

Abstract
Thiazolyl peptides are bacterial secondary metabolites that potently inhibit protein synthesis in Gram-positive bacteria and malarial parasites. Recently, our laboratory and others reported that this class of trithiazolyl pyridine-containing natural products is derived from ribosomally synthesized preproteins that undergo a cascade of posttranslational modifications to produce architecturally complex macrocyclic scaffolds. Here, we report the gene cluster responsible for production of the elongation factor Tu (EF-Tu)-targeting 29-member thiazolyl peptide GE37468 from Streptomyces ATCC 55365 and its heterologous expression in the model host Streptomyces lividans. GE37468 harbors an unusual β-methyl-δ-hydroxy-proline residue that may increase conformational rigidity of the macrocycle and impart reduced entropic costs of target binding. Isotope feeding and gene knockout were employed in the engineered S. lividans strain to identify the P450 monooxygenase GetJ as the enzyme involved in posttranslational transformation of isoleucine 8 to β-methyl-δ-hydroxy-proline through a predicted tandem double hydroxylation/cyclization mechanism. Loss of Ile8 oxygenative cyclization or mutation of Ile8 to alanine via preprotein gene replacement resulted in a 4-fold and 2-fold drop in antibiotic activity, respectively. This report of genetic manipulation of a 29-member thiazolyl peptide sets the stage for further genetic examination of structure activity relationships in the EF-Tu targeting class of thiazolyl peptides.
Keywords: bacterial resistance, bioengineering, thiopeptide
Thiazolyl peptide, or thiopeptide, antibiotics are the most heavily posttranslationally modified ribosomal natural products known to date (1–3). More than a dozen posttranslational modifications transform linear 50–60 residue preproteins, consisting of only canonical amino acids, to molecules rich in thiazole and dehydro amino acids with a pyridine/piperidine core. These rigid scaffolds provide potent inhibition of ribosomal protein synthesis in a broad range of Gram-positive bacteria and malarial parasites. In particular, their activity against Methicillin-resistant Staphylococcus auereus (MRSA) has made them attractive candidates for development as clinical agents to fight emerging resistance to the currently used antimicrobial repertoire. The recent discovery that thiazolyl peptides can function as antitumor agents by inhibiting the 20S proteasome has also heightened interest in exploration of these compounds (4); however, poor pharmacokinetics and low water solubility have kept them from human use. Although laudable synthetic efforts have achieved the total synthesis of several thiazolyl peptides (5–7), their complex scaffolds have prevented synthesis of analogs for extensive structure activity relationship (SAR) studies. An alternate route to diversification, presented in this work, is to exploit the ribosomal synthesis of thiazolyl peptides in a gene replacement approach (8, 9).
Since the discovery of the first thiazolyl peptide, micrococcin, more than half a century ago, nearly 100 poly-azole peptides have been discovered in this class of antibiotics; however, it was not until 2009 that it was reported they are the result of extensive posttranslational modifications on ribosomally generated preproteins (10–13). The mature antibiotic arises from a structural gene encoding a 50–60 amino acid preprotein consisting of a 40–50 amino acid N-terminal leader peptide (residues −50 to −1) and a 14–18 amino acid C-terminal region (residues +1 to +18), which becomes the final product scaffold (3). Flanking the structural gene are encoded enzymes involved in peptide maturation, which appear to mimic the biosynthetic logic for microcin B17, lantibiotic, and cyanobactin antibiotic peptides (14). These enzymes include lantibiotic-type dehydratases that form dehydro amino acids, cyclodehydratase and dehydrogenase enzymes involved in the formation of thiazoles/oxazoles, and a novel enzyme(s) involved in the formation of the central pyridine/piperidine ring. Evidence suggests this could proceed after installation of all thiazoles in the preprotein through an unprecedented cyclization of two dehydroalanine (Dha) residues (derived from serines) in a [4 + 2] aza-Diels–Alder mechanism (15). Placement of the dehydration-fated serine residues in the preprotein sequence ultimately defines the size of the macrocycle and appears to direct the antibiotic mode of targeting to the 50S ribosome or elongation factor Tu (EF-Tu).
Thiazolyl peptides with Ser-derived Dha residues at positions +1 and +10 of the preprotein, which generate at 26-member macrocycle, target the 23S rRNA/L11 protein interface of the 50S ribosome (1, 2). Binding of the antibiotic halts translation by blocking functional association with critical elongation factors. By contrast, thiazolyl peptides with 29-member rings such as GE37468, GE2270, and the thiomuracins (Fig. 1 and SI Appendix, Fig. S1) are formed by cyclization of Dha residues +1 and +11 of the preprotein. These molecules have a distinct mode of action in which protein synthesis is inhibited by binding to EF-Tu and blocking its capacity to chaperone aminoacylated tRNAs to the ribosome. To date, two highly homologous biosynthetic clusters in the 29-member family, GE2270 and thiomuracin from two different strains of the actinomycetes Nonomuraea, have been reported by a natural products team at Novartis (12). Notably, GE2270 is the only thiazolyl peptide in clinical trials for human use (as a topical treatment for acne) (16). Nonomurea are rare actinomycetes without well-established genetic systems, and heterologous expression systems for these clusters in a more tractable genetic host (to enable genetic manipulation for SAR) have not yet been reported.
