

Abstract
An unprecedented enantioselective and general olefin isomerization was realized via biomimetic proton transfer catalysis with a new chiral organic catalyst. A broad range of mono- and di-substituted β,γ-unsaturated butenolides were transformed into the corresponding chiral α,β-unsaturated butenolides in high enantioselectivity and yield in the presence of as low as 0.5 mol% catalyst. Mechanistic studies have revealed the protonation as the rate-determining step.
As exemplified by the Δ5-3-ketosteroid isomerase (KSI)-catalyzed conversion of β,γ- to α,β-unsaturated steroidal ketones (Scheme 1), enzyme-mediated olefin isomerization via a proton transfer from one carbon atom to another in the same substrate molecule constitutes a common and important class of chemical reactions in biology.1 In contrast, only metal-mediated hydride transfer catalysis has been employed in small molecule-catalyzed asymmetric olefin isomerizations, which include enantioselective olefin isomerizations of allylic amines by a chiral Rh+/BINAP complex2 and isomerization of allylic alcohols with a Rh+/planar-chiral phosphaferrocene complex.3 Although substrate-directed diastereoselective olefin isomerizations with either achiral acids or bases have been applied in natural product synthesis,4 only a single example of olefin isomerization by enantioselective proton transfer catalysis, mediated by a bimetallic gadolinium complex, was reported in the literature.5 Herein, we wish to report the realization of a general and highly enantioselective olefin isomerization with a chiral organic catalyst.
Scheme 1.

Isomerization of steroidal ketones catalyzed by KSI
Although the KSI-catalyzed olefin isomerization does not generate a stereocenter, the mechanism underlying the enzymatic proton transfer catalysis is illuminating (Scheme 1). It involves the deprotonation of the C4-β-proton of steroidal ketone by Asp38, while Tyr14 and Asp99 serve as acidic catalytic residues by providing hydrogen bonds to the ketone group. With the oxyanion engaging in hydrogen bonding interactions with Tyr14 and Asp99, the γ-carbon of the dienolate intermediate undergoes a protonation by Asp38 from the β-face. As illustrated in Scheme 2, a 6’-OH cinchona alkaloid in a gauche-open conformation also has a syn-arrangement of acidic and basic active sites, which is similar to how Tyr14 and Asp38 are arranged in the active site of KSI. This observation in combination with the ability of 6’-OH cinchona alkaloids to serve as an acid-base bifunctional catalyst and as a chiral proton donor for asymmetric protonation6,7,8,9 led us to envision the possibility of 6’-OH cinchona alkaloids as a catalyst for an enantioselective isomerization via proton transfer catalysis (Scheme 2).10
Scheme 2.

Proposed isomerization pathway of butenolides catalyzed by cinchona alkaloids
The γ-substituted α,β-unsaturated butenolides constitute a structural motif shared by many biologically interesting natural and synthetic products. Furthermore, they are versatile chiral building blocks for asymmetric synthesis. Thus, the preparation of optically active γ-alkyl α,β-unsaturated butenolides have been a topic of interest. Accordingly, several approaches utilizing easily accessible intermediates from the chiral pool11 or synthetic chiral precursors have been developed.12 Among these only two were catalytic approaches, which involved the Os-mediated asymmetric dihydroxylation of β,γ-unsaturated esters12f,12h, and the Cu-catalyzed heteroallylic asymmetric alkylation, respectively, as the asymmetric induction step.12j,12k Thus, an efficient and general catalytic enantioselective isomerization of γ-substituted β,γ-butenolides to the corresponding α,β-unsaturated butenolides could provide a complementary method that may also expand the scope of chiral butenolides accessible by catalytic asymmetric synthesis (Scheme 2).
