

Abstract
Cellular functions accompanying establishment of premature senescence in methotrexate-treated human colon cancer C85 cells are deciphered in the present study from validated competitive expression microarray data, analyzed with the use of Ingenuity Pathways Analysis (IPA) software. The nitrosative/oxidative stress, inferred from upregulated expression of inducible nitric oxide synthase (iNOS) and mitochondrial dysfunction-associated genes, including monoamine oxidases MAOA and MAOB, β-amyloid precursor protein (APP) and presenilin 1 (PSEN1), is identified as the main determinant of signaling pathways operating during senescence establishment. Activation of p53-signaling pathway is found associated with both apoptotic and autophagic components contributing to this process. Activation of nuclear factor κB (NF-κB), resulting from interferon γ (IFNγ), integrin, interleukin 1β (IL-1β), IL-4, IL-13, IL-22, Toll-like receptors (TLRs) 1, 2 and 3, growth factors and tumor necrosis factor (TNF) superfamily members signaling, is found to underpin inflammatory properties of senescent C85 cells. Upregulation of p21-activated kinases (PAK2 and PAK6), several Rho molecules and myosin regulatory light chains MYL12A and MYL12B, indicates acquisition of motility by those cells. Mitogen-activated protein kinase p38 MAPK β, extracellular signal-regulated kinases ERK2 and ERK5, protein kinase B AKT1, as well as calcium, are identified as factors coordinating signaling pathways in senescent C85 cells.
Electronic supplementary material
The online version of this article (doi:10.1007/s13277-011-0198-x) contains supplementary material, which is available to authorized users.
Keywords: High-dose methotrexate, Colorectal adenocarcinoma, Drug-induced senescence, Expression microarrays, Signaling pathways
Introduction
Cellular senescence is a stable and conditionally reversible growth arrest evoked by stress signals culminating in the induction of DNA damage response [1, 2]. It is viewed as a metabolically active state of hypermitogenic stimulation counterbalanced by induction of cyclin-dependent kinase (CDK) inhibitors [1]. Apart from being originally described as a replicative form of senescence occurring in normal cells that have reached a division limit, senescence induced by activated oncogenes and premature form of senescence, also termed stress-induced or accelerated, or else senescence-like growth arrest, were also identified [3, 4]. Premature senescence may proceed in normal, as well as in cancer cells, and in regard to the latter it is of interest as a consequence of DNA damage brought about by radio- and chemotherapy [4]. Two main issues concerning therapy-induced senescence that require elucidation, before it can be exploited as an anticancer strategy refer to (i) senescence associated secretory phenotype (SASP), i.e., molecules secreted by senescent cells among which growth- and inflammation-promoting as well as extracellular matrix (ECM)-modulating factors are present and (ii) irreversibility of the state of senescence [4, 5]. In view of the reports demonstrating that populations of cancer cells driven into therapy-induced senescence are able to regain proliferative functions, senescence associated with controlled cellular self-destruction process, autophagy, is postulated to underpin tumor dormancy in vivo [6].
Our previous work documented premature senescence as the sole response of human colon cancer C85 cells to the treatment with high dose classic antifolate, methotrexate [7–9]. C85 cells are driven into senescence by the drug treatment despite being also capable of undergoing apoptosis [8]. The present study was aimed at delineation of cellular functions, inferred from the pattern of activated signaling pathways, determining establishment of senescent phenotype in this cellular system. It can be hypothesized that in C85 cells exposed to methotrexate interference with nitric oxide (NO) signaling acts as a molecular trigger for nitrosative/oxidative stress generation upon senescence initiation. Maintenance of senescent phenotype is subsequently executed by autostimulatory action of SASP components and paracrine cytokine stimulation, both determining pro-inflammatory characteristics of those cells. The ability of C85 cells to reconstitute, after methotrexate removal, a cell line displaying the same kinetics of the drug sensitivity [7], correlated with autophagy and expression of ECM degrading SASP factors, indicates that in these cellular settings premature senescence might serve to enable treatment evasion rather than cancer eradication. This corresponds with known inefficacy of methotrexate applied as a single agent for colon cancer treatment.
Pathway-directed intervention is postulated to be a future strategy in cancer treatment, as despite multitude mutations of the genomes of cancer cells, limited is the number of intracellular signaling pathways underlying oncogenic transformation and consequently, tumor response to therapy [10, 11]. Functional validation in various colon cancer cells of the signaling pathways herein identified as pertaining to the establishment of methotrexate-induced senescence in C85 cells may allow us to assess a putative therapeutic strategy aimed at the ultimate destabilization of the state of senescence rather than its maintenance.
Material and methods
Cell culture
Human colorectal adenocarcinoma C85 cells, originating from primary untreated tumor classified as Duke’s stage D disease [12], were maintained in RPMI 1640 medium (Lonza, Belgium), supplemented with 10% fetal bovine serum (Sigma-Aldrich), 100 units/ml penicillin and 100 μg/ml streptomycin (both from LGC Promochem). Cells were exposed for 48 h to 1 μM methotrexate (Schircks Laboratory, Switzerland), a drug concentration corresponding to that clinically attained with the high dose methotrexate therapy [13]. This treatment variant was designated MTX48. Recovery of C85 cells after drug exposure was carried out for 96 h in the regular medium and this treatment variant was designated R96.
Expression microarray analysis
Total RNA was isolated using RNeasy Mini kit (Qiagen). cRNA labeling was performed with the use of Low RNA Input Linear Amplification kit (Agilent Technologies). Two experimental variants of competitive hybridizations were carried out with the Human whole genome expression microarrays (Agilent Technologies): (i) MTX48 cells vs. untreated cells and (ii) R96 cells vs. untreated cells. Two-color hybridizations, run in quadruplicates with dye swap between duplicates of the same variant, were performed according to Agilent Two-Color Microarray-Based Analysis protocol. Axon GenePix 4000B scanner and GenePix software (Molecular Devices) were used for feature extraction. Statistical evaluation employing Student’s t-test was performed in Acuity software (Molecular Devices). Upregulation of particular gene expression level in either variant was inferred from positive log2 ratio value. The data were submitted to ArrayExpress database and are available with E-MEXP-3018 accession at http://www.ebi.ac.uk/arrayexpress.
Signaling pathways analysis
Data sets of statistically evaluated microarray results were loaded into Ingenuity Pathways Analysis (IPA) software (Ingenuity Systems, www.ingenuity.com). Two core analyses were run individually for gene lists corresponding to gene expression profiles of MTX48 and R96 treatment variants. In each analysis expression parameters cut-offs were set at 0.5 for log2 ratio and 0.05 for p value and, as the aim was to identify activated pathways, only upregulated identifiers, considered indicative of those activated pathways, were included. Particular pathway-eligible molecules were deduced from an overlay of IPA canonical pathways, built within the software, with gene lists enriched according to the applied expression parameters cut-offs. Activated signaling pathways predominant for MTX48 and R96 experimental variants based on statistical significance (p value), were inferred from a comparison analysis of the two core analyses.
Quantitative RT-PCR
Total RNA was isolated using RNeasy Mini kit (Qiagen). Reverse transcription was performed using High-Capacity cDNA Reverse Transcription kit and real-time polymerase chain reaction (PCR), using Power SYBR Green PCR Master Mix, both from Applied Biosystems. The primers designed with the use of PrimerExpress software (Applied Biosystems) are listed in Table S1. Target gene expression level, normalized to the expression of cyclophilin D (peptidylprolyl isomerase D, PPID), used as a reference gene, was calculated applying comparative threshold cycle (C T) method.