Fig. 1.

Structure of GE37468 with the macrocyclic ring and central pyridine highlighted in bold. Numbering corresponds to the amino acid alpha carbons from the C-terminal coding region of the preprotein (+1 to +14).
GE37468 is a structurally similar molecule that was first isolated from Streptomyces ATCC 55365 in 1995 by Ferrari et al. in Gerenzano, Italy (from which the “GE” name was derived) (17–19). Both GE37468 and GE2270 were isolated in screens based on the ability of pure EF-Tu to antagonize their antibiotic activity against S. aureus and Moraxella caviae (20). They show similar antibiotic profiles, but notably differ in their inhibition of Streptococcus species (21). They share a triazole-substituted pyridine core and similar primary amino acid sequence with thiomuracin, but GE37468 harbors several distinct modifications in the macrocycle (SI Appendix, Fig. S1), including oxygenative modification of isoleucine 8 (Ile8) to the cyclic hemiaminal β-methyl-δ-hydroxy-proline (mhP). Thiomuracin has also been reported to harbor this unusual residue; however, the modification is stochastic with respect to Ile8, appearing in eight different oxidation states (thiomurcin A–I). Those analogs have similar antibiotic profiles indicating oxidation in the case of thiomuracin has modest impact on activity. Recent work in our laboratory has shown heterocycles within the macrocycle increase conformational rigidity and are required for antibiotic activity in the 26-member thiocillins (9). Because Ile8 is found exclusively as mhP8 in GE37468, it is unknown whether replacement of mhP8 with an acyclic residue would impact antibiotic activity. Manipulation of the preprotein gene sequence and evaluation of the tailoring oxygenations would enable SAR studies in the 29-member subclass for comparison with preprotein gene replacement underway in genetically tractable hosts for ribosome-targeting 26-member thiazolyl peptides (8, 22). With this in mind, we now report the sequencing, stable incorporation, expression, and initial manipulation of the GE37468 cluster in Streptomyces lividans TK24.
Results and Discussion
Sequencing of the GE37468 Biosynthetic Cluster from Streptomyces ATCC 55365.
To identify the biosynthetic gene cluster for GE37468, a high diversity fosmid library (> 105 unique clones) of Streptomyces ATCC 55365 genomic DNA was created in Escherichia coli. Thiazolyl peptide gene clusters have previously been identified in fosmid libraries using primers designed to the cyclodehydratase gene (23, 24), the putative structural gene (25, 26), or unique posttranslational modification genes such as N-dimethyltransferases [in the case of nocathiacin (27)]. The most successful of these methods has been to screen for the cyclodehydratase gene—a homolog of the ycaO gene from bacteriocin biosynthesis that catalyzes the formation of thiazoline rings. This gene has been successfully used to identify the biosynthetic gene clusters responsible for siomycin from Streptomyces sioyaensis, nosiheptide from Streptomyces actuosus, and cyclothiazomycin from Streptomyces hygroscopicus in fosmid libraries (11, 23, 24).
To screen for GE37468’s cyclodehydratase gene in the fosmid library, degenerate primers were designed based on six homologs from the GE2270, thiomuracin, thiocillin, siomycin, nosiheptide, and cyclothiazomycin biosynthetic clusters using the iCODEHOP program (University of Washington) (SI Appendix, Table S1). PCR amplification from crude Streptomyces sp. ATCC 55365 genomic DNA yielded a 540-bp product with approximately 50% identity (in a single translation frame) to the protein sequence of the cyclodehydratase gene from the GE2270 cluster. Primers created directly to this sequence were then used to identify nine unique fosmids from the library that harbored the GE37468 cyclodehydratase gene. The fosmids were sequenced using next-generation Illumina sequencing as described in the SI Appendix. Subsequent contig assembly yielded a 28.2-kbp stretch of genomic DNA containing the cluster described below (SI Appendix, Figs. S2 and S3).
Bioinformatic Analysis of the GE37468 Cluster.
Thirteen open reading frames—annotated as getA–M for GE37468 thiopeptide—in 17.1 kbp of Streptomyces ATCC 55365 genomic DNA were assigned to the GE37468 cluster. The upstream and downstream ends of the cluster were assigned based on coding regions for a putative HEAT repeat type protein and a putative aldehyde dehydrogenase, respectively, which are not expected to participate in GE37468 biosynthesis (Fig. 2). The structural gene and overall organization of the cluster show good similarity to clusters previously reported for GE2270 and thiomuracin (12). GE37468’s structural gene (getA) is located at the upstream end of the cluster and encodes a 57 amino acid preprotein divided into a 42 amino acid leader peptide (residues −42 to −1) and 15 amino acid coding region (residues +1 to +15). Fourteen residues of the coding region give rise to the mature antibiotic. A single asparagine residue positioned at the C terminus of the coding region is not present in the mature antibiotic. As might be expected, the getA peptide has good homology (56% identity) to the structural peptide for thiomuracin. Following getA are two ABC transporter-like genes (getB,C) that putatively enable host resistance.
