

Abstract
DNA stable-isotope probing (DNA-SIP) is a powerful technique for identifying active microorganisms that assimilate particular carbon substrates and nutrients into cellular biomass. As such, this cultivation-independent technique has been an important methodology for assigning metabolic function to the diverse communities inhabiting a wide range of terrestrial and aquatic environments. Following the incubation of an environmental sample with stable-isotope labelled compounds, extracted nucleic acid is subjected to density gradient ultracentrifugation and subsequent gradient fractionation to separate nucleic acids of differing densities. Purification of DNA from cesium chloride retrieves labelled and unlabelled DNA for subsequent molecular characterization (e.g. fingerprinting, microarrays, clone libraries, metagenomics). This JoVE video protocol provides visual step-by-step explanations of the protocol for density gradient ultracentrifugation, gradient fractionation and recovery of labelled DNA. The protocol also includes sample SIP data and highlights important tips and cautions that must be considered to ensure a successful DNA-SIP analysis.
Protocol
1. Preparation of Reagents
DNA-SIP requires the use of reagents that should be prepared in advance of the actual procedure. The directions for preparing each reagent are listed in this section and are modified from a previous SIP protocol1.
Cesium chloride (CsCl) solution for preparing SIP gradients - Prepare a 7.163 M CsCl solution by gradually dissolving 603.0 g of CsCl in distilled and deionized water (ddH2O) to a final volume of 500 mL. Be careful not to exceed 500 mL! Warming the solution slightly while stirring will help dissolve all of the CsCl. Aliquot the final solution in sealed aliquots. In our lab, a common storage practice is to prepare 100-mL aliquots in 125-mL serum vials, which are then crimp-sealed with butyl rubber stoppers. The sealed aliquots can be stored indefinitely at room temperature (20°C). The seals help prevent evaporation and CsCl "crust" formation. Determine the density of the solution by weighing triplicate 100-μL aliquots, or by using a digital refractometer (e.g. Reichert AR200) that has been carefully calibrated for CsCl solutions. Once calibrated successfully, the Reichert AR200 is consistent and provides accurate readings for several years. At room temperature (20°C), the final density of this solution typically ranges from 1.88-1.89 g ml-1. The density varies slightly each time a new stock is prepared.
Cesium chloride solution for preparing gradients with ethidium bromide (EtBr) - Combine 250 g of CsCl with 250 mL of sterile ddH2O water. Aliquot this solution into separate serum vials that have been crimp-sealed with butyl rubber seals as described in 1.1.
Gradient Buffer - Combine 50 ml of 1 M Tris-HCl, 3.75 g KCl and 1 ml of 0.5 M EDTA to 400 ml of water. Dissolve the KCl, then add ddH2O to 500 ml. Filter-sterilize and autoclave. The final solution is 0.1 M Tris, 0.1 M KCl and 1 mM EDTA.
Polyethylene glycol (PEG) solution - Prepare the PEG solution by dissolving 150 g of polyethylene glycol 6000 and 46.8 g of NaCl in sterile ddH2O water to a total volume of 500 mL (30% PEG, 1.6 M NaCl). Autoclave.Note: This solution separates into two phases with autoclaving. Include a stir bar in the autoclaved bottle so that the solution can be properly mixed when this occurs.
TE Buffer - Prepare a solution of 10 mM Tris-HCl (pH 8.0) and 1 mM EDTA (pH 8.0) in sterile ddH2O water, using autoclaved stock solutions of 1 M Tris-HCl (pH 8.0) and 0.5 M EDTA (pH 8.0). Filter sterilize and autoclave.
70% Ethanol - Combine 350 ml of high purity ethanol with 150 ml of sterile ddH2O water.
2. Sample Incubation and DNA Extraction
For DNA-SIP incubations, samples are typically incubated with heavy-isotope carbon (13C) substrate. Incubation periods and conditions (e.g. nutrient supplementation, moisture, light) will vary depending on the type of sample that is incubated and the nature of the substrate. DNA-SIP experiments have been successfully performed using a variety of single carbon compounds 2,3, multi-carbon compounds 4,5,6, and using labelled nitrogen 7,8 or oxygen 9. However, a drawback to using 15N- or 18O-labelled compounds is the decreased physical separation of labelled nucleic acid, primarily due to the presence of fewer nitrogen and oxygen atoms in DNA and RNA relative to carbon atoms.