Results
Validation of the experimental model
C85 cells exposed to methotrexate for 48 h (MTX48) constitute a homologous population of apparently enlarged cells containing irregular nucleoli and staining negative for senescence marker, senescence-associated (SA)-β-galactosidase activity (Fig. 1). These cells are considered to be committed to senesce and thus to correspond to the senescence initiation phase. The cell population recovered for 96 h after methotrexate treatment (R96) consists of two subpopulations, one constituting a small fraction of cells resembling the untreated C85 cells, and the other, being a dominant fraction of large cells, having nucleoli more regular in shape than those in cells of MTX48 variant and staining positive for SA-β-galactosidase activity. The latter subpopulation is considered to represent cells being at the senescence maintenance phase. As in the population of R96 treatment variant, regrowing cells are less represented, the transcriptome corresponding to this population is enriched in those transcripts whose expression is upregulated in senescent cell fraction. Only upregulated genes were included in the analysis of signaling pathways activated during senescence initiation and maintenance phases. Thus, the pathways inferred for C85 cells of R96 treatment variant, in relation to the untreated cells, correspond specifically to the fraction of cells at the senescence maintenance phase. This is supported by the expression pattern of cell cycle checkpoint regulatory genes, cyclins D, CDK inhibitors p21waf1/cip1 and p15 being upregulated, and CDK4, cyclins B, cell division cycle (cdc) cdc2 and cdc25C being downregulated in the population of R96 cells [9]. The signaling pathways specific for each of the senescence establishment phases, initiation and maintenance, respectively, are listed in Table 1.
Fig. 1.
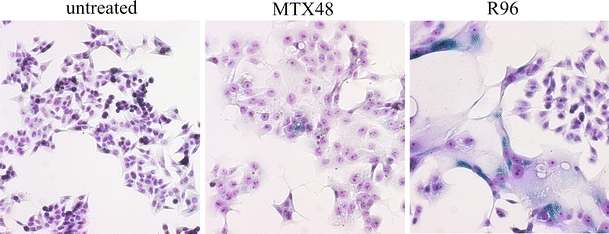
C85 cells populations in the course of establishment of methotrexate-induced senescence. Cells were colorimetrically assayed for senescence-associated β-galactosidase activity performed as previously described [8] and stained with Accustain May–Grunwald solution (Sigma-Aldrich). The images were taken with Olympus IX70 microscope at ×100 magnification
Table 1.
Predominant signaling pathways operating in C85 cells during establishment of methotrexate-induced senescence, as inferred from IPA software
| Signaling pathway | Senescence phase | |
|---|---|---|
| Initiation | Maintenance | |
| p53 signaling | 3.36 × 10−8 | 1.72 × 10−2 |
| Interferon signaling | 1.54 × 10−6 | 6.99 × 10−7 |
| Integrin signaling | 8.72 × 10−5 | 8.58 × 10−9 |
| VDR/RXR activation | 1.57 × 10−4 | 1.98 × 10−3 |
| Germ cell–Sertoli cell junction signaling | 1.82 × 10−4 | 1.41 × 10−8 |
| Regulation of actin-based motility by Rho | 1.96 × 10−4 | 6.00 × 10−6 |
| Apoptosis signaling | 2.31 × 10−4 | 3.24 × 10−3 |
| Caveolar-mediated endocytosis | 1.64 × 10−3 | 7.04 × 10−6 |
| p38 MAPK signaling | 1.95 × 10−3 | 1.91 × 10−3 |
| Xenobiotic metabolism signaling | 2.62 × 10−3 | 3.16 × 10−2 |
| Death receptor signaling | 5.53 × 10−3 | 9.60 × 10−6 |
| ERK/MAPK signaling | 9.87 × 10−3 | 5.93 × 10−6 |
| Semaphorin signaling in neurons | 1.00 × 10−2 | 3.78 × 10−6 |
| Production of nitric oxide and reactive oxygen species in macrophages | 1.93 × 10−2 | 9.75 × 10−6 |
| PI3K/AKT signaling | 2.37 × 10−2 | 6.65 × 10−6 |
| Calcium signaling | 5.47 × 10−2 | 3.53 × 10−2 |
| Clathrin-mediated endocytosis | 1.05 × 10−1 | 3.27 × 10−5 |
| ERK5 signaling | 3.38 × 10−1 | 6.85 × 10−3 |
The p value for statistical significance of each pathway is given.
Apoptosis and autophagy accompany establishment of methotrexate-induced senescence in C85 cells
p53 effectors, including apoptosis-inducing genes and essential for autophagy damage-regulated autophagy modulator gene (DRAM), are upregulated predominantly during senescence initiation phase (Table 2). Other genes involved in autophagy remain upregulated also during senescence maintenance phase. Genes regulating p53 transcriptional activity, including its binding partners (BRCA1 and PLAGL1) and modifiers, acetyltransaferase PCAF and ubiquitin ligase transcript variant MDM2b, are induced during the initiation phase. MDM2 transcript variant is upregulated during the maintenance phase (Table 2). Upregulated throughout the whole process of senescence establishment p53-inducible gene 3 (PIG3), and glycolysis and apoptosis regulator TIGAR, are known to be associated specifically with oxidative stress [14, 15].
Table 2.
Molecules involved in p53-dependent cellular processes and regulation of p53 activity whose expression is upregulated in C85 cells during establishment of methotrexate-induced senescence
| Process | Molecules |
|---|---|
| p53-dependent process | |
| Apoptosis induction | APAF1, BAX, DR4, DR5, FAS, LRDD, NOXA, PERP, PIG3, PUMA, TP53INP1 |
| Angiogenesis inhibition | SERPINB5, THBS1 |
| Autophagy induction | DRAM |
| Cell cycle arrest | CDKN1A, Cyclin G, GADD45A, GADD45B, GADD45G, SFN |
| Cell survival | BCL2L1 |
| DNA repair | PCNA, RRM2B |
| Glycolysis inhibition | TIGAR |
| Senescence | PAI-1 |
| Regulation of p53 activity | BRCA1, MDM2, MDM2b, PCAF, PLAGL1 |
| Autophagy | ATG12, ATG16L1, ATG16L2, ATG4A, ATG4B, ATG4D, BECN1, MAP1LC3B |
| Apoptosis regulation and execution | BAK1, CASP7, CASP9, CASP10, CFLAR, LMNA, SPTAN1 |
The relevant molecules were inferred from the canonical p53 and apoptosis signaling pathways implemented in IPA software and in Ref. [56]. The molecules upregulated during both senescence initiation and maintenance phases are printed bold, upregulated during the initiation phase are printed with a regular font and upregulated during the maintenance phase are underlined.