Fig. 2.

GE37468 biosynthetic gene cluster. The amino acid sequence of the structural gene is shown at bottom with the coding sequence that scaffolds the mature antibiotic underlined.
Downstream of these genes are a cluster of six genes (getD–I) that encode the hallmark “core” posttranslational modification enzymes responsible for peptide maturation. These genes are similar in protein sequence and arrangement to the tpdB–tpdG genes from the GE2270A and thiomuracin clusters (SI Appendix, Table S2). Genes getD and getE encode lantibiotic-type dehydratases putatively responsible for dehydrating serines +1, +11, +13, and +14 to Dhas. Dhas +1 and +11 are subsequently cyclized to form the pyridine core as shown by isotopically labeled serine feeding studies with the thiazolyl peptide nocathiacin (28). The +1 to +11 cyclization generates a 29-membered macrocycle as it forms the central pyridine ring, perhaps through an aza-Diels–Alder reaction catalyzed by the GetF enzyme. This enzyme is homologous to the previously reported TclM enzyme from the thiocillin biosynthetic cluster (15). Genes getG and getI are homologous to enzymes found in the microcin B17 (McbC dehydrogenase) and cyanobactin (YcaO cyclodehydratase) pathways, respectively, and are putatively responsible for the formation of the six thiazole/oxazole/thiazoline rings in GE37468.
Downstream of the “core” proteins are enzymes involved in auxiliary modification (getJ,M), transcriptional regulation (getL), and an additional McbC-like dehydrogenase (getK). getJ presumably encodes a P450 monooxygenase enzyme involved in the conversion of isoleucine 8 to β-methyl-δ-hydroxy-proline (see below). This enzyme shows good homology to tpdQ (43% identity) and tpdJ1 (42% identity) from the GE2270 and thiomuracin gene clusters, respectively. The TpdQ enzyme is hypothesized to β-hydroxylate Phe8 of GE2270 while TpdJ1 is expected to function along with an additional P450 enzyme (TpdJ2) to hydroxylate Phe5 and Ile8 of thiomuracin. This yields an array of oxidations including epoxidation and ketonylation on Ile8 of thiomuracin. In contrast, no alternative oxidation states of Ile8 have been identified in the GE37468 framework.
Transfer of the GE37468 Cluster to Streptomyces lividans TK24.
The GE37468 endogenous producer, Streptomyces ATCC 55365, has been reported to be genetically unstable and highly variable in thiopeptide antibiotic expression (18). In order to create a stable, manipulatable platform for GE37468 expression, the gene cluster was transferred to the model host organism S. lividans TK24. S. lividans has been exploited as a heterologous host for many natural product gene clusters because of its genetic stability, high transformation efficiency, and rapid doubling time (29). To transfer the GE37468 cluster, it was subcloned between the naturally occurring BglII and NheI restriction sites into the E. coli–Streptomyces shuttle vector pSET152 to create pSETGE1 (SI Appendix, Fig. S3). This subcloning strategy also transferred part of a tellurium resistance gene, a putative integral membrane protein, and a putative HEAT repeat type protein located upstream of the gene cluster to the pSETGE1 shuttle vector. Because GE37468 production in E. coli would also be desirable, expression of the pSETGE1 shuttle vector in DH5α E. coli was attempted; however, methanolic extracts from these cultures failed to show any compound by liquid chromatography—mass spectrometry (LCMS) analysis (SI Appendix, Fig. S4).
The pSETGE1 vector was subsequently transferred to S. lividans TK24 via bacterial conjugation and expression carried out in shake flasks under conditions previously described for GE37468 expression in Streptomyces ATCC 55365 (18). After 72 h of growth, the methanolic extract of S. lividans + pSETGE1 was compared with extracts from the endogenous producer (Streptomyces ATCC 55365) and an empty cloning vector as a negative control (S. lividans + pSET152) (Fig. 3 A, B, and F). S. lividans + pSETGE1 exhibited a peak in the UV350 nm trace (2), which coincided with GE37468 from the wild-type producer (1). High-resolution MS confirmed the compound matched the mass of GE37468 from the wild-type producer within 0.5 ppm ([M-OH]+ as previously reported in ref. 17) (SI Appendix, Fig. S5). As expected, no corresponding peak or mass was observed in the pSET152 control sample.
Fig. 3.