A critical control for DNA-SIP experiments is an identical incubation established with native (e.g. 12C) substrate. This incubation provides a subsequent comparison to ensure that any apparent labelling of nucleic acid was not an artifact of the ultracentrifugation or G+C content density differences in DNA contributing to separation 10. It is also important to keep frozen sample material for comparison to 'light' and 'heavy' DNA, and worth including a no-substrate control to assess background population changes throughout the SIP incubation.
Incubate environmental samples in microcosms containing labelled substrate (Figure 1). In our experience, we have found that a minimum incorporation of between 5-500 μmol of 13C carbon per gram of sample will be suitable for samples containing high biomass such as soil samples 1. For aquatic samples containing less biomass than soils, 1-100 μmol of incorporated 13C carbon per liter may yield a detectable heavy-isotopic signature 1. The amount of carbon amendment, proportion of carbon incorporated into biomass and requirement for supplemental nutrient addition for assimilation will all depend on the characteristics of the samples being analyzed and the targeted organisms of interest. A single set of sample incubation guidelines will not be applicable for all samples. Importantly, the substrate concentration used for SIP incubation should ideally be as close as possible to the concentration normally encountered in situ; experimental bias may be a consequence of enrichment culture conditions 10.
Following the incubation of sample with stable-isotope labelled substrate, extract DNA from microcosms using a rigorous extraction protocol (for PCR or small insert cloning) or a trusted enzymatic lysis for high-molecular weight cloning (e.g. large-insert metagenomics). RNA co-extraction does not generally affect analysis, so protocols that yield RNA as well as DNA may be used. The ultracentrifugation of the extracted DNA will not shear fragments shorter than ~50 kb pairs 1.
Quantify extracted DNA prior to setup of the CsCl gradient ultracentrifugation tubes. Quantify DNA using a spectrophotometer (e.g. Nanodrop 2000) if the extraction protocol yields only DNA (e.g. column-based kits). Alternatively, quantify using agarose gel electrophoresis.
3. Preparing Gradient Solutions for Ultracentrifugation
This procedure involves adding DNA to ultracentrifuge tubes. There are more than one type of tube and rotor so the exact protocol will vary and will depend on the manufacturer's instructions. That said, we recommend use of a vertical-well rotor to ensure maximum possible separation of light and heavy DNA. We use a Beckman-Coulter Vti 65.2 rotor with 16 wells for holding 5.1 ml QuickSeal Polyallomer tubes and the protocol will provide the steps and considerations for these conditions.
Using the DNA concentrations determined in step 2.3, calculate the required volume of extracted DNA that is required to provide 0.5 μg - 5 μg of DNA in the ultracentrifuge tubes.
Combine extracted DNA (0.5 - 5 μg) with Gradient Buffer (see step 1.3) and 4.8 ml of 7.163 M CsCl to a total volume of ~6 ml in a sterile disposable 15-ml tube. Note that the density of the CsCl solution can vary even at the same molarity (see step 1.1). The following equation can be used to determine the volume of Gradient Buffer/DNA mixture that is required to generate an appropriate mixing ratio: Gradient buffer and DNA solution volume (ml) = (CsCl stock solution density - desired final density) x volume of CsCl stock solution added x 1.52 Specify the volume of CsCl stock solution at 4.80 ml. The desired final density should be 1.725 g ml-1. The stock solution density was determined in step 1.1. Note also that the relative volumes of CsCl and Gradient Buffer/DNA will result in a combined volume of greater than 5.1 ml. Preparing volumes greater than the maximum volume capacity of the ultracentrifuge tubes (greater than 5.1 ml) will ensure that there is enough solution to completely fill the tube.
Mix by inverting 10 times. DNA is stable at room temperature in CsCl.