APAF1 apoptotic peptidase activating factor, ATG autophagy-related gene, BAK1 BCL2-antagonist/killer 1, BAX BCL2-associated X protein, BCL2L1 BCL2-like 1 nuclear gene encoding mitochondrial protein (Bcl-xL), BECN1 beclin 1, BRCA1 breast cancer 1, DRAM damage-regulated autophagy modulator, CASP7/9/10 caspase 7/9/10, CDKN1A p21waf1/cip1, CFLAR FLAME-1 (FLIP), DR4/5 death receptor 4/5, FAS TNF receptor superfamily, member 6, GADD45A/B/G growth arrest and DNA-damage-inducible alpha/beta/gamma, LMNA lamin A/C, LRDD leucine-rich repeats and death domain containing (PIDD), MAP1LC3B microtubule-associated protein 1 light chain 3 beta, NOXA phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1), PAI-1 plasminogen activator inhibitor type 1 (SERPINE2), PCAF p300/CBP-associated factor, PCNA proliferating cell nuclear antigen, PERP TP53 apoptosis effector, PIG3 tumor protein p53 inducible protein 3 (TP53I3), PLAGL1 pleiomorphic adenoma gene-like 1 (ZAC1), PUMA p53 upregulated modulator of apoptosis (BBC3), RRM2B ribonucleotide reductase M2B, SERPINB5 serpin peptidase inhibitor (maspin), SFN stratifin (14-3-3 sigma), SPTAN1 spectrin; TP53INP1 tumor protein p53 inducible nuclear protein 1 (Teap), TIGAR TP53-inducible glycolysis and apoptosis regulator (C12ORF5), THBS1 thrombospondin 1. Corresponding microarray data are presented in Table S2
Nitrosative/oxidative stress and antioxidant response are manifested during establishment of methotrexate-induced senescence in C85 cells
Upregulation of iNOS (NOS2A) isoform occurs during both senescence initiation and maintenance phases (Table 3). During senescence initiation eNOS (NOS3) isoform is also upregulated. Senescent C85 cells display induced expression of neutrophil cytosolic factor 4 (NCF4, p40 phox), a regulatory molecule of NADPH oxidase complex involved in the production of reactive oxygen species (ROS) on the macrophage plasma or phagosome membrane. Xenobiotic metabolism signaling pathway is not surprisingly activated in C85 cells exposed to methotrexate (Table 1). Among xenobiotic phase I detoxification enzymes, upregulation of MAOA and MAOB genes may provide a putative link to mitochondrial dysfunction and, consequently, intensified ROS formation [16, 17]. Mitochondrial dysfunction may also be potentiated during senescence maintenance due to increased expression of β-amyloid precursor protein (APP) gene, γ-secretase-regulating presenilin 1 (PSEN1) molecule, as well as two other mitochondrial factors being electron donors for complex I, glycerol-3-phosphate and oxoglutarate dehydrogenases (GPD2 and KGDH) (Table 3). Genes encoding cellular antioxidant defense enzymes, being effector molecules in nuclear factor-erythroid 2-related factor 2 (Nrf2)-mediated signaling, which constitutes a part of xenobiotic signaling pathway, are upregulated predominantly during senescence maintenance phase.
Table 3.
Molecules involved in nitrosative/oxidative stress generation and antioxidant response whose expression is upregulated in C85 cells during establishment of methotrexate-induced senescence
| Functional group | Molecules |
|---|---|
| NO producing enzymes | NOS2A, NOS3 |
| NADPH–oxidase complex regulatory molecule (transmembrane) | NCF4 |
| Phase I xenobiotic detoxification enzymes associated with mitochondrial dysfunction | MAOA, MAOB |
| Other mitochondrial dysfunction-associated factors | APP, COX6B2, GPD2, KGDH, NDUFAF1, PSEN1 |
| Nrf-2-dependent antioxidant defense enzymes | FTH1, HMOX1, PRDX1, TRXR1, SOD3, SQSTM1 |
Functional assignment of selected molecules was performed in IPA software. The molecules upregulated during both senescence initiation and maintenance phases are printed bold, upregulated during the initiation phase are printed with a regular font and upregulated during the maintenance phase are underlined APP amyloid β (A4) precursor protein, COX6B2 cytochrome c oxidase subunit VIb polypeptide 2, FTH1 ferritin heavy polypeptide 1, GPD2 glycerol-3-phosphate dehydrogenase 2, HMOX1 heme oxygenase 1 (HO-1), KGDH oxoglutarate dehydrogenase, MAOA/MAOB monoamine oxidase A/B, NCF4 neutrophil cytosolic factor 4 (p40 phox), NDUFAF1 NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, assembly factor 1, NOS2A/NOS3 inducible (iNOS)/endothelial (eNOS) nitric oxide synthase, PRDX1 peroxiredoxin 1, PSEN1 presenilin 1, TRXR1 thioredoxin reductase 1, SOD3 superoxide dismutase 3, SQSTM1 sequestosome 1. Corresponding microarray data are presented in Table S3
Acquisition of pro-inflammatory functions by C85 cells during establishment of methotrexate-induced senescence
iNOS expression is considered a hallmark of immune response [18]. Its upregulation during senescence establishment in C85 cells may proceed due to IFNγ signaling mediated by signal transducer and activator of transcription STAT1 and STAT2 (not shown) molecules, as well as interferon regulatory factor 1 (IRF1), all those factors upregulated during senescence initiation and maintenance phases (Table 5). IFNγ receptor 1 (IFNGR1) is upregulated only during the maintenance phase (Fig. 4). Inducible nitric oxide synthase (iNOS) upregulation during senescence maintenance may also be mediated by nuclear factor κB (NF-κB), activated due to autocrine action of molecules secreted by senescent C85 cells, including IL-1β, TNFSF10, TNFSF15 and an array of growth factors. It may also be executed due to activation of IL4/IL13 and IL22 receptors, as well as integrins and TLRs (Fig. 2). Additionally, activation of transmembrane NADPH oxidase complex (vide supra), points to a property of senescent C85 cells to elicit immunological response. Inflammatory phenotype of C85 cells during senescence establishment is also associated with STAT-mediated upregulation of ECM-modulating enzymes. Those include plasminogen activator urokinase (PLAU), matrix metallopeptidase MMP1 and metallopeptidase inhibitor TIMP3, serving to control the activity of MMPs (Table S6). Other genes involved in ECM degradation whose expression is induced in senescent C85 cells, include NF-κB-dependent tissue plasminogen activator PLAT (Fig. 2), and associated with various types of cancer matrix metallopeptidases MMP7, MMP10 and MMP15 (Table S6). Apart from pro-inflammatory action stimulation of TNF receptors also induces apoptosis [19]. At the point of apoptosis regulation, death receptor signaling interconnects with p53 signaling pathway.
Table 5.
