

Abstract
RNA transcripts are subjected to post-transcriptional gene regulation by interacting with hundreds of RNA-binding proteins (RBPs) and microRNA-containing ribonucleoprotein complexes (miRNPs) that are often expressed in a cell-type dependently. To understand how the interplay of these RNA-binding factors affects the regulation of individual transcripts, high resolution maps of in vivo protein-RNA interactions are necessary1.
A combination of genetic, biochemical and computational approaches are typically applied to identify RNA-RBP or RNA-RNP interactions. Microarray profiling of RNAs associated with immunopurified RBPs (RIP-Chip)2 defines targets at a transcriptome level, but its application is limited to the characterization of kinetically stable interactions and only in rare cases3,4 allows to identify the RBP recognition element (RRE) within the long target RNA. More direct RBP target site information is obtained by combining in vivo UV crosslinking5,6 with immunoprecipitation7-9 followed by the isolation of crosslinked RNA segments and cDNA sequencing (CLIP)10. CLIP was used to identify targets of a number of RBPs11-17. However, CLIP is limited by the low efficiency of UV 254 nm RNA-protein crosslinking, and the location of the crosslink is not readily identifiable within the sequenced crosslinked fragments, making it difficult to separate UV-crosslinked target RNA segments from background non-crosslinked RNA fragments also present in the sample.
We developed a powerful cell-based crosslinking approach to determine at high resolution and transcriptome-wide the binding sites of cellular RBPs and miRNPs that we term PAR-CliP (Photoactivatable-Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation) (see Fig. 1A for an outline of the method). The method relies on the incorporation of photoreactive ribonucleoside analogs, such as 4-thiouridine (4-SU) and 6-thioguanosine (6-SG) into nascent RNA transcripts by living cells. Irradiation of the cells by UV light of 365 nm induces efficient crosslinking of photoreactive nucleoside-labeled cellular RNAs to interacting RBPs. Immunoprecipitation of the RBP of interest is followed by isolation of the crosslinked and coimmunoprecipitated RNA. The isolated RNA is converted into a cDNA library and deep sequenced using Solexa technology. One characteristic feature of cDNA libraries prepared by PAR-CliP is that the precise position of crosslinking can be identified by mutations residing in the sequenced cDNA. When using 4-SU, crosslinked sequences thymidine to cytidine transition, whereas using 6-SG results in guanosine to adenosine mutations. The presence of the mutations in crosslinked sequences makes it possible to separate them from the background of sequences derived from abundant cellular RNAs.
Application of the method to a number of diverse RNA binding proteins was reported in Hafner et al.18
Keywords: Cellular Biology, Issue 41, UV crosslinking, RNA binding proteins, RNA binding motif, 4-thiouridine, 6-thioguanosine
Protocol
The protocol below describes the PAR-CliP procedure for HEK293 cells expressing FLAG/HA-tagged IGF2BP1 upon induction with doxycycline. We will use an anti-FLAG antibody for immunoprecipitation.
PAR-CliP will work with any cell line expressing detectable levels of the endogenous, untagged RNA binding protein (RBP) of interest if an efficient antibody for immunoprecipitation is available.
Expanding Cells
Expand FlpIn-HEK293/TO/FLAG/HA-IGF2BP1 cells in growth medium. We recommend using between 100-400 x 106 cells (approx. 10-40 15 cm cell culture plates) as a starting point. Grow them to approximately 80% confluency.
14 h before crosslinking add a) 4-thiouridine to a final concentration of 100 μM (1:1000 v/v of a 1 M 4-thiouridine stock solution) directly to the cell culture medium and b) induce expression of the FLAG/HA tagged IGF2BP1 by addition of 1 μg/ml of doxycycline (1:10,000 v/v of 10 mg/ml doxycycline stock solution). NOTE: instead of 4-thiouridine you can also use 100 μM of 6-thioguanosine.
UV-Crosslinking
Wash cells once with 10 ml ice-cold PBS per plate and remove PBS completely.