4. Creating an EtBr control Gradient (optional)
Because EtBr is an intercalating dye that complexes with DNA making it visible under UV light, control gradients containing EtBr are helpful because they provide immediate visual confirmation of gradient formation prior to fractionation of sample tubes (e.g. Figure 1). The inclusion of a control tube containing EtBr and a mixture of both 12C-DNA and 13C-DNA (or 14N-DNA and 15N-DNA) allows for immediate visualization of band formation within the tubes upon completion of ultracentrifugation. This is important because a ruptured tube during ultracentrifugation or improperly programmed run conditions can result in failed gradient formation. Bound to DNA, EtBr lowers the density of the DNA and as a result, a different protocol is followed to prepare gradients. Note that other nucleic acid stains can be used instead of EtBr 11 but the protocol will require optimization with other fluorophores.
The control gradient requires two volumes of genomic DNA: one fully labelled with stable-isotope and one without label. We typically use either Sinorhizobium meliloti cultured in media containing 13C- or 12C-glucose as the sole carbon source, or Methylococcus capsulatus strain Bath cultured in the presence of 13C- or 12C-methane as our controls.
Combine a 5 -10 μg quantity of both the 12C-DNA and 13C-DNA with Gradient Buffer to a final volume of 1.00 ml in a disposable 15-ml screw-cap tube.
Add 1.00 g of solid CsCl to the same tube. Mix by inversion.
Add 110 μl of a 10 mg ml-1 EtBr solution and 4.3 ml of a 1 g ml-1 CsCl stock solution to the same screw-cap tube used in step 4.2. The final density of the solution will approximate that of the original CsCl stock solution.
An additional "blank" control solution containing EtBr will also be required to counterbalance the solution created in step 4.4. Combine 1.00 mL of Gradient Buffer, 1.00 g of CsCl, 110 μl of a 10 mg ml-1 EtBr solution and 4.3 ml of a 1 g ml-1 CsCl stock solution in a separate 15 ml screw-cap tube and mix by inversion.
5. Ultracentrifugation
Using a bulb and Pasteur pipette, carefully fill ultracentrifuge tubes with gradient solutions prepared in step 3.2 (or steps 4.4 if preparing an EtBr control gradient). Carefully add the solutions to the tubes using a Pasteur pipette. Label the tubes on the tube shoulder with a fine permanent marker. CAUTION: Ensure that the tubes are filled exactly to the base of the tube neck. Insufficiently filled tubes are likely to burst during ultracentrifugation.
When all of the required tubes are filled with sample solutions, record the precise mass of each tube. Pair tubes and balance them to within 0-10 mg. For balancing, find nearly matched pairs and add or remove minute quantities of solution until they are balanced, keeping the solution level as close to the base of the tube necks as possible. Note that for weighing tubes, we use an inverted 15-ml screw-cap tube that has been cut in half as a tube holder for the balance.
Seal the tubes using a 'tube topper' according to the manufacturer's instructions.
Check that the tubes are sealed properly by inverting them and applying moderate pressure. Weigh the tubes again to check that they are still balanced after sealing to within 0-10 mg.
Check each rotor well carefully to ensure that the wells are clean and free of debris or dust that might puncture the tubes during ultracentrifugation.
Insert the tubes into the rotor with the balanced pairs opposite one another. Record the rotor location of each sample because the ultracentrifugation process can cause marker labels to be damaged or erased. Carefully seal the rotor wells as indicated by the manufacturer.
Load the rotor into the ultracentrifuge. Close the ultracentrifuge door and apply a vacuum. If using a Vti 65.2 rotor, set the rotation speed to 44,100 rpm (~177,000 x gav), the temperature at 20°C, and ultracentrifugation time for 36-40 hours. Select vacuum, maximum acceleration, and turn off the brake (ensures gradient not disrupted by deceleration). Note that turning off the brake will add an additional 1-2 hours to the run time. Also note that shorter run times may not achieve sufficient band resolution. Long ultracentrifugation runs are recommended, as they lead to greater resolution of distinct nucleic acid bands.
