

Abstract
The completion of the human genome sequence, along with that of many other species, has highlighted the challenge of ascribing specific function to non coding sequences. One prominent function carried out by the non coding fraction of the genome is to regulate gene transcription; however, there are no effective methods to broadly predict cis-regulatory elements from primary DNA sequence. We have developed an efficient protocol to functionally evaluate potential cis-regulatory elements through zebrafish transgenesis. Our approach offers significant advantages over cell-culture based techniques for developmentally important genes, since it provides information on spatial and temporal gene regulation. Conversely, it is faster and less expensive than similar experiments in transgenic mice, and we routinely apply it to sequences isolated from the human genome. Here we demonstrate our approach to selecting elements for testing based on sequence conservation and our protocol for cloning sequences and microinjecting them into zebrafish embryos.
Protocol
1. Selection and cloning of conserved sequences for testing
One must first identify potential regulatory sequences for functional testing. We rely on evolutionary sequence conservation as a criterion to select sequences, and most often use the algorithm PhastCons, which is accessible as a track on the UCSC Genome browser (http://genome.ucsc.edu/cgi-bin/hgGateway). However, there are many other algorithms available to evaluate sequence conservation across species.
Once we have selected our sequences, we design primers to amplify each sequence from genomic DNA. Primers should be selected to amplify the conserved region plus 30 or more base pairs on either side.
For amplification of the selected sequences we use the Takara LA TaqTM system. Other polymerases will work, but it is important to use a mix that includes an enzyme with proofreading capability, to minimize the chances of errors introduced during amplification. We verify the amplicon size by agarose gel electrophoresis.
We use the pCR 8/GW/TOPO TA Cloning Kit (Invitrogen) to clone the PCR product into an entry vector containing attL recombination sites, to generate the Gateway entry clone. The cloning reaction is more efficient with fresh PCR product, so it is best to perform the cloning within a day of the PCR. Transformation in DH5α strains is performed followed by spectinomycin resistance selection.
We verify the product of the TA cloning by digestion of ~500ng of plasmid with EcoRI to release the insert, and confirm the size of the insert by agarose gel electrophoresis. We recommended sequencing to verify the insert sequence and orientation; primers used for amplification may also be used for sequencing.
From the entry clone, the non-coding conserved sequence is shuttled by LR recombination to our Gateway ready destination vector, pGW_cfosGFP1, using Gateway LR Clonase II enzyme Mix (Invitrogen). Transformation in DH5α strains is performed followed by ampicillin resistance selection.
We verify the product of the LR recombination by digestion of ~500ng of plasmid with EcoRV to release the insert, and confirm the size of the insert by agarose gel electrophoresis. Once an accurate clone has been identified, we prepare plasmid DNA using the Qiagen HiSpeed® Plasmid Midi Kit. We further purify the plasmid using the QIAquick PCR Purification Kit, according to manufacturer's protocols, and elute the DNA with 30 μL RNase free water. The plasmid is diluted to a concentration of 125 ng/μL and stored at 4°C prior to injections.
RNA encoding functional Tol2 transposase enzyme is transcribed in vitro from the pDB600 plasmid2. We purify the plasmid using the Qiagen Midi-Prep kit, then linearize 10-20 μg with XbaI. The mRNA synthesis is carried out following the mMESSAGE mMACHINE Kit protocol (Ambion).
We purify and precipitate the RNA according to kit instructions. We resuspend RNA to a final concentration of ~1μg/μl, or 20 μl for a single reaction, in RNase free water, and quantify by UV spectrophotometry. We also analyze ~1μg of RNA by agarose gel electrophoresis to verify full length transcription. Although a standard TAE or TBE gel is adequate for this analysis, the denaturing sample buffer included with the transcription kit should be used according to kit instructions.
We test each new batch of transposase RNA for efficacy by performing injections with a known plasmid (Figure 1). After verification, the RNA is divided into small aliquots and stored at -80°C, to avoid repeated thawing and refreezing of individual aliquots.
