receptor activation followed by postinhibitory rebound current. Neuron. 2006;51:113–123. doi: 10.1016/j.neuron.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Pugh JR, Raman IM. Mechanisms of potentiation of mossy fiber EPSCs in the cerebellar nuclei by coincident synaptic excitation and inhibition. J Neurosci. 2008;28:10549–10560. doi: 10.1523/JNEUROSCI.2061-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae MG, Irving AJ. Both mGluR1 and mGluR5 mediate Ca2+ release and inward currents in hippocampal CA1 pyramidal neurons. Neuropharmacology. 2004;46:1057–1069. doi: 10.1016/j.neuropharm.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Raman IM, Gustafson AE, Padgett D. Ionic currents and spontaneous firing in neurons isolated from the cerebellar nuclei. J Neurosci. 2000;20:9004–9016. doi: 10.1523/JNEUROSCI.20-24-09004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara Y, Westbrook GL. Modulation of calcium currents by a metabotropic glutamate receptor involves fast and slow kinetic components in cultured hippocampal neurons. J Neurosci. 1993;13:3041–3050. doi: 10.1523/JNEUROSCI.13-07-03041.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangrey T, Jaeger D. Analysis of distinct short and prolonged components in rebound spiking of deep cerebellar nucleus neurons. Eur J Neurosci. 2010;32:1646–1657. doi: 10.1111/j.1460-9568.2010.07408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Kim YS, Worley PF, Linden DJ. Depolarization-induced slow current in cerebellar Purkinje cells does not require metabotropic glutamate receptor 1. Neuroscience. 2009;162:688–693. doi: 10.1016/j.neuroscience.2009.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengel W, Jainz M, Andreas K. Different potencies of dihydropyridine derivatives in blocking T-type but not L-type Ca2+ channels in neuroblastoma-glioma hybrid cells. Eur J Pharmacol. 1998;342:339–345. doi: 10.1016/s0014-2999(97)01495-7. [DOI] [PubMed] [Google Scholar]
- Swartz KJ, Bean BP. Inhibition of calcium channels in rat CA3 pyramidal neurons by a metabotropic glutamate receptor. J Neurosci. 1992;12:4358–4371. doi: 10.1523/JNEUROSCI.12-11-04358.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadayonnejad R, Anderson D, Molineux ML, Mehaffey WH, Jayasuriya K, Turner RW. Rebound discharge in deep cerebellar nuclear neurons in vitro. Cerebellum. 2010;9:352–374. doi: 10.1007/s12311-010-0168-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telgkamp P, Raman IM. Depression of inhibitory synaptic transmission between Purkinje cells and neurons of the cerebellar nuclei. J Neurosci. 2002;22:8447–8457. doi: 10.1523/JNEUROSCI.22-19-08447.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uusisaari M, Obata K, Knöpfel T. Morphological and electrophysiological properties of GABAergic and non-GABAergic cells in the deep cerebellar nuclei. J Neurophysiol. 2007;97:901–911. doi: 10.1152/jn.00974.2006. [DOI] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. Long-term depression at the mossy fiber-deep cerebellar nucleus synapse. J Neurosci. 2006;26:6935–6944. doi: 10.1523/JNEUROSCI.0784-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Raman IM. Ca currents activated by spontaneous firing and synaptic disinhibition in neurons of the cerebellar nuclei. J Neurosci. 2009;29:9826–9838. doi: 10.1523/JNEUROSCI.2069-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]