Place plates on a tray with ice and irradiate uncovered with 0.15 J/cm2 of 365 nm UV light in a Stratalinker 2400 (Stratagene) or similar device.
Scrape cells off with a rubber policeman in 1 ml PBS per plate, transfer to 50 ml centrifugation tubes and collect by centrifugation at 500 x g for 5 min at 4°C and discard the supernatant. 100 x 106 HEK293 cells (10 15 cm plates) will yield approx. 1 ml of wet cell pellet.
(optional) Unless you want to continue directly with cell lysis, shock freeze the cell pellet in liquid nitrogen and store at -80°C. Cell pellets can be stored for at least 12 months.
Cell lysis and RNaseT1 digest
Take up cell pellet of crosslinked cells in 3 volumes of 1x NP40 lysis buffer and incubate on ice for 10 min.
Clear cell lysate by centrifugation at 13,000 x g for 15 min at 4°C.
Clear the lysate further by filtering it through a 0.2 μm membrane syringe filter (Pall Acrodisc or equivalent).
Add RNase T1 (Fermentas, 10,000 U/μl) to a final concentration of 1 U/μl and incubate in a water bath for 15 min at 22°C. Cool reaction subsequently for 5 min on ice before proceeding.
Immunoprecipitation and recovery of crosslinked target RNA fragments
Using the magnetic separator
Follow these guidelines throughout the sample preparation to prevent the magnetic beads from drying out.
Place the tube containing the beads on the magnetic stand for 1 2 minutes.
Add the buffer to the tube while the tube is on the magnetic separator.
Cap the tube, remove it from the magnetic separator, and resuspend the beads. You can resuspend the beads by flicking the tube with your finger or use a vortexer set at 5 6.
Centrifuge briefly to collect any beads that may remain in the tube cap.
Repeat steps 1 through 4 as required.
Preparation of magnetic beads
Transfer 10 μl of Dynabeads Protein G magnetic particles (Invitrogen) per ml cell lysate (for a typical experiment it should be approx. 40 50 μl of beads) to a 1.5 ml microfuge tube. Wash beads twice with 1 ml of citrate-phosphate buffer.
Resuspend in twice the volume of citrate-phosphate buffer relative to the original volume of bead suspension.
Add 0.25 μg of anti-FLAG M2 monoclonal antibody (Sigma) per ml suspension and incubate on a rotating wheel for 40 min at room temperature.
Wash beads twice in 1 ml of citrate-phosphate buffer to remove unbound antibody.
Resuspend beads in twice the volume of citrate-phosphate buffer relative to the original volume of bead suspension.
Immunoprecipitation (IP), second RNase T1 digestion, and dephosphorylation
Add 20 μl of freshly prepared antibody-conjugated magnetic beads per ml of partial RNase T1 treated cell lysate and incubate in 15 ml centrifugation tubes on a rotating wheel for 1 h at 4°C.
Collect magnetic beads on a magnetic particle collector for 15 and 50 ml centrifugation tubes (Invitrogen) and transfer to 1.5 ml microfuge tubes.
Wash beads 3 times in 1 ml of IP wash buffer.
Add RNaseT1 (Fermentas, 10,000 U/μl) to a final concentration of 100 U/μl and incubate the bead suspension in a water bath for 15 min at 22 °C. Cool subsequently on ice for 5 min.
Wash beads 3 times in 1 ml of high-salt wash buffer.
Resuspend beads in 1 volume of dephosphorylation buffer
Add calf intestinal alkaline phosphatase (CIAP, NEB) to a final concentration of 0.5 U/μl, and incubate the suspension for 10 min at 37°C.
Wash beads twice in 1 ml of phosphatase wash buffer
Wash beads twice in polynucleotide kinase (PNK) buffer without DTT (the DTT concentration necessary for the enzymatic reaction is high enough to damage the magnetic beads).
Resuspend beads in one original bead volume of PNK buffer
Radiolabeling of RNA segments crosslinked to immunoprecipitated proteins
To the bead suspension described above, add Υ-32P-ATP to a final concentration of 0.5 μCi/μl and T4 PNK (NEB) to 1 U/μl in one original bead volume. Incubate the suspension for 30 min at 37°C.