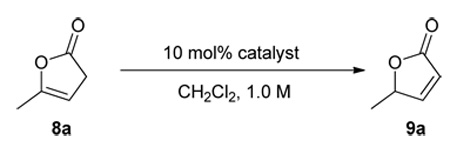
Guided by these considerations we investigated a series of existing 6’-OH cinchona alkaloids and other structurally distinct cinchona alkaloids reported in the literature for their ability to promote the enantioselective isomerization of the commercially available γ-methyl β,γ-butenolide 8a to the corresponding γ-methyl α,β-unsaturated butenolide 9a in dichloromethane at room temperature (Table 1). To our surprise, the catalytic activity of these cinchona alkaloids for this olefin isomerization was found to be critically dependent on not only whether a hydrogen bond donor is presented, but also where it is located in the cinchona alkaloid skeleton. Specifically, while 6’-OH cinchona alkaloids 1a afforded promising activity, cinchona alkaloid 2a, which bears no hydrogen bond donor, was found to be inactive as a catalyst. Although hydroquinine (2b), a 9-OH cinchona alkaloid was active, its activity was significantly lower than that of 1a (entry 3 vs. 1, Table 1). Moreover, the 9-thiourea cinchona alkaloid 4 failed to catalyze this reaction. Even in the presence of both cinchona alkaloid 2a and 6’-OH quinoline 5, no isomerization occurred. These data indicated that the specific spatial relationship between the hydrogen bond donor and acceptor is largely responsible for the outstanding activity of 1a toward this isomerization.
Table 1.
Isomerization of 8a to 9a catalyzed by cinchona alkaloidsa
 | |||||
|---|---|---|---|---|---|
| entry | catalyst | T(°C) | Time | conv.(%)b | ee(%)b |
| 1 | 1a | rt | 4h | 80 | 47c |
| 2 | 2a | rt | 4h | 0 | - |
| 3 | 2b | rt | 4h | 20 | 32 |
| 4 | 4 | rt | 4h | 0 | - |
| 5 | 5 | rt | 4h | 0 | - |
| 6 | 2 + 5 | rt | 4h | <5 | 0 |
Unless noted, reactions were run with 0.05 mmol of 8a in 0.05 mL of CH2Cl2 with 10 mol% catalyst. See supporting information for details.
Determined by HPLC analysis.
10 min, 20% conv., 67% ee.
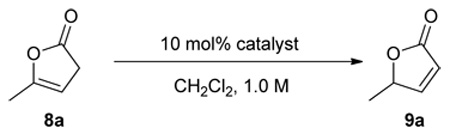
We next focused our attention to the optimization of enantioselectivity by screening various 6’-OH cinchona alkaloids (1 and 3) already reported in the literature (Table 2). As the optical purity of chiral α,β-butenolide 9a was found to gradually decrease at room temperature in the presence of catalyst 1a (table 1, footnote c), the enantioselectivity afforded by different catalysts was evaluated by ee of 9a obtained at 10 min after the initiation of the isomerization. To our disappointment, 6’-OH cinchona alkaloids bearing 9-aryl and alkyl groups of various bulk (1a, 1b, 3a, 3b) afforded similarly modest selectivity (entries 1–5, Table 2). Interestingly, the enantioselectivity was improved with cuperidine 1c (entry 3, Table 2), albeit not to a synthetically useful level. We also investigated a 6’-thiourea cinchona alkaloid 6. In contrast to the completely inactive 9-thiourea cinchona alkaloid 4, 6 proved to be remarkably active but furnished only a modest enantioselectivity. Following the hypothesis that a perturbation of the acidity of the 6’-OH might improve the catalytic activity and enantioselectivity of the 6’-OH cinchona alkaloids,13 we prepared a novel cinchona alkaloid derivative Q-7 in which the quinoline ring was oxidized into the corresponding N-oxide analogue as 6-hydroxyquinoline-N-Oxide was determined to be more acidic than 6-hydroxyquinoline.14 To our delight, it was shown to be a more active as well as selective catalyst in comparison to 6’-OH cinchona alkaloids 1 and 3 (entry 7 vs. entries 1–5, Table 2). Importantly, QD-7 afforded even higher activity and selectivity while providing the expected opposite sense of asymmetric induction (entry 8, Table 2). By decreasing the reaction temperature to −20 °C, the QD-7-mediated isomerization of 8a afforded 9a in 92% ee (entry 9, Table 2). At this temperature, the racemization of 9a was found to be minimal, if not completely suppressed (entry 10 vs. 9, Table 2).