Immediately upon completion of the ultracentrifugation procedure, remove the rotor carefully. Avoiding any tilting or bumping of the rotor, gently remove tubes from the rotor to avoid disturbing the gradients within the tubes. In rare circumstances, a tube will burst during the run. If so, there is a chance that the gradients in the other tubes did not form properly. If a control gradient was included, check this tube carefully under UV light to confirm gradient formation. If the gradient has not formed properly in the control tube, it is best to repeat all of step 5. Note that the EtBr control tube and its blank control may be stored in the dark and reused for up to six months. Take care to clean the rotor carefully according to the manufacturer's instructions once the burst tube has been removed. Do not use metal brushes or abrasive cleaners to clean rotor wells in order to avoid scratches to the rotor wells! Rotor-specific brushes and cleaning solution can be purchased from Beckman.
6. Gradient Fractionation
There are two methods that are currently used to recover DNA from the ultracentrifuge tubes: fractionation and needle extraction. This protocol will only describe the process of extracting DNA using the fractionation technique. This is because for most SIP experiments, labelled DNA cannot be visualized with EtBr and must instead be detected by comparing equivalent light and heavy fractions from multiple sample tubes. A syringe pump is highly recommended to retrieve equal density gradient fractions from ultracentrifuge tubes. We use a BSP model infusion pump (Braintree Scientific Inc.). A low-flow peristaltic pump or an HPLC pump may also be used.
Fill a sterile 60-ml syringe with sterile ddH2O containing sufficient bromophenol blue dye to provide a dark blue color. Place the syringe on the loading arm of the syringe pump. Attach pump tubing fitted with a 23-gauge 1" needle and turn on the pump until some ddH2O has come through the end of the needle. Note that any air bubbles in this ddH2O supply will negatively affect the fractionation process.
Fix one of the ultracentrifugation tubes to a clamp stand. Ensure that the clamp is sufficiently tight to prevent the tube from being displaced but not such that pressure on the tube would cause a release of CsCl solution when the tube is pierced. Pierce the very bottom of the tube along the tube seam using a fresh 23 gauge 1" needle. For the best results, pierce the tube in a controlled, quick, and confident manner. This is very difficult to do well, practice several times before this is first attempted with sample tubes.
For each sample, prepare 12 sterile 1.5 ml microcentrifuge tubes with labels indicating the sample number and fraction (1-12; heavy to light). Using the needle attached to the pump tubing (step 6.1), pierce the top of the tube on the upper tube shoulder, along the seam. Collect the gradient solution using the microcentrifuge tubes. As performed for the bottom of the tube, pierce the tube in a quick and controlled manner. Practice beforehand and be very careful to use a controlled pulling motion to prevent the forced needle from passing through the tube and into a finger! Use a previously calibrated pump rate that will yield 12 x 425 μl fractions in 12 minutes (425 μl min-1).
Use a digital refractometer (e.g. Reichert AR200; recommended) or an analytical balance to check the density of fractions from one gradient to confirm proper gradient formation. You will need to use ~50 μl of sample for this test. We often include pure culture DNA in one tube (as described for preparing the EtBr control gradients) to serve as a control for fractionation and use this for density determination. Expect the densities to range from ~1.690-1.760 g ml-1, with a median density of ~1.725 g ml-1.
7. DNA Precipitation
Precipitate DNA from all fractions by first adding 20 μg of linear polyacrylamide as a carrier for precipitation. Mix by inversion. Add 2 volumes of PEG solution (see step 1) and mix by inversion. Note that a carrier for precipitation (e.g. glycogen or linear polyacrylamide) is critical for quantitative recovery of DNA from gradient factions, but caution should be used if glycogen is used as a carrier for precipitation for this protocol. Glycogen preparations have been shown to be contaminated with bacterial nucleic acid and contamination can easily confuse the interpretation of SIP gradient fractions 12.
Leave the tubes at room temperature for 2 hours to allow the DNA to precipitate. If desired, tubes can be left overnight at room temperature.
Centrifuge at 13,000 g for 30 minutes with the back of the tubes facing outwards for a consistent tube orientation in the rotor. Carefully aspirate and discard the supernatant. A pellet should be visible but can be very difficult to see at this stage. Work under a bright light source (e.g. desk lamp) to assist in visualizing the pellet.
Wash the pellet with 500 μl of 70% ethanol. Centrifuge at 13,000 g for 10 minutes. Carefully aspirate and discard the supernatant. The pellet will usually be more visible for this step, but will dissociate from the tube wall more easily.