Add non-radioactive ATP to obtain a final concentration of 100 μM and incubate for another 5 min at 37°C.
Wash the magnetic beads 5 times with 800 μl of PNK buffer without DTT.
Resuspend the beads in 70 μl of SDS-PAGE loading buffer.
SDS-PAGE and electroelution of crosslinked RNA-protein complexes from gel slices
Incubate the radiolabeled suspension for 5 min in a heat block at 95°C to denature and release the immunoprecipitated RBP with crosslinked RNA and vortex.
Remove the magnetic beads on the separator and transfer the supernatant to a clean 1.5 ml microfuge tube.
Load 40 μl of the supernatant per well of a Novex Bis-Tris 4-12% (Invitrogen) precast polyacrylamide gel and run the gel for 55 min at 200 V.
Disassemble the gel chamber and dismantle the gel, leaving it mounted on one plate. To facilitate the alignment of the gel to the phosphorimager paper printout, we recommend implanting three tiny radioactive gel pieces asymmetrically at three of the four corners of the gel. Radioactive gel pieces can be collected from gels that were previously used to purify radiolabeled synthetic oligonucleotides. Wrap the gel in plastic film (e.g. Saran wrap) to avoid contamination.
Expose the gel to a blanked phosphorimager screen for 1 h and visualize on a phosphorimager.
Align the gel on top of the phosphorimager printout using the implanted gel pieces for orientation. Cut out the bands that correspond to the expected size of RBP (IGF2BP1, approx. 75 kDa) and transfer to a D-Tube Dialyzer Midi Tube and add 800 μl 1x SDS running buffer.
Electroelute the crosslinked RNA-RBP complex in 1x SDS running buffer at 100 V for 2 h. excised from the gel and electroeluted in a D-Tube Dialyzer Midi (Novagen) in 800 μl SDS running buffer according to the instructions of the manufacturer.
Proteinase K digestion
Add an equal volume of 2x Proteinase K Buffer with respect to the electroeluate, followed by the addition of Proteinase K (Roche) to a final concentration of 1.2 mg/ml. Incubate for 30 min at 55°C.
Recover the RNA by acidic phenol/chloroform/IAA extraction (25:24:1, pH 4.0) followed by a chloroform extraction. Add 1 μl of glycogen (10 mg/ml stock) precipitate the RNA by adding 3 volumes of ethanol. Dissolve the pellet in 10.5 μl water.
cDNA library preparation and deep sequencing
Carry the recovered RNA through a standard cDNA library preparation protocol originally described for cloning of small regulatory RNAs 19. The first step, 3' adapter ligation, was carried out as described on a 20 μl scale using 10.5 μl of the recovered RNA. Use the Solexa sequencing adapter sets described. Depending on the amount of RNA recovered, 5'-adapter-3'-adapter products without inserts may be detected after amplification of the cDNA as additional PCR bands. In such cases, excise the longer PCR product of expected size from a 3% NuSieve low-melting point agarose gel, elute the PCR product from the gel pieces using the GelElute kit (Qiagen) and sequence using the Solexa technology. One Solexa sequencing run usually affords between 6 and 10 million sequence reads that are enough for a transcriptome wide coverage of the binding sites of RNA binding proteins.
Bioinformatic analysis
Careful bioinformatic analysis of the sequence reads needs to be done to obtain meaningful insights into the RNA binding sites for the examined RBP, such as the RNA recognition element, the preferred binding regions the RBP has (exonic vs. intronic, coding sequence vs. untranslated sequence). The sequence reads need to be aligned against the genome and EST databases. We usually use reads mapping uniquely to the genome with up to one mismatch, insertion or deletion to build clusters of sequence reads that can then be further analyzed. The frequency of characteristic mutations in the clustered sequenced reads, T to C transitions when using 4-SU and G to A transitions when using 6-SG, are indicative of successfully crosslinked sequences. In our experience uncrosslinked RNAs labeled with 4-SU show a background mutation rate of approximately 20%. This rate is increases to approx. 50-80% upon crosslinking.