Table 2.
Asymmetric isomerization of 8a to 9a by a cinchona alkaloid bearing a 6' hydrogen bonding donora
 | |||||
|---|---|---|---|---|---|
| entry | catalyst | T(°C) | Time | conv.(%)b | ee(%)b |
| 1 | 1a | rt | 10min | 20 | 67 |
| 2 | 1b | rt | 10min | 16 | 60 |
| 3 | 1c | rt | 10min | 12 | 74 |
| 4 | 3a | rt | 10min | 5 | 60 |
| 5 | 3b | rt | 10min | 5 | 63 |
| 6 | 6 | rt | 10min | 55 | 64 |
| 7 | Q-7 | rt | 10min | 33 | 84 |
| 8 | QD-7 | rt | 10min | 57 | −86 |
| 9 | QD-7 | −20 | 24h | 36 | −92 |
| 10 | QD-7 | −20 | 72h | 80 | −90 |
Unless noted, reactions were run with 0.05 mmol of 8a in 0.05 mL of CH2Cl2 with 10 mol% catalyst. See supporting information for details.
Determined by HPLC analysis.
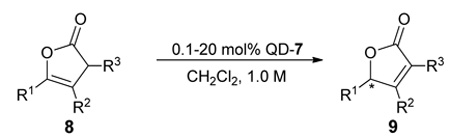
With 7 as the catalyst we explored the scope of the asymmetric olefin isomerization (table 3). With either QD- or Q-7 the isomerization of 8a produced 9a in 90% ee. The conversion of this isomerization reached the maximum at near 80% due to a reverse isomerization. Importantly, the separation of 8a and 9a could be achieved via standard chromatographic procedure, thereby allowing the isolation of optically active 9a in moderate yields (entry 1, Table 3). The enantioselectivity of the reactions promoted by 7 was insensitive to the alteration of the steric bulk of the alkyl substituent (entries 2–4). Due to minimal racemization of 9d, the isomerization of γ-isopropyl β,γ-butenolide 8d could be carried out at room temperature at which the reaction proceeded rapidly (entry 4). Moreover, the isomerization of butenolide 8e, which bears a free hydroxyl group on the alkyl chain, was also tolerated by QD-7 (entry 5, Table 3).
Table 3.
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | 8 | T(°C) | t | Yield (%)c | ee(%)d | ||
| 1 | 8a | −20 | 60h | 63(56) | 90h(90) | ||
| 2e |  |
8b | −20 | 3d | 69 | 91 | |
| 3e | 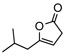 |
8c | −20 | 3d | 64 | 90 | |
| 4 | 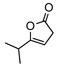 |
8d | rt | 1h | 73(69) | 90(88) | |
| 5 | 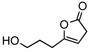 |
8e | −20 | 36h | 64 | 87 | |
| 6 | 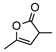 |
8f | rt | 1h | 95(92) | 90h(90) | |
| 7 | 12hf | 95 | 90 | ||||
| 8 | 24g | 91 | 86 | ||||
| 9f | 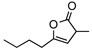 |
8g | rt | 12h | 94 | 90 | |
| 10f | 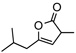 |
8h | rt | 12h | 95 | 90 | |
| 11f | 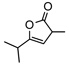 |
8i | rt | 12h | 95 | 94 | |
| 12f | 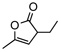 |
8j | rt | 12h | 94 | 92 | |
| 13f | 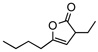 |
8k | rt | 12h | 95 | 91 | |
| 14 | 8l | −20 | 24h | 95 | 81h | ||
| 15 | 8m | −20 | 24h | 83 | 82 | ||
Unless noted, reactions were run with 0.10 mmol of 8 in 0.10 mL of CH2Cl2 with 10 mol% catalyst.
Results in parentheses were obtained with Q-7.
Isolated yield.
Determined by HPLC analysis.
Reaction was run with 20 mol% catalyst.
Reaction was run with 0.5 mol% catalyst.