Relative gene expression levels determined by quantitative RT-PCR in C85 cells subject to methotrexate treatment and recovery
| Gene name (GeneBank ID) | Treatment variant | ||
|---|---|---|---|
| Untreated | MTX48 | R96 | |
| CCND2 (NM_001759) | 1.93 ± 0.05 | 2.63 ± 0.14 (−/1.4) | 6.09 ± 0.61 (3.6/3.2) |
| CDKN1A (NM_000389) | 3.25 ± 0.21 | 28.03 ± 1.52 (4.2/8.6) | 28.75 ± 0.19 (6.2/8.9) |
| GADD45A (NM_001924) | 0.70 ± 0.062 | 4.31 ± 0.10 (4.5/6.2) | 2.21 ± 0.09 (2.4/3.2) |
| IL1R2 (NM_004633) | 0.20 ± 0.023 | 0.45 ± 0.010 (2.4/2.3) | 0.33 ± 0.019 (1.4/1.7) |
| IL1RN (NM_173842) | 0.14 ± 0.001* | 5.05 ± 0.41 (3.6/36) | 5.35 ± 0.21 (3.0/38) |
| IRF1 (NM_002198) | 0.47 ± 0.022 | 1.82 ± 0.07 (1.7/3.9) | 2.08 ± 0.09 (2.3/4.4) |
| ITGB4 (NM_000213) | 0.53 ± 0.052 | 1.40 ± 0.01 (1.8/2.6) | 0.77 ± 0.006 (1.7/1.5) |
| MAP2K4 (NM_003010) | 0.51 ± 0.03 | 0.75 ± 0.026 (−/1.5) | 0.70 ± 0.054 (1.9/1.4) |
| MAPK11 (NM_002751) | 0.43 ± 0.018 | 1.37 ± 0.09 (1.8/3.2) | 0.96 ± 0.017 (1.5/2.2) |
| MDM2 (NM_006879, NM_002392) | 1.54 ± 0.16 | 8.28 ± 0.28 (2.3/5.4; 1.6/5.4) | 4.79 ± 0.21 (1.6/3.1) |
| MICB (NM_05931) | 0.23 ± 0.020 | 0.80 ± 0.018 (2.4/3.5) | 0.35 ± 0.013 (2.0/1.5) |
| NOS2A (NM_000625) | undetected | 1.63 ± 0.09 (2.0/ ) | 16.60 ± 1.86 (10/ ) |
| NOS3 (NM_000603) | 0.11 ± 0.009* | 20.62 ± 0.65 (1.5/188) | 6.89 ± 0.05 (1.3/63) |
| PDGFA (NM_002607) | 0.24 ± 0.013 | 4.72 ± 0.47 (4.7/20) | 2.15 ± 0.10 (6.2/9.0) |
| PLA2G4C (NM_003706) | 0.48 ± 0.061 | 2.21 ± 0.28 (1.9/4.6) | 1.73 ± 0.07 (1.8/3.6) |
| PSMB8 (NM_004159) | 0.83 ± 0.053 | 2.31 ± 0.22 (1.4/2.8) | 2.00 ± 0.12 (1.8/2.4) |
| RHOF (NM_019034) | 0.75 ± 0.070 | 3.08 ± 0.18 (3.6/4.1) | 2.38 ± 0.09 (2.3/3.2) |
| RRAS (NM_006270) | 0.29 ± 0.014 | 0.49 ± 0.011 (1.6/1.7) | 0.25 ± 0.014 (−/0.9) |
| STAT1 (NM_007315) | 0.67 ± 0.062 | 3.69 ± 0.05 (1.9/5.5) | 2.76 ± 0.12 (1.9/4.1) |
| TP53 (NM_000546) | 1.81 ± 0.07 | 1.46 ± 0.08 (−/0.8) | 1.03 ± 0.04 (−/0.6) |
| UBD (NM_006398) | 0.34 ± 0.025* | 1.14 ± 0.011 (−/3.4) | 39.90 ± 1.47 (25.2/117) |
| VCL (NM_014000) | 3.30 ± 0.30 | 5.06 ± 0.25 (−/1.5) | 4.55 ± 0.06 (2.3/1.4) |
Average 2exp−ΔC T values ± SD from triplicates are presented. Asterisks indicate gene expression level detected below the 32nd cycle. Correlations between fold change in gene expression level for particular treatment variant, determined by microarray vs. quantitative RT-PCR approach, are shown in parentheses by two values, respectively. Hyphen indicates that gene expression level was not found changing by microarray approach. Two GeneBank accession numbers provided at MDM2 gene refer to MDM2b and MDM2 transcript variants as primers used were complementary to both. The following pathway-eligible molecules were included in the analysis: (i) cell cycle: CCND2 cyclin D2; CDKN1A p21waf1/cip1; GADD45A growth arrest and DNA-damage-inducible, alpha; MDM2 p53 binding protein, transcript variants MDM2b and MDM2; PDGFA platelet-derived growth factor, alpha; RRAS related RAS viral (r-ras) oncogene homolog; TP53 tumor protein p53; (ii) cytoskeletal dynamics: ITGB4 integrin beta 4; RHOF ras homolog gene family member F; VCL vinculin; (iii) IFN signaling: IRF1 interferon regulatory factor 1; MICB MHC class I polypeptide-related sequence B; PSMB8 proteasome subunit, beta type, 8; STAT1 signal transducer and activator of transcription 1; (iv) IL-1 signaling: IL1R2 interleukin 1 receptor, type II; IL1RN interleukin 1 receptor antagonist; (v) MAP kinase signaling: MAP2K4 mitogen-activated protein kinase kinase 4 (MKK4); MAPK11 mitogen-activated protein kinase 11 (p38 MAPK β); PLA2G4C phospholipase A2, group IVC; (vi) nitric oxide production: NOS2A inducible NOS (iNOS); NOS3, endothelial NOS (eNOS); (vii) protein ubiquitination: UBD ubiquitin D
Fig. 4.
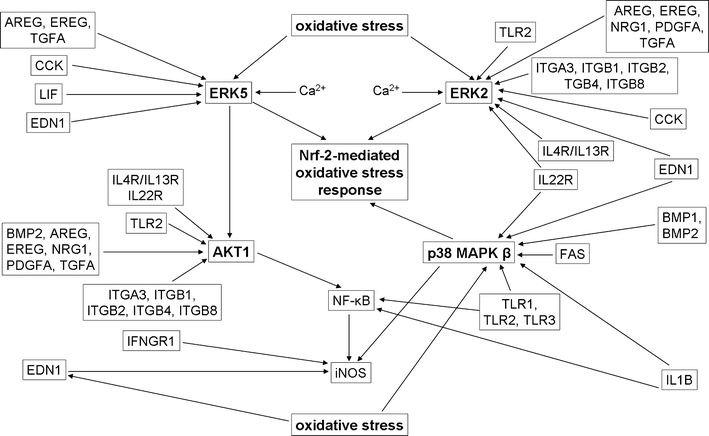
Major intracellular signaling pathways coordinating maintenance of methotrexate-induced senescence in C85 cells. The autocrine action of SASP factors as well as stimulation of FAS, IL4R/IL13R, IL22R, TLR1-3 and integrins are proposed to activate p38 MAPK β, ERK2, ERK5 and AKT1. p38 MAPK β, ERK2 and ERK5 can also be activated by ROS. ERK2 and ERK5 can be activated by Ca2+. The effector pathways include Nrf-2-mediated oxidative stress response and NF-κB-mediated survival and NO production. The latter can be potentiated due to EDN1 action and IFNGR1 stimulation. The interactions shown were inferred from the following IPA canonical signaling pathways: calcium signaling, cholecystokinin/gastrin-mediated signaling, colorectal cancer metastasis signaling, endothelin 1 signaling, ERK1/2 signaling, ERK5 signaling, IL-4 signaling, IL-22 signaling, neuregulin signaling, Nrf-2-mediated oxidative stress response, production of nitric oxide and reactive oxygen species in macrophages, PI3K/AKT signaling, p38 MAPK signaling and xenobiotic signaling. CCK cholecystokinin, EDN1 endothelin 1, IFNGR1 interferon gamma receptor 1, LIF leukemia inhibitory factor. Other abbreviations are used as in Figs. 2 and 3. Corresponding microarray data are presented in Table S7
Fig. 2.