Allow the pellet to dry at room temperature for 15 minutes.
Suspend each pellet in 50 μl of TE buffer (see step 1.5). Run 5 μl of each fraction on an agarose gel according to standard lab protocols.
8. Fraction Characterization
The method used to characterize gradient fractions to assess the success of a SIP incubation will vary depending on the lab and availability of equipment. Using a fingerprinting method for targeting the 16S rRNA gene is a common approach and methods such as terminal restriction fragment length polymorphism (T-RFLP) or denaturing gradient gel electrophoresis (DGGE) are appropriate (Figure 1). Following the protocol described above, expect the light DNA to be associated with fractions 9-11 (~1.705-1.720 g ml-1) and the heavy DNA fingerprints to be associated within fractions 5-8 (~1.720-1.735 g ml-1). Unique fingerprints associated with fractions 5-8 of stable-isotope incubated samples, but not with native-substrate incubated controls provides strong evidence linking specific organisms with the metabolism of particular labelled substrate. If insufficient labelled DNA remains for some applications (hybridization, metagenomics), multiple displacement amplification may be used to produce greater quantities 13-15 but this can introduce chimeras into the amplified DNA 14,16.
9. Results
Typical DNA-SIP results will demonstrate a separation of labelled and unlabelled DNA in the gradient formed by ultracentrifugation. Ideally, complete resolution of high molecular weight genetic material (e.g. 13C, 15N) from unlabelled materials will be achieved. Resolution can be witnessed visually by observing band formation in EtBr control tubes. The concentrations of retrieved genomic DNA contained in the individual gradient fractions may also be used to confirm proper gradient formation.
For this protocol, we include representative results of gradient ultracentrifugation performed using nucleic acid from two pure cultures (Figure 2). The fractionated gradient included here was prepared using genomic DNA extracted from S. meliloti (ATCC 1021), and 13C-labelled M. capsulatus str. Bath. Following ultracentrifugation, fractionation and DNA recovery, labelled and unlabelled genomic DNA separate into respective gradient fractions with differing densities (Figure 2A). Heavy-isotope labelled DNA can be observed in fractions 4-5, whereas unlabelled DNA is found at high concentrations in fractions 9-10. The DNA from each fraction was characterized with denaturing gradient gel electrophoresis 17 and the PCR-amplified products generated discrete banding patterns corresponding to the two organisms included in the gradient (Figure 2B). The density of the fractions ranged from ~1.580 - 1.759 g ml-1, and they are shown in order of decreasing density from left to right.
Although the separation of pure 13C- and 12C-DNA can be pronounced (Figure 2), environmental sample incubations may be more difficult to interpret. For example, we incubated tundra soils from Resolute Bay (Nunavut, Canada) with either 12C- or 13C-labelled glucose for a 14-day period at 15°C. The agarose gels of purified gradient fraction DNA demonstrated that genomic DNA was 'smeared' across fractions 7-10 for both 12C- and 13C-incubations (Figure 3A and 3C, respectively). In this case, 13C-enrichment of biomass from particular microbial taxa can only be determined with an approach such as DGGE of 16S rRNA genes. The 12C-glucose incubated soil DNA generates similar patterns across all gradient fractions (Figure 3B), but the 13C-glucose incubated sample generated DGGE fingerprints that are uniquely associated with fractions 5-8 (Figure 3D). Of particular interest are the conserved bands indicated by the arrows. This dominant 'phylotype' is consistent across all gradient fractions but shifts to heavier fractions for DNA obtained from 13C-glucose incubated soil. Subsequent DNA sequencing of this band and/or clone library analysis would confirm the identity of this particular 16S rRNA gene and guide subsequent metagenomic or cultivation-based approaches.
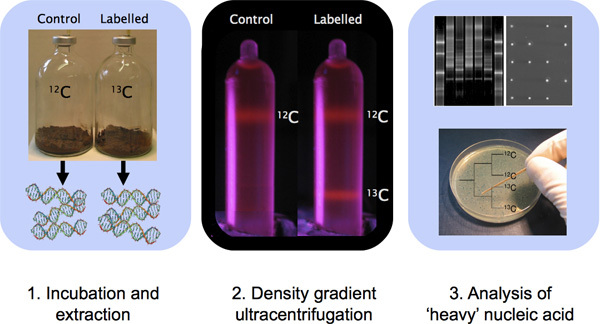 Figure 1. Outline of a DNA-SIP experiment involving sample incubation, DNA extraction, CsCl density gradient ultracentrifugation and DNA characterization with molecular techniques.