Reaction was run with 0.10 mmol of 8 in 20 uL of CH2Cl2 with 0.1 mol% catalyst.
Absolute configuration was determined to be S; see Supporting Information for details.
Interestingly, the isomerizations with α,γ-disubstituted β,γ-unsaturated butenolides (8f–k) proceeded readily to completion in a highly enantioselective fashion without detectable racemizations of the corresponding α,β-unsaturated butenolide (9f–k) even at room temperature. Consequently, rapid asymmetric isomerizations of high yield and enantioselectivity could be achieved. For example, with 10 mol% QD-7 the isomerization of 8f was complete within 1.0 h to afford 9f in 95% yield and 90% ee at room temperature (entry 6, Table 3). The catalyst loading could be reduced to as low as 0.5 mol% without sacrificing either the enantioselectivity or the yield (entry 7, Table 3), though a longer reaction time of 12h was required. As exemplified with the isomerization of 8f, a complete and high yielding reaction could still be attained even with 0.1 mol% catalyst, although the enantioselectivity was slightly decreased from 90% to 86% ee (entry 8, Table 3). Importantly, such catalytic efficiency could be extended to a series of substrates bearing various α- and γ-alkyl groups (entries 9–13, Table 3). The β,γ-disubstituted β,γ-unsaturated butenolides (8l–m) turned out to be a more challenging class of substrates. The isomerization of these butenolides proceeded in lower yet still useful level of enantioselectivity. In light of the lack of an efficient approach toward optically active β,γ-disubstituted α,β-butenolides,12f,12i,15 the current isomerization represents a valuable access toward these chiral building blocks.
To gain insights into the reaction mechanism, we carried out a preliminary kinetic study of the QD-7-mediated isomerization of 8f. The reaction was found to show first-order dependence on the QD-7 and 8f, respectively.16 In addition we measured the carbon isotope effect on carbon 2–6 of 8f employing Singleton’s NMR technique at natural abundance to discern the rate-limiting step of the olefin isomerization.16,17 A pronounced carbon isotope effect was observed on the γ carbon when the 13C ratio of recovered 8f at 71% conversion was compared to that of the virgin sample (13C(recovered)/13C(virgin) at Cγ = 1.023, average of three runs) (eq. 1). This kinetic isotope effect indicates the γ-protonation step is the rate-limiting step of the isomerization reaction.
 |
(1) |
Based these results from our mechanistic studies and the drastic difference in catalytic activity between 6’-OH cinchona alkaloid 1a and the corresponding 6’-OMe congener 2a (entry 1 vs. 2, Table 1), we propose a catalytic cycle for this enantioselective isomerization reaction (Scheme 3). In this mechanism, the deprotonation of β,γ-butenolide 8 occurs after the hydrogen bond-based complexion of 8 with catalyst 7, which is followed by the rate-determining γ-protonation. It should be noted that in the protonation step, either the protonated quinuclidine or the 6’-OH could serve as the proton donor.
Scheme 3.

Proposed catalytic cycle for the enantioselective isomerization by QD-7
In summary, we have realized an unprecedented enantioselective olefin isomerization via biomimetic proton transfer catalysis with a new chiral organic catalyst. This asymmetric transformation is applicable to a broad range of β,γ-butenolides bearing one or more substituents. With a low catalyst loading and a simple experimental protocol, this reaction should provide a valuable method for the asymmetric synthesis of chiral α,β-butenolides. Mechanistic studies have revealed that the protonation step is the rate-determining step of this organocatalytic olefin isomerization. Interestingly, this stands in contrast to the enzyme-catalyzed olefin isomerizations, which feature a rate-determining deprotonation.1a
Supplementary Material
Figure 1.

Structure of cinchona alkaloids and achiral acids.
