

Abstract
Allenylsilanes are used as carbon nucleophiles in highly stereoselective Lewis acid-promoted C-glycosidations, resulting in the introduction of an internal alkyne with an adjacent stereocenter. Both achiral and chiral allenylsilanes form the desired products with high diastereoselectivity, where the nucleophile adds exclusively to the α-face of the intermediate oxonium ion. Reactions with glucal and galactal afford dihydropyran products, while reactions with a ribose derivative yield dihydrofuran products.
The Ferrier glycal allylic rearrangement allows for the selective modification of complex carbohydrates.1 Glycosides bearing a C-glycosidic bond are important building blocks for synthetic chemistry, since they can be subunits of biologically active natural products, or potential inhibitors of enzymes that use carbohydrates as substrates.2
Organosilane reagents have proven to be versatile carbon nucleophiles for the modification and functionalization of carbohydrates.3 These reactions favor addition to the α-face to the sugar, resulting in an axial orientation of the new carbon bond.
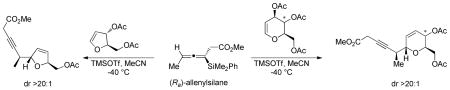
Danishefsky’s initial report on the C-glycosidation of glycals with allyltrimethylsilane documented that the nucleophile approached the oxonium ion predominantly from the α-face.4 When chiral crotylsilane reagents were used a double stereodifferention was observed, wherein the stereochemistry of the silane nucleophile affected the diastereomeric ratio of the C-glycosidation products (Scheme 1).5
Scheme 1.

Additions of Silane Nucleophiles to Glucal
Recently allenylsilanes have reemerged as an important class of carbon nucleophiles. These allenes have demonstrated their versatility in nucleophilic additions to oxonium and iminum ions, leading to the stereospecific formation of functionalized alkynes.6 Despite the recent advances exploring the synthesis and reactivity of allenylsilanes, there are no reports of these nucleophiles (or similar allenylmetal reagents) in C-glycosidation reactions. Herein we report an efficient and highly stereoselective C-glycosidation of glycals with allenylsilanes, forming glycosides containing an internal alkyne.7
We have recently reported the multigram synthesis of both enantiomers of allenylsilane 1.6d The C-glycosidations of tri-O-acetyl-D-glucal with allenylsilanes (Ra)−1 and (Sa)−1, mediated by TMSOTf in MeCN8, gave the desired α-C-glycoside products in good yields as single diastereomers (Scheme 2). Both the (Ra) and (Sa) enantiomers display exceptional face selectivity, as the axial chirality of the allene overrides the inherent chirality of the glycal. In other words, the “matched” or “mismatched” reaction partners, which was observed with chiral crotylsilanes, was not observed with the allenes.5 The relative and absolute stereochemistry of the products was assigned based on comparison to known products, confirming the expected α-addition to the carbohydrate.9
Scheme 2.