A detailed description of the bioinformatic analysis can be found in the Supplementary material of the publication by Hafner et al.18
Optional Steps
Determination of incorporation levels of 4-thiouridine into total RNA
Isolate total RNA from the cell line stably expressing the RBP of interest after growing in medium supplemented with 100 μM 4SU 16 h prior to harvest. As a control, harvest cells grown without 4SU addition. Isolate total RNA by addition of 3 volumes of Trizol reagent (Sigma) to the washed cell pellets following the manufacturer s instructions. was Further purify total RNA using Qiagen RNeasy according to the manufacturer's protocol. To prevent oxidization of 4SU during RNA isolation and analysis, add 0.1 mM dithiothreitol (DTT) to the wash buffers and subsequent enzymatic steps. Digest and dephosphorylated total RNA to single nucleosides for HPLC analysis as described before 20. Briefly, in a 30 μl volume, incubate 40 μg of purified total RNA were for 16 h at 37°C with 0.4 U bacterial alkaline phosphatase (Worthington Biochemical) and 0.09 U snake venom phosphodiesterase (Worthington Biochemical). As a reference standard, use a synthetic 4SU-labeled RNA, (we standardly use CGUACGCGGAAUACUUCGA(4SU)U) and also subject it to complete enzymatic digestion. Separate the resulting mixtures of ribonucleosides by HPLC on a Supelco Discovery C18 (bonded phase silica 5 μM particle, 250 x 4.6 mm) reverse phase column (Bellefonte PA, USA). HPLC buffers are 0.1 M TEAA in 3% acetonitrile (A) and 90% acetonitrile in water (B). Use an isocratic gradient: 0% B for 15 min, 0 to 10 % B for 20 min, 10 to 100% B for 30 min. Apply a 5 min 100 % B wash applied between runs to clean the HPLC column.
Representative Results
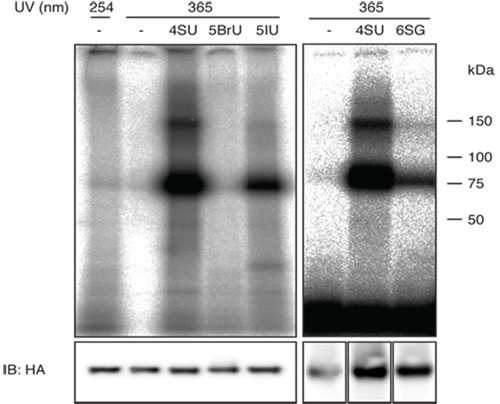 Figure 1 (right panel) shows a representative result of a PAR-CliP performed with cell lines expressing FLAG/HA-tagged IGF2BP1 with 4-SU and 6-SG. Note that the crosslinking efficiency of 6-SG for IGF2BP1 is lower than the crosslinking efficiency for 4-SU. The lower crosslinking efficiency will result in a higher background of sequences derived from fragments of abundant cellular RNAs and therefore you should consider scaling up the experiment when using less efficient photoreactive nucleosides.
Figure 1 (right panel) shows a representative result of a PAR-CliP performed with cell lines expressing FLAG/HA-tagged IGF2BP1 with 4-SU and 6-SG. Note that the crosslinking efficiency of 6-SG for IGF2BP1 is lower than the crosslinking efficiency for 4-SU. The lower crosslinking efficiency will result in a higher background of sequences derived from fragments of abundant cellular RNAs and therefore you should consider scaling up the experiment when using less efficient photoreactive nucleosides.
The left panel of Figure 1 shows a comparison of using different photoreactive uridine analogs that could be potentially used for PAR-CliP compared to traditional UV 254 nm crosslinking.