LCMS analysis of GE37468 and analogs expression in S. lividans and Streptomyces ATCC 55365. UV350 chromatogram traces of methanolic extracts from (A) Streptomyces ATCC 55365 (GE37468, 1, red), (B) S. lividans + pSETGE1 (GE37468, 2, blue), (C) S. lividans + pSETGE-getJ::FRT (dihydro GE37468mhp8I, 3, green; GE37468mhP8I, 4, green), (D) pSETGE-getAI8A (GE37468mhP8A, 5, orange), (E) S. lividans + pSETGE-getA∷FRT, and (F) S. lividans + pSET152 control.
1H-NMR of purified GE37468 confirmed the compound produced by S. lividans + pSETGE1 was indistinguishable from GE37468 produced by the wild-type host (SI Appendix, Fig. S6). A time-course shake flask expression showed GE37468 production was detectable after 24 h in S. lividans and peaked at 72 h (SI Appendix, Fig. S7). Maximum yields of GE37468 from S. lividans (approximately 2–3 mg/L) were 40% the yields of Streptomyces ATCC 55365 per gram of cell paste (SI Appendix, Fig. S8). Minor amounts of other GE37468 products were also detectable in the extracts. These alternate products resulted from cleavage of the poly-Dha tail and were previously reported by Stella et al. as GE37468 B and C (30). A product corresponding to a dihydro GE37468 was also detectable in small amounts in the extracts from both Streptomyces ATCC 55365 and S. lividans (SI Appendix, Fig. S9). Despite the presence of export pumps in the biosynthetic cluster (getB and getC), no compound was detectable in cleared S. lividans media (SI Appendix, Fig. S10).
Biosynthetic Origins of β-Methyl-δ-Hydroxy-Proline.
The mhP residue is found only in two thiazolyl peptides, GE37468 and thiomuracin I (12). The structural genes in both biosynthetic clusters suggest mhP8 arises from hydroxylation and cyclization of Ile8 rather than methylation and hydroxylation of proline as previously hypothesized (17). In thiomuracin, the side chain of Ile8 actually occurs in eight distinct forms [the unmodified Ile and seven oxygenated forms (Fig. 4)], whereas only the cyclic hemiaminal oxidation state of Ile8 is observed in GE37468. The absolute stereochemistry of mhp8 in thiomuracin I has been assigned based on NOE correlations (12); however, stereochemistry has not yet been assigned in GE37468.
Fig. 4.

Oxidation states of Ile8 observed in the 29-member thiazolyl peptides GE37468 and thiomuracin. Letters below each residue correspond to the thiomuracin variant in which they are found. The proposed mechanism for mhP biosynthesis is shown below.
To unambiguously confirm this unique biosynthetic transformation, we carried out feeding studies with 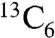 -L-isoleucine in S. lividans + pSETGE1. Gratifyingly, S. lividans + pSETGE1 grew and expressed well in minimal media with > 92% incorporation of labeled isoleucine in GE37468, further demonstrating the utility of the S. lividans expression platform. MS showed GE37468 from cultures fed
-L-isoleucine in S. lividans + pSETGE1. Gratifyingly, S. lividans + pSETGE1 grew and expressed well in minimal media with > 92% incorporation of labeled isoleucine in GE37468, further demonstrating the utility of the S. lividans expression platform. MS showed GE37468 from cultures fed 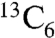 -L-isoleucine was +6 m/z heavier than cultures fed unlabeled L-isoleucine, indicating the incorporation/modification of a single isoleucine in the mature antibiotic (Fig. 5). Tandem MS/MS was employed to localize the labeled carbons to the mhP8 portion of GE37468 macrocycle (SI Appendix, Fig. S11).
-L-isoleucine was +6 m/z heavier than cultures fed unlabeled L-isoleucine, indicating the incorporation/modification of a single isoleucine in the mature antibiotic (Fig. 5). Tandem MS/MS was employed to localize the labeled carbons to the mhP8 portion of GE37468 macrocycle (SI Appendix, Fig. S11).
Fig. 5.

Expression of GE37468 in S. lividans + pSETGE1 with L-isoleucine (Upper) or  -L-isoleucine (Lower). Gray circles in the lower structure indicate 13C-labeled carbons localized by MS/MS.
-L-isoleucine (Lower). Gray circles in the lower structure indicate 13C-labeled carbons localized by MS/MS.