Figure 1. Outline of a DNA-SIP experiment involving sample incubation, DNA extraction, CsCl density gradient ultracentrifugation and DNA characterization with molecular techniques.
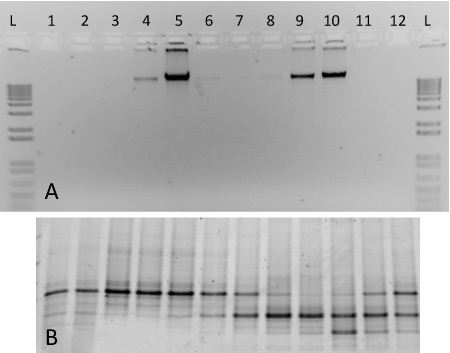 Figure 2. Expected results for a SIP gradient fractionation including DNA from two pure cultures. (A) Aliquots of DNA from gradient fractions 1-12 were run on a 1% agarose gel from a gradient containing 13C-labelled M. capsulatus strain Bath (fractions 4-6) and 12C-labelled S. meliloti (fractions 8-10). A 1-kb ladder is included for comparison (B) PCR-amplified DNA from the same fractions were run on a 10% DGGE gel. Fingerprint patterns reveal distinct differences between fractions 5 and 9, for example.
Figure 2. Expected results for a SIP gradient fractionation including DNA from two pure cultures. (A) Aliquots of DNA from gradient fractions 1-12 were run on a 1% agarose gel from a gradient containing 13C-labelled M. capsulatus strain Bath (fractions 4-6) and 12C-labelled S. meliloti (fractions 8-10). A 1-kb ladder is included for comparison (B) PCR-amplified DNA from the same fractions were run on a 10% DGGE gel. Fingerprint patterns reveal distinct differences between fractions 5 and 9, for example.
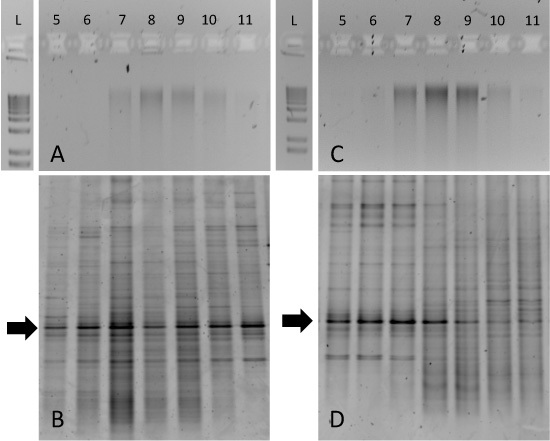 Figure 3. Expected results for SIP gradient fractionations from soil sample incubations. Aliquots of gradient fractions from both 12C-glucose amended soil (A) and 13C-glucose amended soil (C) were run on 1% agarose gels and a 1-kb ladder is included for comparison. Corresponding DGGE fingerprints for each of these samples are shown in (B) and (D). Fingerprinting of fractions reveals enrichment of particular bacterial taxa in the 13C-glucose amended sample in fractions 5-8 (D).
Figure 3. Expected results for SIP gradient fractionations from soil sample incubations. Aliquots of gradient fractions from both 12C-glucose amended soil (A) and 13C-glucose amended soil (C) were run on 1% agarose gels and a 1-kb ladder is included for comparison. Corresponding DGGE fingerprints for each of these samples are shown in (B) and (D). Fingerprinting of fractions reveals enrichment of particular bacterial taxa in the 13C-glucose amended sample in fractions 5-8 (D).