ACKNOWLEDGMENT
We are grateful for financial support from National Institute of Health (GM-61591). We thank Keith Bartelson for his assistance with the 13C KIE study.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental procedures and characterization of the products. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Xue L, Talalay P, Mildvan AS. Biochemistry. 1990;29:7491. doi: 10.1021/bi00484a019. [DOI] [PubMed] [Google Scholar]; (b) Austin JC, Kuliopulos A, Mildvan AS, Spiro TG. Protein Sci. 1992;1:259. doi: 10.1002/pro.5560010208. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hawkinson DC, Pollack RM, Ambulos NP. Biochemistry. 1994;33:12172. doi: 10.1021/bi00206a021. [DOI] [PubMed] [Google Scholar]; (d) Zhao Q, Abeygunawardana C, Talalay P, Mildvan AS. Proc. Natl. Acad. Sci. U. S. A. 1996;93:8220. doi: 10.1073/pnas.93.16.8220. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhao QJ, Abeygunawardana C, Gittis AG, Mildvan AS. Biochemistry. 1997;36:14616. doi: 10.1021/bi971549m. [DOI] [PubMed] [Google Scholar]; (f) Massiah MA, Abeygunawardana C, Gittis AG, Mildvan AS. Biochemistry. 1998;37:14701. doi: 10.1021/bi981447b. [DOI] [PubMed] [Google Scholar]
- 2.(a) Tani K, Yamagata T, Otsuka S, Akutagawa S, Kumobayashi H, Taketomi T, Takaya H, Miyashita A, Noyori R. J. Chem. Soc., Chem. Commun. 1982:600. [Google Scholar]; (b) Tani K, Yamagata T, Akutagawa S, Kumobayashi H, Taketomi T, Takaya H, Miyashita A, Noyori R, Otsuka S. J. Am. Chem. Soc. 1984;106:5208. [Google Scholar]
- 3.(a) Tanaka K, Qiao S, Tobisu M, Lo MMC, Fu GC. J. Am. Chem. Soc. 2000;122:9870. [Google Scholar]; (b) Tanaka K, Fu GC. J. Org. Chem. 2001;66:8177. doi: 10.1021/jo010792v. [DOI] [PubMed] [Google Scholar]
- 4.(a) Suenaga K, Takayanagi Y, Yamaura M, Kigoshi H. Chem. Lett. 2004;33:918. [Google Scholar]; (b) Bao G, Zhao L, Burnell DJ. Org. Biomol. Chem. 2005;3:3576. doi: 10.1039/b509777k. [DOI] [PubMed] [Google Scholar]; (c) Pandey SK, Orellana A, Greene AE, Poisson J-F. Org. Lett. 2006;8:5665. doi: 10.1021/ol062419l. [DOI] [PubMed] [Google Scholar]
- 5.Saga Y, Motoki R, Makino S, Shimizu Y, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2010;132:7905. doi: 10.1021/ja103183r. [DOI] [PubMed] [Google Scholar]
- 6.For a recent review on asymmetric protonation, see: Mohr JT, Hong AY, Stoltz BM. Nat. Chem. 2009;1:359. doi: 10.1038/nchem.297.
- 7.For cinchona alkaloid-catalyzed asymmetric protonation, see: Wang Y, Liu X, Deng L. J. Am. Chem. Soc. 2006;128:3928. doi: 10.1021/ja060312n. Wang B, Wu F, Wang Y, Liu X, Deng L. J. Am. Chem. Soc. 2007;129:768. doi: 10.1021/ja0670409. Poisson T, Dalla V, Marsais F, Dupas G, Oudeyer S, Levacher V. Angew. Chem., Int. Ed. 2007;46:7090. doi: 10.1002/anie.200701683.
- 8.Selected references for other organocatalyzed asymmetric protonations: Fehr C, Galindo J. Angew. Chem., Int. Ed. 1994;33:1888. Vedejs E, Kruger AW. J. Org. Chem. 1998;63:2792. doi: 10.1021/jo990119u. Mitsuhashi K, Ito R, Arai T, Yanagisawa A. Org. Lett. 2006;8:1721. doi: 10.1021/ol0603007. Cheon CH, Yamamoto H. J. Am. Chem. Soc. 2008;130:9246. doi: 10.1021/ja8041542. Leow D, Lin S, Chittimalla SK, Fu X, Tan C-H. Angew. Chem., Int. Ed. 2008;47:5641. doi: 10.1002/anie.200801378. Vora HU, Rovis T. J. Am. Chem. Soc. 2010;132:2860. doi: 10.1021/ja910281s.