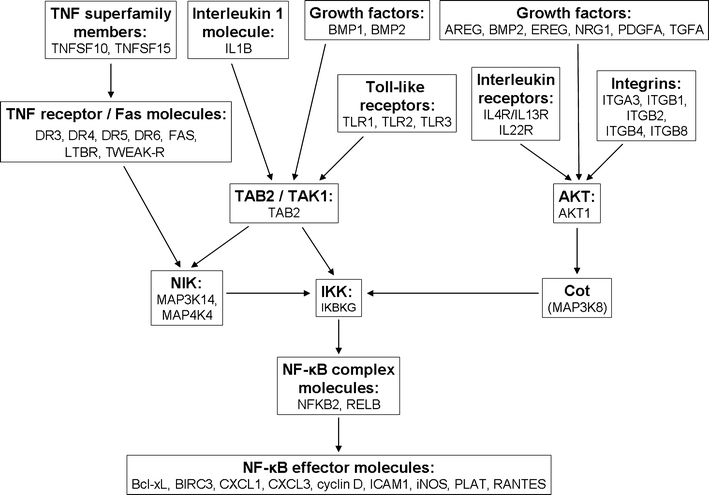
Summary of interactions leading to NF-κB activation in C85 cells during maintenance phase of methotrexate-induced senescence. Listed are selected molecules upregulated in senescent C85 cells whose functional assignment was performed in IPA software. AKT protein kinase B; AREG amphiregulin; Bcl-xL BCL2-like 1 nuclear gene encoding mitochondrial protein (BCL2L1); BIRC3 baculoviral IAP repeat-containing 3 apoptosis inhibitor; BMP1/2 bone morphogenetic protein 1/2; CXCL1/3 chemokine ligand 1/3; DR3/4/5/6 death receptor 3/4/5/6; EREG epiregulin; FAS TNF receptor superfamily, member 6; ICAM1 intracellular adhesion molecule 1; IKBKG inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase gamma; iNOS inducible nitric oxide synthase (NOS2A); LTBR lymphotoxin beta receptor; NFKB2 nuclear factor of kappa light polypeptide gene enhancer in B-cells 2; NIK NFKB-inducing kinase; NRG1 neuregulin 1; PDGFA platelet-derived growth factor alpha; PLAT tissue plasminogen activator; RANTES chemokine ligand 5; RELB v-rel reticuloendotheliosis viral oncogene homolog B; TAB2 MAP3K7-interacting protein 2; TAK1 TGF beta-activated kinase 1 (MAP3K7); TGFA transforming growth factor alpha; TWEAK-R TNF receptor superfamily member 12A. Corresponding microarray data are presented in Table S5
Cytoskeleton remodeling and endocytic pathways underlie motile functions of C85 cells during maintenance phase of methotrexate-induced senescence
Signaling pathways specifying cell adhesive interactions and cytoskeletal dynamics, are activated in C85 cells predominantly during the maintenance phase of senescence, indicating acquisition of motility (Table 1, Fig. 3). Cell-to-cell interactions also play a role in senescent C85 cells, as molecules involved in germ cell–Sertoli cell [20] and also general tight-junction formation, including catenins CTNNA1 and CTNND1, claudins (CLDN1,4,7 and 23), cingulin, occludin and zona occludens proteins TJP1 and TJP3, are also upregulated (Table S6). Importance of intercellular connections points to putative interaction between senescent cells and small descendant, regrowing cells present in the population recovering after methotrexate treatment (Fig. 1). Activation of two endocytic pathways, caveolar- and clathrin-mediated endocytosis, occurs in senescent C85 cells (Table 1). Caveolin 1 and clathrin B genes are upregulated during senescence initiation and maintenance phases (Table S6). Of note is that oxidative stress-induced premature senescence of human epithelial cells was shown to depend on upregulation of caveolin 1 gene transcription [21]. Under oxidative stress, phosphorylated caveolin 1 associated with epidermal growth factor receptor was found to determine the prolonged activation of the latter [22]. Apart from endocytosis, clathrin was also shown to participate in early autophagosome formation [23].
Fig. 3.
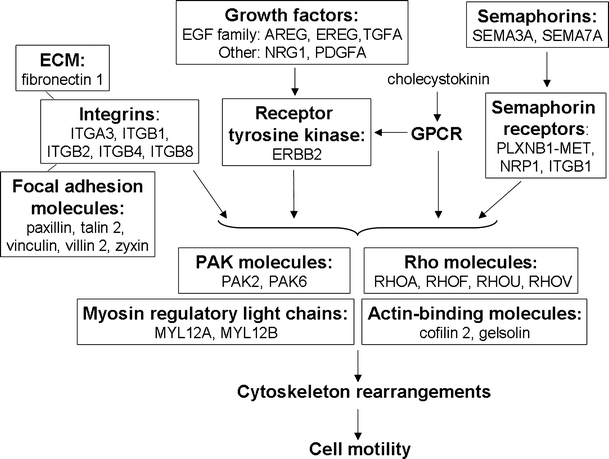
Summary of interactions determining cytoskeletal rearrangements and cell motility in C85 cells during maintenance phase of methotrexte-induced senescence. The interactions of the “outside-in” signaling direction are marked. ECM, integrins and focal adhesion molecules are shown as a complex. Listed are selected molecules upregulated in senescent C85 cells whose functional assignment was performed in IPA software. ERBB2 v-erb-b2 erythroblastic leukemia viral oncogene homolog 2, GPCR G-protein-coupled receptor, MET met proto-oncogene, MYL12A/B myosin regulatory light chain, NRP1 neuropilin 1, PAK2/6 p21-activated kinase 2/6, PLXNB1 plexin B1, Rho ras homologue gene family. Other abbreviations are used as in Fig. 2. Corresponding microarray data are presented in Table S6
p38 MAPK β, ERK2, ERK5, AKT1 and calcium mediate the main intracellular signaling pathways operating in C85 cells during establishment of methotrexate-induced senescence
Signaling cascades, described above as pertaining to senescence establishment in C85 cells treated with methotrexate converge on p38 MAPK signaling, important during both senescence initiation and maintenance phases, as well as ERK1/2, ERK5 and PI3K/AKT signaling, important predominantly during the maintenance phase (Table 1, Fig. 4). Additionally, vitamin D is identified as an important signaling factor leading to upregulated expression of platelet-derived growth factor alpha (PDGFA), several growth-suppressive molecules and calcium transporting transient receptor potential cation channel gene (TRPV6) (Tables 1 and 4). At the senescence initiation phase, upregulated expression of inositol 1,4,5-trisphosphate (IP3) receptors indicates the importance of calcium as a second messenger. It is further supported during the senescence maintenance phase by the upregulated expression of TRPV6, as well as other transmembrane calcium channels and an array of calcium signaling-involved molecules (Table 4).
Table 4.
Molecules involved in vitamin D receptor/retinoid receptor (VDR/RXR) and calcium signaling whose expression is upregulated in C85 cells during establishment of methotrexate-induced senescence
| Functional group | Molecules |
|---|---|
| VDR receptor | VDR |
| VDR/RXR effector molecules | CDKN1A, CST6, GADD45A, IGFBP3, IGFBP6, MXD1, PDGFA, PPARD, SEMA3B, SERPINB1, TRPV6 |
| Calcium transmembrane transporter and channels | ATP2B4, CACNA1C, GRIN2C, TRPV6 |
| Sarcoplasmic and endoplasmic reticulum calcium transporters | ATP2A2, ITPR1, ITPR3 |
| Calmodulin molecules | CALM2, CALM3 |
| Calcineurin molecule | PPP3CC |
| Calcium/calmodulin-dependent protein kinases (CaMK II) | CAMK2D, CAMK2G |
| cAMP-dependent protein kinases | PRKAG2, PRKAR1B |
The relevant molecules were inferred from the canonical VDR/RXR and calcium signaling pathways implemented in IPA software. The molecules upregulated during both senescence initiation and maintenance phases are printed bold, upregulated during the initiation phase are printed with a regular font and upregulated during the maintenance phase are underlined
ATP2A2/ATP2B4 ATPase calcium transporting cardiac muscle slow twitch 2/plasma membrane 4, CACNA1C voltage-dependent calcium channel L type, CDKN1A p21waf1/cip1, CST6 cystatin E/M, GADD45A growth arrest and DNA-damage-inducible alpha, GRIN2C glutamate receptor (NMDAR2C), IGFBP3/6 insulin-like growth factor binding proteins 3/6, ITPR1/3 inositol 1,4,5-triphosphate receptor type 1/3, MXD1 MAX dimerization protein 1, PDGFA platelet-derived growth factor alpha, PPARD peroxisome proliferator-activated receptor delta, PPP3CC protein phosphatase 3 catalytic subunit (calcineurin A gamma), PRKAG2 protein kinase AMP-activated gamma 2 non-catalytic subunit, PRKAR1B protein kinase cAMP-dependent regulatory type I beta, SEMA3B semaphorin 3B, SERPINB1 serpin peptidase inhibitor, clade B (ovalbumin) member 1, TRPV6 transient receptor potential cation channel subfamily V member 6. Corresponding microarray data are presented in Table S4
Validation of microarray data
Twenty-two genes upregulated in methotrexate-treated C85 cells of MTX48 and/or R96 treatment variant and identified by IPA software as eligible for the activated pathways, were chosen for their expression level measurement by quantitative RT-PCR (Table 5). All genes tested were confirmed upregulated. As only upregulated identifiers served for pathways delineation, the study is thus considered validated.