To investigate the P450 monooxygenase GetJ as the responsible oxygenation/cyclization catalyst for transformation of Ile8 to mhP8, we replaced getJ with a chloramphenicol antibiotic resistance cassette (cat) in the GE37468 cluster to create pSETGE-getJ∷cat using λ Red recombination in E. coli (31). Conjugative transfer and expression of pSETGE-getJ∷cat in S. lividans failed to yield any thiazolyl peptide product detectable by LCMS in methanolic extracts. This was thought to be a result of polar effects of the cat cassette on the GE37468 cluster; therefore, it was removed in E. coli using flippase recognition target (FRT) sites included in the original cat cassette (31). Expression of this new plasmid (pSETGE-getJ∷FRT) in S. lividans yielded a product consistent by MS with a GE37468 analog harboring a nonoxygenated Ile8 (GE37468mhP8I) (Fig. 3C and SI Appendix, Fig. S5). Tandem MS/MS was used to confirm the difference in mass was localized to the Ile8 portion of the molecule; however, low yields, possibly due to residual polar effects of the 102-bp FRT scar, prevented analysis by NMR. Two compounds corresponding to GE37468mhP8I with truncated poly-Dha tails and a dihydro GE37468mhP8I were also detectable (SI Appendix, Fig. S9); the latter product was present in greater amounts than GE37468mhP8I in extracts (peaks 3 vs. 4, respectively, in Fig. 3C).
Preprotein Gene Replacement and Antibiotic Activity of Analogs.
To demonstrate the tractability of the S. lividans + pSETGE1 expression platform with the interest of investigating the role of the mhP residue in GE37468’s antibiotic activity, we manipulated the preprotein gene in E. coli (SI Appendix, Fig. S12). A 2.2-kb section of the pSETGE1 plasmid encoding the preprotein gene getA was replaced with a preprotein gene harboring an Ile8Ala mutation to create pSETGE-getAI8A. Expression of pSETGE-getAI8A in S. lividans yielded the predicted GE37468mhP8A analog as confirmed by LCMS and tandem MS/MS fragmentation (peak 5 in Fig. 3D and SI Appendix, Figs. S5 and S11). Minor truncation and a dihydro product were also detectable in small amounts but were not isolated (SI Appendix, Fig. S9). As a control, the getA gene was also knocked out and marker removed with FRT sites (using the λ Red recombination method described above) to create pSETGE-getA∷FRT. As expected, pSETGE-getA∷FRT failed to yield any thiazolyl peptide compound by LCMS analysis of methanolic extracts (Fig. 3E).
With the GE37468mhP8I and GE37468mhP8A analogs in hand, we carried out an initial evaluation of how loss of the mhP residue affects bactericidal potency. Antibiotic activity was quantified against MRSA strain MW2 or Bacillus subtilis 168 using HPLC-purified compounds in serial-dilution liquid culture assays (SI Appendix, Table S3). Activity of wild-type GE37468 from both the endogenous host and our engineered host against MRSA MW2 (0.047 μg/mL, 36 nM) was similar to values reported by Stella and coworkers against the S. aureus strain L165 (0.03 μg/mL, 23 nM) (19, 30). GE37468 was equally active against B. subtilis in our assay. Variants GE37468mhP8I and GE37468mhP8A led to a 4-fold (0.19 μg/mL, 144 nM) and 2-fold (0.093 μg/mL, 71 nM) drop, respectively, in antibiotic potency against MRSA.
Conclusions
Whereas genetically tractable host systems such as Bacillus cereus and Streptomyces laurentii have previously been exploited to produce analogs of 26-member thiazolyl peptides, the heterologous expression platform presented in this work enables manipulation of EF-Tu-targeting 29-member variants in S. lividans. Using this system, we have begun to explore how the unique mhP residue is installed in the thiazolyl peptide scaffold and its role in GE37468’s antibiotic activity. GetJ likely carries out a regiospecific tandem double oxygenation of the δ-CH3 of the Ile8 side chain, first to the δ-CH2OH, and then the δ-CH(OH)2, which is in equilibrium with the aldehyde (Fig. 4). Subsequent intramolecular attack by the amide nitrogen forms the cyclic hemiaminal mhP residue that is the accumulating form of GE37468. The homogenous modification of Ile8 in GE37468 stands in contrast to thiomuracin’s seven oxygenated forms of Ile8. To determine how this oxygenation chemistry is controlled, the tpdJ1 and tpdJ2 P450 homologs from the thiomuracin gene cluster could be used to replace getJ and determine if promiscuously oxygenated Ile8 forms of GE37468 are produced.
Here, we show that loss of oxidation of Ile8 to mhP8 in GE37468 leads to a 4-fold decrease in antibiotic potency against MRSA, whereas the corresponding change in thiomuracin (thiomuracin I vs. C, respectively) yields no variation in antibiotic activity against S. aureus (12). It may be that oxygenative transformation of Ile8 to mhP8 is a requirement in GE37468 because of the structural constraint it introduces to the scaffold. Studies from our group on thiocillin analogs have shown conformational rigidity imposed by thiazole sp2 α-carbons impart a structural rigidity to the macrocycle that is required for thiocillin’s activity. This is presumably conveyed through a reduced entropic cost of target binding for a molecule with fewer conformational states (9). It is plausible that in the case of GE37468 the incomplete oxidation of thiazoline 6 introduces conformational flexibility that is compensated for by the mhP residue. Conversely, thiomuracin harbors a thiazole at position 6, which may create a rigid molecule that makes stochastic oxygenation of Ile8 permissible with retention of activity. If this is correct, synthetic oxidation (32) of thiazoline 6 in GE37468I8A and GE37468mhP8I should rescue antibiotic activity of these mutants; this is currently under investigation.