Discussion
Proper design of stable-isotope probing experiments is of critical importance for obtaining labelled DNA above the background unlabelled community. Considerations related to sample incubation times, substrate concentrations, incubation conditions (e.g. nutrients, soil moisture content), cross-feeding and replication have been discussed elsewhere 10,18 and we recommend the reader consult these publications when designing a SIP incubation. Related to the current protocol, it is worth commenting on additional considerations related to the interpretation of data from SIP gradients. Due to the nature of the ultracentrifugation process, it is additionally important to include controls such as pure cultures and native-substrate incubated samples to ensure that bands appearing or disappearing in particular fractions are not artifacts of the protocol itself. For example, DNA within an ultracentrifuge gradient may not be visible in an agarose gel (Figure 2A), but may still contaminate the full length of the gradient (Figure 2B). Although M. capsulatus patterns are most distinct in the dense fractions (5-7) of the gel shown in Figure 2B, the same DGGE pattern was still observed in the lightest fraction (12). With carefully considered controls, interpretation of SIP gradient fraction data is possible.
Due to the nature of some well-designed SIP experiments (e.g. near in situ substrate concentrations, short incubation times), isotope incorporation can be very low10. In addition, most microorganisms in terrestrial or aquatic environments have long generation times compared to growth in the laboratory, and require extended incubation times to reach detectable levels of isotopic enrichment. Other populations may be capable of metabolizing a variety of substrates, and may be not grow fully on labelled substrate. There are also communities (e.g. groundwater) that may be associated with low biomass levels and generate low yields of extracted nucleic acid. In all of these cases, the quantitative retrieval of labelled nucleic acids may be challenging.
To circumvent these limitations, a variety of natural and synthetic carrier molecules exist that assist in the precipitation and recovery of DNA from CsCl gradients. Carrier molecules can be biological in origin such as glycogen or DNA from an archaeal organism 19, or synthetic in nature, such as linear polyacrylamide. The benefit of using carrier molecules such as these when performing DNA-SIP is that they can enable visualization of bands in the CsCl gradients that would not normally be visible and ensure quantitative recovery of low DNA concentrations. Successful recovery of low nanogram amounts of DNA from CsCl gradients actually requires the use of a carrier molecule 1,12. Recent research has indicated that carrier molecules obtained from biological sources can often be contaminated with DNA from the source organism 12 and the results are very difficult to distinguish from patterns associated with 13C-labelled DNA (data not shown). Therefore it is recommended that synthetic carrier molecules such as linear polyacrylamide be used for DNA-SIP. In addition, the use of multiple-displacement amplification (MDA) can generate high fidelity yields of labelled DNA for downstream molecular analyses 13,14, although chimeras may be generated by the amplification and detected in downstream molecular analyses 14.
One of the most powerful applications of DNA-SIP that has yet to be fully exploited is the potential recovery of DNA from active community members for metagenomic library analysis. We expect that major advances in enzyme discovery will result from the integration of stable-isotope probing into existing metagenomic surveys from diverse terrestrial and aquatic environments. The protocol visualized here will produce labelled DNA of sufficient quality for these discovery-based applications.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by Strategic Project and Discovery Grants to J.D.N. from the Natural Sciences and Engineering Research Council of Canada (NSERC).