Discussion
Functional analysis of gene expression pattern of human colon cancer C85 cells driven into drug-induced senescence by methotrexate treatment is described. The nitrosative/oxidative stress associated with inflammatory functions is inferred to determine establishment of senescent phenotype in this cellular system. p53-dependent cell-fate decisive programs, apoptosis and autophagy, are found to contribute to this process. Cytoskeletal dynamics inferred to occur in senescent C85 cells indicate motility, a phenomenon corresponding with specificity of inflammatory cells. SASP of senescent C85 cells consists predominantly of growth factors and pro-inflammatory cytokines whose autocrine action is likely to stimulate maintenance of their characteristics.
Upon genotoxic insult, p53 controls cell-fate decisive processes, including senescence [15, 24]. An effector of p53 signaling, p21waf1/cip1, was shown previously to mediate methotrexate-induced senescence in C85 cells [8, 9]. Herein identified activation of p53 signaling pathway at the senescence initiation phase, correlated with apparent manifestation of oxidative stress, is indicative of DNA damage caused by ROS. Induction of oxidative stress and accumulation of DNA damage are established manifestations of methotrexate action on various cell types [25–27]. However, in regard to methotrexate used as an anticancer agent, depletion in DNA precursors pool, resulting from inhibition of dihydrofolate reductase (DHFR), an enzyme being the primary intracellular target for that drug, is considered a prevalent cause of DNA damage [28]. As oxidative stress in living systems is underpinned by excessive generation of both ROS and reactive nitrogen species (RNS), it is referred to as nitrosative/oxidative stress, especially in regard to cells containing activated NOS [29]. All NOS isoforms have a property to secrete, apart from NO, also a superoxide anion [30]. In endothelial cells under diseased states, a phenomenon of eNOS uncoupling, i.e., superoxide production by activated eNOS, is attributed to limited bioavailability of one of NOS cofactors, tetrahydrobiopterin (BH4) [31]. BH4 is recycled within a cell from dihydrobiopterin in the reaction catalyzed by DHFR [32]. In endothelial cells, methotrexate was demonstrated to cause, due to DHFR inhibition, eNOS uncoupling leading to increased eNOS-dependent superoxide production [33]. Numerous human tumors were shown to express different NOSs and, consequently, to secrete NO that may show either tumor-promoting or tumor-regressive action, with the latter proposed to be exerted by high NO concentrations evoking DNA damage [34]. Activities of constitutive NOSs (cNOSs), i.e., eNOS and nNOS (neuronal NOS), and iNOS are inversely regulated. cNOSs produce lower NO levels than iNOS, maintaining transcriptional repression of the latter. cNOSs expression decline with aging and inflammation, leading to decrease in NO level below the threshold sufficient to relieve iNOS expression, the latter also known to be mediated by NF-κB signaling [29, 35]. Analogous regulatory pattern of eNOS and iNOS expression seems to operate in C85 cells progressing into methotrexate-induced senescence (Table 5). Detection of eNOS expression in untreated C85 cells indicates NO to play a signaling role. At the senescence maintenance phase, eNOS expression decreases by 3-fold in relation to the initiation phase, and iNOS expression undetected in the untreated cells, increases by 10-fold in relation to the cells at the initiation phase. It is therefore reasonable to consider that methotrexate acting as an inhibitor of BH4 recycling leads to eNOS uncoupling and superoxide, instead of NO, generation. Upon NO level dropping below the threshold and due to pro-inflammatory signaling (Figs. 2 and 4), upregulation of iNOS expression may bring about production of toxic NO amounts. Nitrosative/oxidative stress thus generated may be further potentiated due to mitochondrial dysfunction (Table 3). Thus, toxic effects of ROS/RNS may be ultimately responsible for cellular damage and maintenance of senescent phenotype of methotrexate-treated C85 cells.
The role of ROS in senescence establishment is well documented [36–38]. According to a proposed model of a positive feedback regulatory loop between DNA damage and ROS generation, DNA damage-triggered p53 signaling leads to cell cycle arrest exerted by p21waf1/cip1, and to mitochondrial dysfunction resulting in ROS production, the latter exerted by GADD45 transmitting the signal via p38 MAPK α and transforming growth factor β (TGFβ), and providing perpetual DNA damage stimulation [39]. p38 MAPK was proposed to define a common senescence signaling pathway [40]. A signaling cascade mediated by p38 MAPK β, instead of p38 MAPK α, and independent on TGFβ, also seems to underlie maintenance of methotrexate-induced senescence in C85 cells (Table 2, Fig. 4). Apart from causing detrimental effects, ROS are also known to act as secondary messengers [41]. ROS produced by membrane NADPH oxidases are proposed to play a primary role in propagation of growth factor- and inflammatory cytokine-mediated signaling [41, 42]. In accord, putative activation of NDPH–oxidase complex by p40 phox during senescence maintenance in methotrexate-treated C85 cells (Table 3), points to ROS playing a signaling role. Inhibition of apoptosis was demonstrated to occur due to ROS-mediated inactivation of caspase 3 [43]. In summary, in C85 cells driven into senescence by methotrexate, ROS may (i) induce DNA damage, (ii) propagate growth factor and cytokine signaling and (iii) constitute one of the means to inhibit apoptosis (vide infra).
The expression profile of p53 effector genes identified in this study (Table 2) indicates that components of two cell-fate decisive programs, apoptosis and autophagy, contribute to the establishment of senescent phenotype in the cellular system studied. Apoptosis in C85 cells treated with methotrexate is inferred to be induced due to death receptor signaling, as well as by oxidative stress and/or DNA damage [14, 19]. Oxidative stress and activation of TLR may induce autophagy [44, 45]. As senescence remains the sole C85 cells outcome in response to methotrexate [8], pro-apoptotic functions are apparently counteracted by anti-apoptotic factors including Bcl-xL and cIAP member BIRC3, or ROS (vide supra) (Table 2, Fig. 2). Involvement of autophagy in the establishment of oncogene-induced form of senescence was documented for human fibroblasts [46]. In accord with both apoptosis- and autophagy-related factors participating in the establishment of senescence in C85 cells exposed to methotrexate, remains activation of p38 MAPK and ERK1/2 signaling. p38 MAPK signaling is known to mediate both cellular responses, apoptosis and senescence [47]. ERK1/2 signaling cascade is also known to mediate apoptosis and autophagy, as well as to underlie senescence [48].