Among the most intriguing features of the GE37468 posttranslational maturation is how conversion of Cys6 to a thiazoline is controlled while four other Cys residues are fully desaturated to thiazoles in GE37468. Notably, the 26-member thiazolyl peptide thiostrepton also contains a single thiazoline residue in the presence of four thiazoles. This has been hypothesized to be a result of ring strain driven nonenzymatic epimerization of the thiazoline to the D-isomer, which may protect it from further oxidation by dehydrogenases because the “unnatural” isomer is not expected to be an enzyme substrate (33). It is unknown if this strategy is also employed by the GE37468 cluster to maintain thiazoline 6; however, the putatively reduced ring strain of the mhP8I and mhP8A analogs could be instrumental in yielding insights into how this chemistry is controlled and how macrocyclic architecture controls and affects function.
Methods
Additional details on cluster sequencing, media compositions, reagents, minimal inhibitory concentration assays, and MS parameters used in this work are listed in the SI Appendix.
Construction of Fosmid Library and Sequencing.
Streptomyces ATCC 55365 was grown in seed media for 3–5 d, and genomic DNA was isolated via the Pospiech and Neumann “salting out procedure” (34). Fosmid libraries were constructed in the pCC2FOS vector following the “CopyControl Fosmid Library Production Kit” protocol from Epicentre Biotechnology. DNA was cleaned with Genomic DNA Clean and Concentrator (Zymo) prior to ligation. Library pools were screened for the cyclodehydratase gene using primers VNTI F and TSY R (SI Appendix, Table S1). Nine unique fosmids were identified (SI Appendix, Fig. S2), combined, and prepared for Illumina sequencing with the Nextera DNA Sample Prep Kit (Epicenter Biotech) using the low molecular weight buffer without removal of < 300 bp fragments. Details on cluster assembly can be found in SI Appendix, Fig. S2.
Subcloning in pSET152 and Transfer to S. lividans.
To subclone the GE37468 cluster to the integrative pSET152 vector, the NheI restriction sites were removed using site-directed PCR mutagenesis with primers pSET NheI QC F and pSET NheI QC R. The pSET152 backbone was amplified using primers pSET BglII F and pSET NheI R, and the resulting PCR product was digested with NheI and BglII and ligated to the similarly digested pCC2FOS G8-G12 fosmid to create pSETGE1 (SI Appendix, Fig. S3).
getA and getJ genetic knockouts were created using λ Red recombination in E. coli following protocols described by Datsenko and Wanner (31). Briefly, the cat resistance cassette was PCR amplified from pKD3 using primers GetA prim 2F and GetA hygroR or primers GetJ alt F and GetJ alt R to knock out the getA and getJ genes, respectively. Knockouts were carried out using the pKD46 plasmid in E. coli BW25113 to create pSETGE-getA∷cat and pSETGE-getJ∷cat. Markers were removed using E. coli BT340 (pCP20) expressing the FLP recombinase, which left the FRT scar and created plasmids pSETGE-getA::FRT and pSETGE-getJ∷FRT. To create a preprotein gene with the Ile8Ala mutation, a 2.2-kbp fragment of the pSETGE1 plasmid harboring the getA gene was removed using AclI and Scal restriction sites. An identical 2.2-kbp fragment containing getAI8A was inserted using “Gibson/Venter” cloning (described in SI Appendix, Fig. S12) to create pSETGE-getAI8A (35). pSET-derived plasmids were transformed into E. coli ET12567 and transferred to S. lividans TK24 using protocols found in the REDIRECT technology PCR-targeting system in Streptomyces coelicolor (John Innes Center).
Expression and Isolation of GE37468 and Analogs.
Briefly, expression was carried out as previously described by Marinelli et al. (18). S. lividans or Streptomyces ATCC 55365 starter cultures were initiated from spores on soya flour mannitol plates into 30–50 mL starter cultures (seed media or J media) and were grown for 2–3 d (3–5 d for Streptomyces ATCC 55365) at 28–30 °C, 250 rpm in baffled flasks containing ColiRollers (Novagen) glass beads. Starter cultures were inoculated into AF/MS at 1% culture volume. Expression (100–150 mL culture in 500 mL baffled flasks containing glass beads) was carried out for 3 d. For labeling experiments, cultures were started in J media and inoculum was washed twice with filtered MG Base media prior to inoculation at 2% culture volume. Uniformly labeled 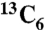 -L-isoleucine (Cambridge Isotopes) was added to 2 mM at 0, 1, and 2 d of expression.
-L-isoleucine (Cambridge Isotopes) was added to 2 mM at 0, 1, and 2 d of expression.