The intensity of the radioactive band of the correct length gives you a good idea whether the PAR-CliP experiment has worked and you have isolated sufficient RNA to carry through a small RNA sequencing protocol (step-by-step description for cDNA library preparation of small RNAs sequencing can be found in 19). The frequency of characteristic mutations in the sequenced reads, T to C transitions when using 4-SU and G to A transitions when using 6-SG, are indicative of successfully crosslinked sequences. In our experience uncrosslinked RNAs labeled with 4-SU show a background mutation rate of approximately 20%. This rate is increases to approx. 50-80% upon crosslinking.
Disclosures
T.T. is a cofounder and scientific advisor to Alnylam Pharmaceuticals and an advisor to Regulus Therapeutics.
Acknowledgments
We thank members of the Tuschl laboratory for helpful discussions. M.H. is supported by the Deutscher Akademischer Austauschdienst (DAAD). This work was supported by the Swiss National Fund Grant #3100A0-114001 to M.Z.; T.T. is an HHMI investigator, and work in his laboratory was supported by NIH grants GM073047 and MH08442 and the Starr Foundation.
References
- Keene JD. RNA regulons: coordination of post-transcriptional events. Nat. Rev. Genet. 2007;8(7):533–533. doi: 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- Tenenbaum SA. Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc. Nat. Acad. Sci. 2000;97(26):14085–14085. doi: 10.1073/pnas.97.26.14085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber AP. Genome-wide identification of mRNAs associated with the translational regulator PUMILIO in Drosophila melanogaster. Proc. Nat. Acad. Sci. 2006;103(12):4487–4487. doi: 10.1073/pnas.0509260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Silanes I. Identification of a target RNA motif for RNA-binding protein HuR. Proc. Nat. Acad. Sci. 2004;101(9):2987–2987. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg JR. Ultraviolet light-induced crosslinking of mRNA to proteins. Nucl. Acids Res. 1979;6(2):715–715. doi: 10.1093/nar/6.2.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenmakers AJ. Cross-linking of mRNA to proteins by irradiation of intact cells with ultraviolet light. Eur. J. Biochem. 1980;112(2):323–323. doi: 10.1111/j.1432-1033.1980.tb07207.x. [DOI] [PubMed] [Google Scholar]
- Mayrand S. Structure of nuclear ribonucleoprotein: identification of proteins in contact with poly(A)+ heterogeneous nuclear RNA in living HeLa cells. The Journal of Cell Biology. 1981;90(2):380–380. doi: 10.1083/jcb.90.2.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss G. Characterization of heterogeneous nuclear RNA-protein complexes in vivo with monoclonal antibodies. Mol. Cell. Biol. 1984;4(6):1104–11. doi: 10.1128/mcb.4.6.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam SA, Dreyfuss G. Adenovirus proteins associated with mRNA and hnRNA in infected HeLa cells. J. Virol. 1987;61(10):3276–3276. doi: 10.1128/jvi.61.10.3276-3283.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302(5648):1212–1212. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- Licatalosi DD. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456(7221):464–464. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo GW. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat. Struct. Mol. Biol. 2009;16(2):130–130. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford JR. Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009;19(3):381–381. doi: 10.1101/gr.082503.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granneman S. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proc. Nat. Acad. Sci. 2009. [DOI] [PMC free article] [PubMed]
- Guil S, Caceres JF. The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat. Struct. Mol. Biol. 2007;14(7):591–591. doi: 10.1038/nsmb1250. [DOI] [PubMed] [Google Scholar]
- Chi SW. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460(7254):479–479. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zisoulis DG. Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans. Nat. Struct. Mol. Biol. 2010. [DOI] [PMC free article] [PubMed]
- Hafner M. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010. [DOI] [PMC free article] [PubMed]
- Hafner M. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods. 2008;44(1):3–3. doi: 10.1016/j.ymeth.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrus A, Kuimelis RG. Base composition analysis of nucleosides using HPLC. Current Protocols in Nucleic Acid Chemistry. 2001;Chapter 10(Unit 10.6) doi: 10.1002/0471142700.nc1006s01. [DOI] [PubMed] [Google Scholar]