References
- Neufeld JD. DNA stable-isotope probing. Nat. Protocols. 2007;2:860–866. doi: 10.1038/nprot.2007.109. [DOI] [PubMed] [Google Scholar]
- Neufeld JD, Boden R, Moussard H, Schäfer H, Murrell JC. Substrate-specific clades of active marine methylotrophs associated with a phytoplankton bloom in a temperate coastal environment. Appl. Environ. Microbiol. 2009;74:7321–7328. doi: 10.1128/AEM.01266-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nercessian O, Noyes E, Kalyuzhnaya MG, Lidstrom ME, Chistoserdova L. Bacterial populations active in metabolism of C1 compounds in the sediment of Lake Washington, a freshwater lake. Appl. Environ. Microbiol. 2005;71:6885–6899. doi: 10.1128/AEM.71.11.6885-6899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan P. Respiration of 13C-labelled substrates added to soil in the field and subsequent 16S rRNA gene analysis of 13C-labelled soil DNA. Appl. Environ. Microbiol. 2003;69:1614–1622. doi: 10.1128/AEM.69.3.1614-1622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard L. Dynamics and identification of soil microbial populations actively assimilating carbon from 13C-labelled wheat residue as estimated by DNA- and RNA-SIP techniques. Environ. Microbiol. 2007;9:752–764. doi: 10.1111/j.1462-2920.2006.01197.x. [DOI] [PubMed] [Google Scholar]
- Haichar elZahar, F Identification of cellulolytic bacteria in soil by stable isotope probing. Environ. Microbiol. 2007;9:625–634. doi: 10.1111/j.1462-2920.2006.01182.x. [DOI] [PubMed] [Google Scholar]
- Addison S, McDonald I, Lloyd-Jones G. Stable isotope probing: Technical considerations when resolving 15N-labelled RNA in gradients. J. Microbiol. Meth. 2009;80:70–75. doi: 10.1016/j.mimet.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Buckley DH, Huangyutitham V, Hsu S-F, Nelson TA. Stable isotope probing with 15N achieved by disentangling the effects of genome G + C content and isotope enrichment on DNA density. Appl. Environ. Microbiol. 2007;73:3189–3195. doi: 10.1128/AEM.02609-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz E. Characterization of growing microorganisms in soil by stable isotope probing with H218O. Appl. Environ. Microbiol. 2007;73:2541–2546. doi: 10.1128/AEM.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld JD, Dumont MG, Vohra J, Murrell JC. Methodological considerations for the use of stable isotope probing in microbial ecology. Microb. Ecol. 2007;53:435–442. doi: 10.1007/s00248-006-9125-x. [DOI] [PubMed] [Google Scholar]
- Martineau C, Whyte L, Greer C. Development of a SYBR safe technique for the sensitive detection of DNA in cesium chloride density gradients for stable isotope probing assays. J. Microbiol. Meth. 2008;73:199–202. doi: 10.1016/j.mimet.2008.01.016. [DOI] [PubMed] [Google Scholar]
- Bartram AK, Poon C, Neufeld JD. Nucleic acid contamination of glycogen used in nucleic acid precipitation and assessment of linear polyacrylamide as an alternative co-precipitant. Biotechniques. 2009;47:1019–1022. doi: 10.2144/000113276. [DOI] [PubMed] [Google Scholar]
- Chen Y. Revealing the uncultivated majority: combining DNA stable-isotope probing, multiple displacement amplification and metagenomic analyses of uncultivated Methylocystis in acidic peatlands. Environ. Microbiol. 2008;10:2609–2622. doi: 10.1111/j.1462-2920.2008.01683.x. [DOI] [PubMed] [Google Scholar]
- Neufeld JD, Chen Y, Dumont MG, Murrell JC. Marine methylotrophs revealed by stable-isotope probing, multiple displacement amplification and metagenomics. Environ. Microbiol. 2008;10:1526–1535. doi: 10.1111/j.1462-2920.2008.01568.x. [DOI] [PubMed] [Google Scholar]
- Kalyuzhnaya M. High-resolution metagenomics targets specific functional types in complex microbial communities. Nat. Biotechnol. 2008;26:1029–1034. doi: 10.1038/nbt.1488. [DOI] [PubMed] [Google Scholar]
- Binga EK, Lasken RS, Neufeld JD. Something from (almost) nothing: the impact of multiple displacement amplification on microbial ecology. ISME J. 2008;2:233–241. doi: 10.1038/ismej.2008.10. [DOI] [PubMed] [Google Scholar]
- Green SJ, Leigh MB, Neufeld JD. In: Microbiology of Hydrocarbon and Lipid Microbiology. Timmis KN, editor. Berlin Heidelberg: Springer-Verlag; 2010. pp. 4137–4158. [Google Scholar]
- Neufeld JD, Wagner M, Murrell JC. Who eats what, where and when? Isotope-labelling experiments are coming of age. ISME J. 2007;1:103–110. doi: 10.1038/ismej.2007.30. [DOI] [PubMed] [Google Scholar]
- Gallagher E, McGuinness L, Phelps C, Young LY, Kerkhof LJ. DNA shortens the incubation time needed to detect benzoate-utilizing denitrifying bacteria by stable-isotope probing. Appl. Environ. Microbiol. 71:5192–5196. doi: 10.1128/AEM.71.9.5192-5196.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]