Isolation of GE37468 was carried out following methods previously described by Bowers et al. (9). Cultures were pelleted, media decanted, and pellets resuspended 10–20% culture volume of methanol containing anhydrous sodium sulfate. After 30 min of frequent vortexing, the slurry was filtered through Whatman filter paper, rinsed with excess methanol, and solvent removed in vacuo. The resulting oil was resuspended with 2.5% the initial culture volume of 50∶50 acetonitrile/water and filtered through a 0.45-μm filter for LCMS analysis (SI Appendix, Fig. S5) or further purification by HPLC (described in SI Appendix, Fig. S6).
Supplementary Material
Acknowledgments.
We thank Michael Acker and Albert Bowers for helpful discussion on thiazolyl peptide biosynthesis and Jared Parker and Steve Malcolmson for careful reading of this manuscript. This work was supported by a New England Regional Center of Excellence grant and National Institutes of Health Grant GM20011 (to C.T.W.). T.S.Y. is supported by a National institutes of Health F32 postdoctoral fellowship.
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database, http://www.ncbi.nlm.nih.gov/genbank/ (accession no. JN052143).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1110435108/-/DCSupplemental.
References
- 1.Arndt HD, Schoof S, Lu JY. Thiopeptide antibiotic biosynthesis. Angew Chem Int Ed Engl. 2009;48:6770–6773. doi: 10.1002/anie.200901808. [DOI] [PubMed] [Google Scholar]
- 2.Bagley MC, Dale JW, Merritt EA, Xiong X. Thiopeptide antibiotics. Chem Rev. 2005;105:685–714. doi: 10.1021/cr0300441. [DOI] [PubMed] [Google Scholar]
- 3.Walsh CT, Acker MG, Bowers AA. Thiazolyl peptide antibiotic biosynthesis: A cascade of post-translational modifications on ribosomal nascent proteins. J Biol Chem. 2010;285:27525–27531. doi: 10.1074/jbc.R110.135970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhat UG, Halasi M, Gartel AL. Thiazole antibiotics target FoxM1 and induce apoptosis in human cancer cells. PLoS One. 2009;4:e5592. doi: 10.1371/journal.pone.0005592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lefranc D, Ciufolini MA. Total synthesis and stereochemical assignment of micrococcin P1. Angew Chem Int Ed Engl. 2009;48:4198–4201. doi: 10.1002/anie.200900621. [DOI] [PubMed] [Google Scholar]
- 6.Muller HM, Delgado O, Bach T. Total synthesis of the thiazolyl peptide GE2270 A. Angew Chem Int Ed Engl. 2007;46:4771–4774. doi: 10.1002/anie.200700684. [DOI] [PubMed] [Google Scholar]
- 7.Nicolaou KC, et al. Total synthesis of thiostrepton. Retrosynthetic analysis and construction of key building blocks. J Am Chem Soc. 2005;127:11159–11175. doi: 10.1021/ja0529337. [DOI] [PubMed] [Google Scholar]
- 8.Acker MG, Bowers AA, Walsh CT. Generation of thiocillin variants by prepeptide gene replacement and in vivo processing by Bacillus cereus. J Am Chem Soc. 2009;131:17563–17565. doi: 10.1021/ja908777t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowers AA, Acker MG, Koglin A, Walsh CT. Manipulation of thiocillin variants by prepeptide gene replacement: Structure, conformation, and activity of heterocycle substitution mutants. J Am Chem Soc. 2010;132:7519–7527. doi: 10.1021/ja102339q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelly WL, Pan L, Li CX. Thiostrepton biosynthesis: Prototype for a new family of bacteriocins. J Am Chem Soc. 2009;131:4327–4334. doi: 10.1021/ja807890a. [DOI] [PubMed] [Google Scholar]
- 11.Liao RJ, et al. Thiopeptide biosynthesis featuring ribosomally synthesized precursor peptides and conserved posttranslational modifications. Chem Biol. 2009;16:141–147. doi: 10.1016/j.chembiol.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris RP, et al. Ribosomally synthesized thiopeptide antibiotics targeting elongation factor Tu. J Am Chem Soc. 2009;131:5946–5955. doi: 10.1021/ja900488a. [DOI] [PubMed] [Google Scholar]