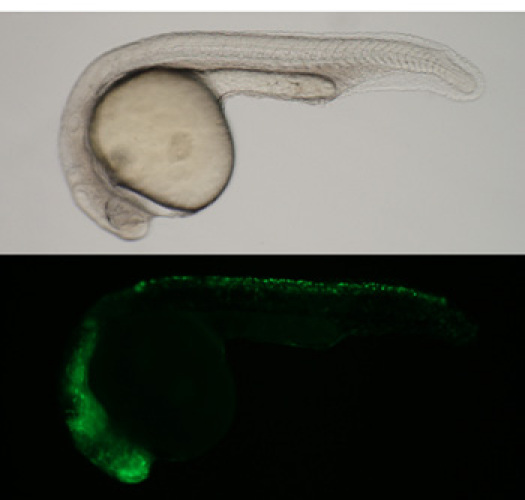 Figure 1. Zebrafish embryo injected with the mouse Sox10 enhancer as a control.(Top) Brightfield image (Bottom) GFP fluorescence image. Each new batch of transposase RNA is tested for efficacy.
Figure 1. Zebrafish embryo injected with the mouse Sox10 enhancer as a control.(Top) Brightfield image (Bottom) GFP fluorescence image. Each new batch of transposase RNA is tested for efficacy.
2. Microinjection of zebrafish embryos for mosaic transgenic analysis
We pull the needles for microinjection from 1.2 mm O.D. filament capillary glass on a Sutter P-97 Micropipette Puller, with a program designed to yield a strong tip with a fairly sharp taper to penetrate intact chorions. We break the tips by hand under a stereomicroscope to an outer diameter of approximately 15 μm, using a clean razor blade and a micrometer slide. The needles can be made the day before injections, and stored in a covered holding dish to keep clean.
We maintain our zebrafish on a regular light dark cycle, with 14 hours of light, under standard conditions3. The day prior to performing microinjections, we set up fish for timed matings in small breeding tanks, each consisting of a base tank, a slotted insert, and a plastic lid. We place three females or two males per tank in parallel rows.
On the morning of the microinjections, shortly after the light cycle begins, we start combining males and females in clean system-treated water for egg production. It is important to check on the fish frequently, and collect the eggs within a few minutes of laying, to get well synchronized batches. Egg production can be maintained for two to three hours, by continuing to combine fresh groups of fish throughout the morning.
With a wide bore, 5 1/4" glass Pasteur pipette fitted with a latex bulb, we sort the collected embryos into 60 x 15 mm Petri dishes, partially filled with Embryo Medium2, in groups of 50 embryos, and mark the time collected on the lid of each dish.
- Once the fish have begun laying, we prepare fresh injection solution by mixing the following in a microfuge tube on ice:
Component Transposon plasmid stock (125ng/μl) 1μl Transposase RNA stock (175ng/μl) 1μl Phenol red stock (2% in H2O) 0.5μl RNase free water 2.5μl The filled needle is loaded into the hand held needle holder of a Pneumatic Pico-Pump or similar pressure injector, configured and connected to a N2 tank per manufacturer's instructions. We calibrate injection volumes by measuring the diameter of droplets expelled into mineral oil on a micrometer slide. Typically, an injection time of 120 ms with a pressure of 20 p.s.i. will yield a droplet of approximately 1 nl, but slight variations in needle diameter will affect these parameters and recalibration may be required between needles.
The injections are performed under a stereomicroscope at 6-10X magnification. We use a pair of fine forceps to hold the embryo in place, push the injection needle with steady pressure through the chorion and into the yolk of an embryo at the one cell or early two cell stage. Ideally, we position the needle tip in the yolk just below the blastomeres. Approximately 1 nl of injection solution is expelled and the needle withdrawn. For each construct we inject ≥ 200 embryos. It is also very important, for each clutch of embryos collected, to save a dish of uninjected control embryos.
After injections are completed we sort the embryos by removing unfertilized eggs, damaged embryos, and failed injections (embryos with no phenol red in blastomeres). We then incubate the embryos in embryo medium at 28.5°C until the appropriate time for observations.
3. Analysis of mosaic expression patterns
Following an appropriate incubation time, we examine the injected embryos for fluorescence. The timing depends on the normal expression pattern of the endogenous zebrafish gene, but we usually look daily through the first 5 days post fertilization.
We examine the embryos for fluorescence on an Olympus MVX10 macroscope fitted for epifluorescence, but any similar microscope capable of low power imaging will work. If there are a large number of embryos that died or developed abnormally in the first day, look at the uninjected controls for that clutch to see if they look normal. If the controls look normal, the most common problem is insufficient purity of the injected DNA. Also, the embryos at early stages are sensitive to the total amount of injected plasmid, so it is possible that the quantification was inaccurate and too much DNA was injected.