Additions of Enantioenriched Allenylsilanes to Tri-O-acetyl-D-glucal and galactala
a.Reaction conditions: TMSOTf (1.0 equiv) was added to a solution of allenylsilane (1.0 equiv) and carbohydrate (1.2 equiv) in MeCN (0.5 M) at −40 °C and stirred for 1 hour. b. Isolated yields after chromatographic purification. Diastereomeric ratios determined by 1H NMR analysis of crude material.
Enantioenriched allenylsilanes 1 also underwent C-glycosidation reactions with tri-O-acetyl-D-galactal, providing the diastereomeric dihydropyran products in slightly lower yield than the analogous glucal additions (Scheme 2). As before, the products were formed as a single observed diastereomer, with both allene enantiomers exibiting similar levels of diastereoselectivity. However, it is interesting to note that the Sa-enantiomer provided lower yields in both additions, so it is possible that the “mismatched” enantiomer is less reactive than the “matched” counterpart. The relative and absolute stereochemistry of the products were assigned by analogy to known products.9
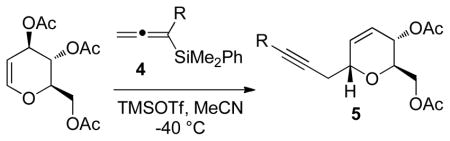
Achiral allenylsilanes 4a–4c were prepared using a Fleming SN2′ displacement of the appropriate propargyl mesylate,10 while 4d was obtained by a Johnson orthoester Claisen rearrangement.6g These achiral allenylsilanes underwent C-glycosidation with tri-O-acetyl-D-glucal, giving the desired dihydropyrans in moderate to high yield (Table 1). The products of these reactions were again formed as a single diastereoisomer, with preferential addition to the α-face.
Table 1.
Additions of Achiral Allenylsilanes to Tri-O-acetyl-D-glucal
 | ||||
|---|---|---|---|---|
| Allene | R | Yielda | drb | product |
| 4a | Me | 93 | >20:1 | 5a |
| 4b | Et | 88 | >20:1 | 5b |
| 4c | Ph | 54 | >20:1 | 5c |
| 4d | CH2CO2Me | 65 | >20:1 | 5d |
Isolated yield after chromatographic purification.
Diastereomeric ratios determined by 1H NMR analysis of crude material.
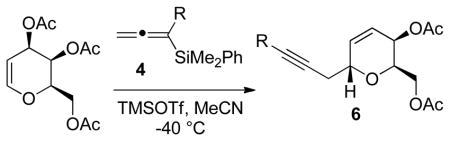
Achiral allenylsilanes 4a–4d also provided the desired C-glycosidation adducts when added to tri-O-acetyl-D-galactal in the presence to TMSOTf (Table 2). The galactal-derived products were isolated in slightly lower yields than the corresponding glucal products, but the desired pyran diastereomer was the exclusive product in all cases.
Table 2.
Additions of Achiral Allenylsilanes to Tri-O-acetyl-D-galactal
 | ||||
|---|---|---|---|---|
| Allene | R | Yielda | drb | product |
| 4a | Me | 82 | >20:1 | 6a |
| 4b | Et | 86 | >20:1 | 6b |
| 4c | Ph | 39 | >20:1 | 6c |
| 4d | CH2CO2Me | 63 | >20:1 | 6d |
Isolated yield after chromatographic purification.
Diastereomeric ratios determined by 1H NMR analysis of crude material.
While C-glycosidation reactions with commercially available glucal and galactal have been well developed, there are fewer examples that utilize furanose derivitives as the electrophile.11 While bis-O-acetyl-D-ribose derivitive 7 is a known compound, previous syntheses report that it is unstable and readily decomposes during synthesis. Consequently, it has not been used as an electrophiis reaction partner in C-glycosidations.12 Herein we describe a modified and reproducible procedure for the synthesis of furanose 7 in 3 steps from D-ribose (Scheme 3). While the product yield is moderate (33% over 3 steps), the material is stable to chromatographic purification, and can be fromed from readily available starting materials.
Scheme 3.

Synthesis of Dihydrofuran 7
a.Isolated yield after chromatographic purification.
C-glycosidation reactions of 2,3-dihydrofuran 7 with both enantiomers of allenylsilane 1 provided the desired trans-dihydrofuran products in moderate yields (Scheme 4). These reactions displayed excellent diastereoselectivity as the isolated products were diastereomerically pure when either allene enantiomer was employed.
Scheme 4.