- 13.Wieland Brown LC, Acker MG, Clardy J, Walsh CT, Fischbach MA. Thirteen posttranslational modifications convert a 14-residue peptide into the antibiotic thiocillin. Proc Natl Acad Sci USA. 2009;106:2549–2553. doi: 10.1073/pnas.0900008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McIntosh JA, Donia MS, Schmidt EW. Ribosomal peptide natural products: Bridging the ribosomal and nonribosomal worlds. Nat Prod Rep. 2009;26:537–559. doi: 10.1039/b714132g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowers AA, Walsh CT, Acker MG. Genetic interception and structural characterization of thiopeptide cyclization precursors from Bacillus cereus. J Am Chem Soc. 2010;132:12182–12184. doi: 10.1021/ja104524q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butler MS. Natural products to drugs: Natural product-derived compounds in clinical trials. Nat Prod Rep. 2008;25:475–516. doi: 10.1039/b514294f. [DOI] [PubMed] [Google Scholar]
- 17.Ferrari P, Colombo L, Stella S, Selva E, Zerilli LF. Antibiotic GE37468 A: A novel inhibitor of bacterial protein synthesis. II. Structure elucidation. J Antibiot (Tokyo) 1995;48:1304–1311. doi: 10.7164/antibiotics.48.1304. [DOI] [PubMed] [Google Scholar]
- 18.Marinelli F, Gastaldo L, Toppo G, Quarta C. Antibiotic GE37468A: A new inhibitor of bacterial protein synthesis. III. Strain and fermentation study. J Antibiot (Tokyo) 1996;49:880–885. doi: 10.7164/antibiotics.49.880. [DOI] [PubMed] [Google Scholar]
- 19.Stella S, et al. Antibiotic GE37468 A: A new inhibitor of bacterial protein synthesis. I. Isolation and characterization. J Antibiot (Tokyo) 1995;48:780–786. doi: 10.7164/antibiotics.48.780. [DOI] [PubMed] [Google Scholar]
- 20.Selva E, et al. Targeted screening for elongation factor Tu binding antibiotics. J Antibiot (Tokyo) 1997;50:22–26. doi: 10.7164/antibiotics.50.22. [DOI] [PubMed] [Google Scholar]
- 21.Zuurmond AM, et al. GE2270A-resistant mutations in elongation factor Tu allow productive aminoacyl-tRNA binding to EF-Tu. GTP.GE2270A complexes. J Mol Biol. 2000;304:995–1005. doi: 10.1006/jmbi.2000.4260. [DOI] [PubMed] [Google Scholar]
- 22.Li CX, Zhang FF, Kelly WL. Heterologous production of thiostrepton A and biosynthetic engineering of thiostrepton analogs. Mol Biosyst. 2011;7:82–90. doi: 10.1039/c0mb00129e. [DOI] [PubMed] [Google Scholar]
- 23.Wang J, et al. Identification and analysis of the biosynthetic gene cluster encoding the thiopeptide antibiotic cyclothiazomycin in Streptomyces hygroscopicus 10–22. Appl Environ Microbiol. 2010;76:2335–2344. doi: 10.1128/AEM.01790-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu Y, et al. Nosiheptide biosynthesis featuring a unique indole side ring formation on the characteristic thiopeptide framework. ACS Chem Biol. 2009;4:855–864. doi: 10.1021/cb900133x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engelhardt K, Degnes KF, Zotchev SB. Isolation and characterization of the gene cluster for biosynthesis of the thiopeptide antibiotic TP-1161. Appl Environ Microbiol. 2010;76:7093–7101. doi: 10.1128/AEM.01442-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei MC, Deng J, Wang SZ, Liu N, Chen YJ. A simple reverse genetics approach to elucidating the biosynthetic pathway of nocathiacin. Biotechnol Lett. 2011;33:585–591. doi: 10.1007/s10529-010-0460-0. [DOI] [PubMed] [Google Scholar]
- 27.Ding Y, et al. Moving posttranslational modifications forward to biosynthesize the glycosylated thiopeptide nocathiacin I in Nocardia sp. ATCC202099. Mol Biosyst. 2010;6:1180–1185. doi: 10.1039/c005121g. [DOI] [PubMed] [Google Scholar]
- 28.Singh SB, Herath K, Yu NX, Walker AA, Connors N. Biosynthetic studies of nocathiacin-I. Tetrahedron Lett. 2008;49:6265–6268. [Google Scholar]
- 29.Ziermann R, Betlach MC. Recombinant polyketide synthesis in Streptomyces: Engineering of improved host strains. Biotechniques. 1999;26:106–110. doi: 10.2144/99261st05. [DOI] [PubMed] [Google Scholar]
- 30.Stella S, et al. Antibiotics GE 37468 A, B, and C. 5,618,724. US Patent. 1997
- 31.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schoof S, et al. Antiplasmodial thiostrepton derivatives: Proteasome inhibitors with a dual mode of action. Angew Chem Int Ed Engl. 2010;49:3317–3321. doi: 10.1002/anie.200906988. [DOI] [PubMed] [Google Scholar]
- 33.Schoof S, Arndt HD. D-cysteine occurrence in thiostrepton may not necessitate an epimerase. Chem Commun (Camb) 2009:7113–7115. doi: 10.1039/b912733j. [DOI] [PubMed] [Google Scholar]
- 34.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. Norwich, UK: Crowes; 2000. p. 613. [Google Scholar]
- 35.Gibson DG, et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science. 2008;319:1215–1220. doi: 10.1126/science.1151721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.