When examining the embryos, keep in mind that they will all be mosaic for the injected construct. It will be necessary to look carefully at all of the embryos, especially if the expected domain of expression is small. Also remember that the expression could be dynamic; strong fluorescence on day 1 might disappear by day 2, and embryos negative for expression the first 3 days might show something on day 4.
We choose some embryos with more extensive expression, and use these to document the pattern of fluorescence by photomicroscopy. After 5 days of observations, the embryos should be returned to the facility and raised as a stock. In several months, the adults can be screened for germline transmission of the transgenes.
4. Results
We see a wide variety of patterns and levels of expression, depending on the enhancer element being analyzed. We are usually interested in very tissue specific expression patterns, and are often focusing on genes expressed in the skeleton. and are often focusing on genes expressed in the skeleton. Figure 2 shows examples of mosaic expression patterns we have observed in our injected embryos, with enhancers regulating expression in cartilage and bone. Finally, a substantial portion of tested sequences do not regulate tissue specific expression. For these, we most often see very little fluorescence, perhaps a few scattered cells per embryo. Less often, we might see fluorescence, but only in two common sites for ectopic expression, the epidermis and the skeletal muscle (Figure 3).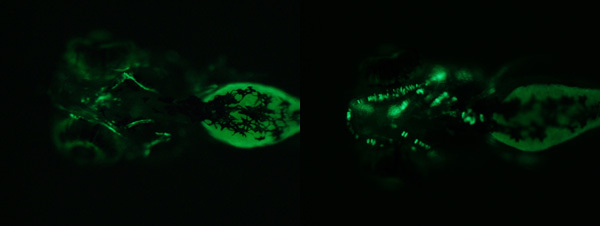 Figure 2. Example of cartilage and bone expression patterns observed in injected embryos.
Figure 2. Example of cartilage and bone expression patterns observed in injected embryos.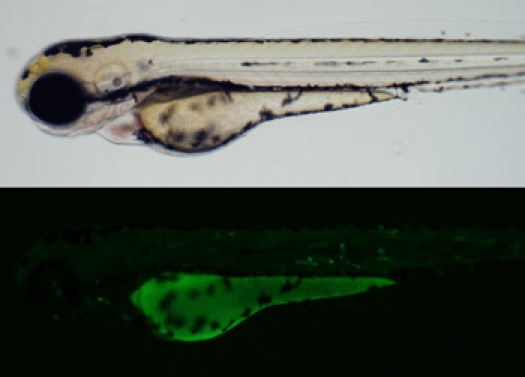 Figure 3. There is little correlation between location of the enhancer relative to the gene and regulation of expression.A substantial portion of tested sequences do not regulate tissue specific expression. These often have very little fluorescence, or may have two common sites for ectopic expression: the epidermis and skeletal muscle. (Top) Brightfield image (Bottom) GFP fluorescence image.
Figure 3. There is little correlation between location of the enhancer relative to the gene and regulation of expression.A substantial portion of tested sequences do not regulate tissue specific expression. These often have very little fluorescence, or may have two common sites for ectopic expression: the epidermis and skeletal muscle. (Top) Brightfield image (Bottom) GFP fluorescence image.
Discussion
We have demonstrated the approach used in our laboratory for the efficient analysis of potential enhancers using zebrafish transgenesis. Most often, we use this approach to look for tissue-specific enhancers associated with human genes. Despite the lack of obvious sequence homology most of the time between human and fish non-coding sequences, we find that this approach is usually successful in identifying enhancers for the genes of interest to us. The critical factors for success are taking care in the preparation of the plasmid DNA and the transposase RNA, and injecting and examining a sufficient number of embryos for each construct. The general methods of transgene construction, embryo microinjections, and mosaic expression analysis would be applicable to generating transgenic zebrafish lines for many other purposes.
Acknowledgments
We thank Ms. Paula Roy for excellent fish husbandry, and Steve Ekker for the kind gift of the pDB600 plasmid. These studies were funded by a grant to S.F. from NHGRI.
References
- Fisher S. Evaluating the biological relevance of putative enhancers using Tol2 transposon-mediated transgenesis in zebrafish. Nat Protoc. 2006;1(3):1297–12. doi: 10.1038/nprot.2006.230. [DOI] [PubMed] [Google Scholar]
- Balciunas D. Harnessing a high cargo-capacity transposon for genetic applications in vertebrates. PLoS Genet. 2006;2(11):e169–169. doi: 10.1371/journal.pgen.0020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M, editor. The Zebrafish Book. 3 ed. Eugene, OR: University of Oregon Press; 1995. [Google Scholar]