Additions of Chiral Allenysilanes to Dihydrofuran 7
a.Isolated yield after chromatographic purification. Diastereomeric ratios determined by 1H NMR analysis of crude material.
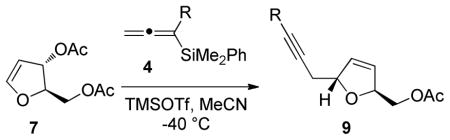
Reactions with achiral allenylsilanes and 2,3-dihydrofuran 7 also resulted in the formation of the desired 3,4-dihydrofuran products in moderate to high yield (Table 3). All of the cases examined exhibited very high diastereoselectivity, further demonstrating the utility of this electrophile as a route to the stereoselective formation of functionalized 2,5-trans-dihydrofurans. The stereochemistry of the products were assigned based on 2D NMR studies.9
Table 3.
Additions of Achiral Allenylsilanes to Dihydrofuran 7
 | ||||
|---|---|---|---|---|
| Allene | R | Yielda | drb | product |
| 4a | Me | 93 | >20:1 | 9a |
| 4b | Et | 88 | >20:1 | 9b |
| 4c | Ph | 45 | >20:1 | 9c |
| 4d | CH2CO2Me | 54 | >20:1 | 9d |
Isolated yield after chromatographic purification.
Diastereomeric ratios determined by 1H NMR analysis of crude material.
In conclusion, we have reported the stereoselective C-glycosidation of glycal derivitives with both achiral and enantioenriched allenylsilanes. The reactions all proceed with moderate to high yield with excellent diastereoselectivity, with complete addition to the α-face of the oxonium ion regardless of the nucleophile. The products of these glycosidations will be exploited as building blocks for complex molecules and library synthesis of biologically relavant compounds.
Supplementary Material
Acknowledgments
The authors would like to thank Paul Ralifo (Boston University) for assistance with structural assignments. RAB is grateful to AstraZeneca for a 2009–2010 graduate fellowship. Financial support was provided by the National Institutes of Health (NIH CA53604).
Footnotes
Supporting Information Available Experimental data and selected spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ferrier RJ. Top Curr Chem. 2001;215:153–175. [Google Scholar]
- 2.(a) Danishefsky SJ, Bilodeau MT. Angew Chem Int Ed Engl. 1996;35:1380–1419. [Google Scholar]; (c) Faul MM, Huff BE.Chem Rev 20001002047–2060.11749283 [Google Scholar]; (d) Štambasky J, Hocek M, Kočovskŷ P. Chem Rev. 2009;109:6729–6764. doi: 10.1021/cr9002165. [DOI] [PubMed] [Google Scholar]
- 3.(a) Lewis MD, Cha JK, Kishi Y. J Am Chem Soc. 1982;104:4976–4978. [Google Scholar]; (b) Panek JS, Sparks MA. J Org Chem. 1989;54:2034–2038. [Google Scholar]; (c) Larsen CH, Ridgway BH, Shaw JT, Woerpel KA. J Am Chem Soc. 1999;121:12208–12209. [Google Scholar]; (d) Romero JAC, Tobacco SA, Woerpel KA. J Am Chem Soc. 2000;122:168–169. [Google Scholar]
- 4.(a) Danishefsky SJ, Kerwin JF. J Org Chem. 1982;47:3803–3805. [Google Scholar]; (b) Danishefksy SJ, Armistead DM, Wincott FE, Selnick HG, Hungate R. J Am Chem Soc. 1987;109:8117–8119. [Google Scholar]
- 5.Panek JS, Schaus JV. Tetrahedron. 1997;53:10971–10982. [Google Scholar]
- 6.(a) Danheiser RL, Carini DJ. J Org Chem. 1980;45:3927–3929. [Google Scholar]; (b) Danheiser RL, Carini DJ, Kwasigroch CA. J Org Chem. 1986;51:3870–3878. [Google Scholar]; (c) Marshall JA, Maxson K. J Org Chem. 2000;65:630–633. doi: 10.1021/jo991543y. [DOI] [PubMed] [Google Scholar]; (d) Brawn RA, Panek JS. Org Lett. 2007;9:2689–2692. doi: 10.1021/ol070936d. [DOI] [PubMed] [Google Scholar]; (e) Felzmann W, Castagnolo G, Rosenbeiger D, Mulzer J. J Org Chem. 2007;72:2182–2186. doi: 10.1021/jo062502m. [DOI] [PubMed] [Google Scholar]; (f) Brawn RA, Panek JS. Org Lett. 2009;11:4362–4365. doi: 10.1021/ol901692n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Brawn RA, Welzel M, Lowe JT, Panek JS. Org Lett. 2010;12:336–339. doi: 10.1021/ol902681t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Marchart S, Gromov A, Mulzer J. Angew Chem Int Ed. 2010;49:2050–2053. doi: 10.1002/anie.200906453. [DOI] [PubMed] [Google Scholar]
- 7.For the synthesis of C-glycosides with an allene or alkyne functionality see: Ichikawa Y, Isobe M, Goto T. Tetrahedron Lett. 1984;25:5049–5052.Ichikawa Y, Isobe M, Konobe M, Goto T. Carbohydr Res. 1987;171:193–199.Tsukiyama S, Isobe M. Tetrahedron Lett. 1992;33:7911–7914.Saeeng R, Isobe M. Org Lett. 2005;7:1585–1588. doi: 10.1021/ol050265o.Vieira AS, Fiorante PF, Hough TLS, Ferreira FP, Lüdtke DS, Stefani HA. Org Lett. 2008;10:5215–5218. doi: 10.1021/ol8022177.
- 8.Reactions carried out in other solvents (DCM, THF, toluene) gave poor yields.
- 9.See supporting information for assignment of relative and absolute stereochemistry.
- 10.(a) Fleming I, Terrett NK. J Organomet Chem. 1984;264:99–118. [Google Scholar]; (b) Fleming I, Newton TW. J Chem Soc, Perkin Trans. 1984;1:1805–1808. [Google Scholar]
- 11.For some examples of C-glycosidation reactions with furanose derivitives see: (a) Ref. 1d. (b) Ref. 3c. Cameron MA, Cush SB, Hammer RP. J Org Chem. 1997;62:9065–9069.Singh I, Seitz O. Org Lett. 2006;8:4319–4322. doi: 10.1021/ol061701p.
- 12.Strauss CR, Scott JL, Saylik D, Malic N. Resolution of chiral alcohols via transacetalization with enantiomerically pure chiral auxiliaries. 2005070911 A1. PCT Int Appl WO. 2005 Aug 4;
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.