- 9.Selected references for metal catalyzed asymmetric protonations: Ishihara K, Nakamura S, Kaneeda M, Yamamoto H. J. Am. Chem. Soc. 1996;118:12854. Hodous BL, Ruble JC, Fu GC. J. Am. Chem. Soc. 1999;121:2637. Mohr JT, Nishimata T, Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2006;128:11348. doi: 10.1021/ja063335a. Morita M, Drouin L, Motoki R, Kimura Y, Fujimori I, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2009;131:3858. doi: 10.1021/ja9005018.
- 10.(a) Oku M, Arai S, Katayama K, Shioiri T. Synlett. 2000;2000:493. [Google Scholar]; (b) Liu H, Leow D, Huang K-W, Tan C-H. J. Am. Chem. Soc. 2009;131:7212. doi: 10.1021/ja901528b. [DOI] [PubMed] [Google Scholar]; (c) Liu H, Feng W, Kee CW, Leow D, Loh W-T, Tan C-H. Adv. Synth. Catal. 2010;352:3373. [Google Scholar]
- 11.Camps P, Cardellach J, Corbera J, Font J, Ortuño RM, Ponsatí O. Tetrahedron. 1983;39:395. [Google Scholar]
- 12.Selected references: Hanessian S, Hodges PJ, Murray PJ, Sahoo SP. J. Chem. Soc., Chem. Commun. 1986:754. Solladie G, Frechou C, Demailly G, Greck C. J. Org. Chem. 1986;51:1912. Bloch R, Gilbert L. J. Org. Chem. 1987;52:4603. Corbera J, Font J, Monsalvatje M, Ortuno RM, Sanchez-Ferrando F. J. Org. Chem. 1988;53:4393. Hoye TR, Humpal PE, Jiménez JI, Mayer MJ, Tan L, Ye Z. Tetrahedron Lett. 1994;35:7517. Harcken C, Brückner R. Angew. Chem., Int. Ed. 1997;36:2750. Harcken C, Bruckner R. New J. Chem. 2001;25:40. Braukmüller S, Brückner R. Eur. J. Org. Chem. 2006:2110. Kapferer T, Brückner R. Eur. J. Org. Chem. 2006:2119. Geurts K, Fletcher SP, Feringa BL. J. Am. Chem. Soc. 2006;128:15572. doi: 10.1021/ja065780b. Mao B, Geurts K, Fañanás-Mastral Mn, van Zijl AW, Fletcher SP, Minnaard AJ, Feringa BL. Org. Lett. 2011;13:948. doi: 10.1021/ol102994q.
- 13.(a) Vedejs E, Kruger AW, Suna E. J. Org. Chem. 1999;64:7863. doi: 10.1021/jo990119u. [DOI] [PubMed] [Google Scholar]; (b) Jensen KH, Sigman MS. Angew. Chem., Int. Ed. 2007;46:4748. doi: 10.1002/anie.200700298. [DOI] [PubMed] [Google Scholar]; (c) Jensen KH, Sigman MS. J. Org. Chem. 2010;75:7194. doi: 10.1021/jo1013806. [DOI] [PubMed] [Google Scholar]
- 14.Solntsev KM, Clower CE, Tolbert LM, Huppert D. J. Am. Chem. Soc. 2005;127:8534. doi: 10.1021/ja0514545. [DOI] [PubMed] [Google Scholar]
- 15.For an exmaple of preparing chiral 9k, see: Bekish AV, Prokhorevich KN, Kulinkovich OG. Eur. J. Org. Chem. 2006:5069.
- 16.See supporting information for details.
- 17.(a) Singleton DA, Thomas AA. J. Am. Chem. Soc. 1995;117:9357. [Google Scholar]; (b) Kwon K-H, Lee DW, Yi CS. Organometallics. 2010;29:5748. doi: 10.1021/om100764c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.