Activation of motile functions in senescent C85 cells driven into senescence by methotrexate treatment (Fig. 3), corresponds well with morphology adapted by senescent cells of all types when cultured in vitro [5]. Also common to all senescent cells is SASP [49]. Although its composition varies among cellular systems, it contains molecules exerting growth-promoting, growth-suppressive and pro-inflammatory roles [3]. SASP of senescent C85 cells consists of molecules whose autocrine action is likely to provide growth and inflammatory stimulation (Fig. 4). Pro-inflammatory SASP factors identified for various senescent systems, include IL-1 and TNF molecules, as is also the case for senescent C85 cells, as well as a set of other molecules including IFNγ, IFNβ, IL-6 or IL-8 [50, 51]. The ultimate role of SASP is proposed to create a self-sustaining signaling loop that assures maintenance of senescent phenotype [52], and this is also apparent for senescent C85 cells (Fig. 4). Upregulation of TLRs, known to elicit immune response not only due to stimulation by pathogen-specific molecular patterns [53], but also by endogenous ligands [54], indicates the latter way of signaling to operate in senescent C85 cells. ECM-modulating factors facilitating cell movement are also present in SASP of those cells (Table S6). The growth-suppressive role is ascribed to secreted by senescent C85 cells, IGFBP3 and IGFBP6 molecules (Table 4), whose action is to inhibit insulin-like growth factor signaling pathway.
Senescent cells are characterized by common morphology and a common set of molecular markers [3]. Despite considerable variability among senescent cellular systems, a commonality among signaling pathways regulating senescence was deciphered to occur [55]. Activation of p53, IFN, TLR, cytoskeleton reorganization, MAP kinase and oxidative stress signaling and inhibition of insulin growth factor-related signaling by IGFBPs, were proposed to be critical for senescence establishment. The results of the present study conform to that general senescence signaling network and allow us to identify the sets of molecules specifically involved in each pathway execution in C85 cells driven into senescence by methotrexate treatment. Among other signaling components, nitric oxide, calcium, a set of cytokines including TNF superfamily members, and hormones, are identified as contributing to the maintenance of senescent phenotype in those cells. Interference of methotrexate with nitric oxide signaling emerges as a constituent of the drug cytostatic action on colon cancer C85 cells.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Sequences of primers designed with the use of PrimerExpress software (Applied Biosystems), used in real-time PCR. (DOC 49 kb)
Molecules involved in p53-dependent cellular processes and regulation of p53 activity whose expression is upregulated in C85 cells during establishment of methotrexate-induced senescence. (DOC 94 kb)
Molecules involved in nitrosative/oxidative stress generation and antioxidant response whose expression is upregulated in C85 cells during establishment of methotrexate-induced senescence. (DOC 54 kb)
Molecules involved in vitamin D receptor/retinoid receptor (VDR/RXR) and calcium signaling whose expression is upregulated in C85 cells during establishment of methotrexate-induced senescence. (DOC 68 kb)
Molecules involved in NF-κB activation in C85 cells during establishment of methotrexate-induced senescence. (DOC 100 kb)
Molecules involved in cytoskeletal rearrangements, cell motility and cell-to-cell interactions upregulated in C85 cells during establishment of methotrexte-induced senescence. Growth factors and integrins are listed in Supplementary Table 5. (DOC 90 kb)
Molecules involved in the main intracellular signaling pathways coordinating establishment of methotrexate-induced senescence in C85 cells. Listed are the molecules that were not included in Supplementary Tables 5 and 6. (DOC 46 kb)
Acknowledgments
Conflicts of interest
None
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003;4:358–62. doi: 10.1038/sj.embor.embor806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.d’Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–22. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 3.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 4.Gewirtz DA, Holt SE, Elmore LW. Accelerated senescence: an emerging role in tumor cell response to chemotherapy and radiation. Biochem Pharmacol. 2008;76:947–57. doi: 10.1016/j.bcp.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 5.Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–7. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gewirtz DA. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy. 2009;5:1232–4. doi: 10.4161/auto.5.8.9896. [DOI] [PubMed] [Google Scholar]
- 7.Dabrowska M, Hendrikx PJ, Skierski J, Malinowska M, Bertino JR, Rode W. EGFP fluorescence as an indicator of cancer cells response to methotrexate. Eur J Pharmacol. 2007;555:93–9. doi: 10.1016/j.ejphar.2006.10.052. [DOI] [PubMed] [Google Scholar]
- 8.Dabrowska M, Mosieniak G, Skierski J, Sikora E, Rode W. Methotrexate-induced senescence in human adenocarcinoma cells is accompanied by induction of p21waf1/cip1 expression and lack of polyploidy. Cancer Lett. 2009;284:95–101. doi: 10.1016/j.canlet.2009.04.015. [DOI] [PubMed] [Google Scholar]
- 9.Dabrowska M, Skoneczny M, Mosieniak G, Sikora E, Rode W. Expression of cell cycle checkpoints regulatory genes during methotrexate-induced senescence in human adenocarcinoma cells. Pteridines. 2009;20:143–7. [Google Scholar]
- 10.Copeland NG, Jenkins NA. Deciphering the genetic landscape of cancer-from genes to pathways. Trends Genet. 2009;25:455–62. doi: 10.1016/j.tig.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 11.Swanton C, Caldas C. Molecular classification of solid tumors: towards pathway-driven therapeutics. Br J Cancer. 2009;100:1517–22. doi: 10.1038/sj.bjc.6605031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Longo GSA, Izzo J, Gorlick JR, Banerjee D, Jhanwar SC, Bertino JR. Characterization and drug sensitivity of four newly established colon adenocarcinoma cell lines to antifolate inhibitors of thymidylate synthase. Oncol Res. 2000;12:309–14. doi: 10.3727/096504001108747756. [DOI] [PubMed] [Google Scholar]
- 13.Zelcer S, Kellick M, Wexler LH, Gorlick R, Meyers PA. The Memorial Sloan Kettering Cancer Center experience with outpatient administration of high dose methotrexate with leucovorin rescue. Pediatr Blood Cancer. 2008;50:1176–80. doi: 10.1002/pbc.21419. [DOI] [PubMed] [Google Scholar]
- 14.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein A. A model for p53-induced apoptosis. Nature. 1997;389:300–5. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 15.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–83. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 16.Kumar MJ, Nicholls DG, Andersen JK. Oxidative α-ketoglutarate dehydrogenase inhibition via subtle elevations in monoamine oxidase B levels results in loss of spare respiratory capacity. J Biol Chem. 2003;278:46432–9. doi: 10.1074/jbc.M306378200. [DOI] [PubMed] [Google Scholar]
- 17.Manoli I, Le H, Alesci S, McFann KK, Su YA, Kino T, Chrousos GP, Blackman MR. Monoamine oxidase-A is a major target gene for glucocorticoids in human skeletal muscle cells. FASEB J. 2005;19:1359–61. doi: 10.1096/fj.04-3660fje. [DOI] [PubMed] [Google Scholar]
- 18.Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–42. [PubMed] [Google Scholar]
- 19.Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Apoptosis signaling by death receptors. Eur J Biochem. 1998;254:439–59. doi: 10.1046/j.1432-1327.1998.2540439.x. [DOI] [PubMed] [Google Scholar]
- 20.Mruk DD, Cheng CY. Sertoli–Sertoli and Sertoli–germ cell interactions and their significance in germ cell movement in the seminiferous epithelium during spermatogenesis. Endocr Rev. 2004;25:747–806. doi: 10.1210/er.2003-0022. [DOI] [PubMed] [Google Scholar]
- 21.Dasari A, Bartholomew JN, Volonte D, Galbiati F. Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res. 2006;66:10805–14. doi: 10.1158/0008-5472.CAN-06-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khan EM, Heidinger JM, Levy M, Lisanti MP, Ravid T, Goldkorn T. Epidermal growth factor receptor exposed to oxidative stress undergoes src- and caveolin-1-dependent perinuclear trafficking. J Biol Chem. 2006;281:14486–93. doi: 10.1074/jbc.M509332200. [DOI] [PubMed] [Google Scholar]
- 23.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–57. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H. Molecular signaling and genetic pathways of senescence: its role in tumorigenesis and ageing. J Cell Physiol. 2007;210:567–74. doi: 10.1002/jcp.20919. [DOI] [PubMed] [Google Scholar]
- 25.Herman S, Zurgil N, Deutsch M. Low dose methotrexate induces apoptosis with reactive oxygen species involvement in T lymphocytic cell lines to a greater extent than in monocytic lines. Inflamm Res. 2005;54:273–80. doi: 10.1007/s00011-005-1355-8. [DOI] [PubMed] [Google Scholar]
- 26.Martin SA, McCarthy A, Barber LJ, Burgess DJ, Parry S, Lord CJ, Ashworth A. Methotrexate induces oxidative damage and is selectively lethal to tumour cells with defects in the DNA mismatch repair gene MSH2. EMBO Mol Med. 2009;1:323–37. doi: 10.1002/emmm.200900040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robaey P, Krajinovic M, Marcoux S, Moghrabi A. Pharmacogenetics of the neurodevelopmental impact of anticancer chemotherapy. Dev Disabil Res Rev. 2008;14:211–20. doi: 10.1002/ddrr.29. [DOI] [PubMed] [Google Scholar]
- 28.Kinsella AR, Smith D, Pickard M. Resistance to chemotherapeutic antimetabolites: a function of salvage pathway involvement and cellular response to DNA damage. Br J Cancer. 1997;75:935–45. doi: 10.1038/bjc.1997.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mariotto S, Miscusi M, Persichini T, Colasanti M, Suzuki H. Ageing-related role of nitric oxide in the brain. In: Straub RH, Mochegiani E, editors. The neuroendocrine immune network in ageing. Elsevier B.V., Amsterdam, The Netherlands; 2004. pp 291–300.
- 30.Stuehr D, Pou S, Rosen GM. Oxygen reduction by nitric-oxide synthases. J Biol Chem. 2001;276:14533–6. doi: 10.1074/jbc.R100011200. [DOI] [PubMed] [Google Scholar]
- 31.Cosentino F, Luscher TF. Tetrahydrobiopterin and endothelial nitric oxide synthase activity. Cariovascular Res. 1999;43:274–8. doi: 10.1016/S0008-6363(99)00134-0. [DOI] [PubMed] [Google Scholar]
- 32.Hasegawa H, Sawabe K, Nakanishi N, Wakasugi OK. Delivery of exogenous tetrahydrobiopterin (BH4) to cells of target organs: role of salvage pathway and uptake of its precursor in effective elevation of tissue BH4. Mol Genet Metab. 2005;86:S2–10. doi: 10.1016/j.ymgme.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 33.Crabtree MJ, Tatham AL, Hale AB, Alp NJ, Channon KM. Critical role for tetrahydrobiopterin recycling by dihydrofolate reductase in regulation of endothelial nitric-oxide synthase coupling: relative importance of the de novo biopterin synthesis versus salvage pathways. J Biol Chem. 2009;284:28128–36. doi: 10.1074/jbc.M109.041483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumor progression. Nat Rev Cancer. 2006;6:521–34. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 35.Colasanti M, Suzuki H. The dual personality of NO. Trends Pharmacol Sci. 2000;21:249–52. doi: 10.1016/S0165-6147(00)01499-1. [DOI] [PubMed] [Google Scholar]
- 36.Passos JF, von Zglinicki T. Oxygen free radicals in cell senescence: are they signal transducers? Free Radic Res. 2006;40:1277–83. doi: 10.1080/10715760600917151. [DOI] [PubMed] [Google Scholar]
- 37.Passos JF, Saretzki G, Ahmed S, Richter T, Peters H, Wappler I, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLOS. 2007;5:e110. doi: 10.1371/journal.pbio.0050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramsey MR, Sharpless NE. ROS as a tumor suppressor? Nat Cell Biol. 2006;8:1213–5. doi: 10.1038/ncb1106-1213. [DOI] [PubMed] [Google Scholar]
- 39.Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010;6:347. doi: 10.1038/msb.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iwasa H, Han J, Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells. 2003;8:131–44. doi: 10.1046/j.1365-2443.2003.00620.x. [DOI] [PubMed] [Google Scholar]
- 41.Sauer H, Wartenberg M, Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem. 2001;11:173–86. doi: 10.1159/000047804. [DOI] [PubMed] [Google Scholar]
- 42.Woo HA, Yim SH, Shin DH, Kang D, Yu DY, Rhee SG. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell. 2010;140:517–28. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 43.Choi K, Ryu SW, Song S, Choi H, Kang SW, Choi C. Caspase-dependent generation of reactive oxygen species in human astrocytoma cells contributes to resistance to TRAIL-mediated apoptosis. Cell Death Differ. 2010;17:833–45. doi: 10.1038/cdd.2009.154. [DOI] [PubMed] [Google Scholar]
- 44.Delgado MA, Deretic V. Toll-like receptors in control of immunological autophagy. Cell Death Differ. 2009;16:976–83. doi: 10.1038/cdd.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007;17:422–7. doi: 10.1016/j.tcb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 46.Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15:11–8. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- 48.Cagnol S, Chambard JC. ERK and cell death: Mechanisms of ERK-induced cell death—apoptosis, autophagy and senescence. FEBS J. 2009;277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]
- 49.Fumagali M, d’Adda di Fagagna F. SASPense and DDRama in cancer and ageing. Nat Cell Biol. 2009;11:921–3. doi: 10.1038/ncb0809-921. [DOI] [PubMed] [Google Scholar]
- 50.Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, et al. Persistent DNA damage signaling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–81. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Novakova Z, Hubackova S, Kosar M, Janderova-Rossmeislova L, Dobrovolna J, Vasicova P, et al. Cytokine expression and signaling in drug-induced cellular senescence. Oncogene. 2010;29:273–84. doi: 10.1038/onc.2009.318. [DOI] [PubMed] [Google Scholar]
- 52.Bartek J, Hodny Z, Lukas J. Cytokine loops driving senescence. Nat Cell Biol. 2008;10:887–9. doi: 10.1038/ncb0808-887. [DOI] [PubMed] [Google Scholar]
- 53.Abreu MT. Toll-like receptor signaling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 2010;10:131–43. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 54.Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, Kroemer G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2008;15:3–12. doi: 10.1038/sj.cdd.4402269. [DOI] [PubMed] [Google Scholar]
- 55.Fridman AL, Tainsky MA. Critical pathways in cellular senescence and immortalization revealed by gene expression profiling. Oncogene. 2008;27:5975–87. doi: 10.1038/onc.2008.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequences of primers designed with the use of PrimerExpress software (Applied Biosystems), used in real-time PCR. (DOC 49 kb)
Molecules involved in p53-dependent cellular processes and regulation of p53 activity whose expression is upregulated in C85 cells during establishment of methotrexate-induced senescence. (DOC 94 kb)
Molecules involved in nitrosative/oxidative stress generation and antioxidant response whose expression is upregulated in C85 cells during establishment of methotrexate-induced senescence. (DOC 54 kb)
Molecules involved in vitamin D receptor/retinoid receptor (VDR/RXR) and calcium signaling whose expression is upregulated in C85 cells during establishment of methotrexate-induced senescence. (DOC 68 kb)
Molecules involved in NF-κB activation in C85 cells during establishment of methotrexate-induced senescence. (DOC 100 kb)
Molecules involved in cytoskeletal rearrangements, cell motility and cell-to-cell interactions upregulated in C85 cells during establishment of methotrexte-induced senescence. Growth factors and integrins are listed in Supplementary Table 5. (DOC 90 kb)
Molecules involved in the main intracellular signaling pathways coordinating establishment of methotrexate-induced senescence in C85 cells. Listed are the molecules that were not included in Supplementary Tables 5 and 6. (DOC 46 kb)