

Abstract
Introduction
Alzheimer’s disease (AD) is characterized by the accumulation and extensive deposition of amyloid beta in the parenchyma of the brain. This accumulation of amyloid is associated with perturbations in synaptic function, impairments in energy metabolism and induction of a chronic inflammatory response, which acts to promote neuronal loss and cognitive impairment.
Areas Covered
Currently, there are no drugs that target the underlying mechanisms of Alzheimer’s disease. Here, we propose that a class of nuclear receptors are novel and promising new therapeutic targets for Alzheimer’s disease. This review summarizes the literature on nuclear receptors and their effects on AD-related pathophysiology.
Expert opinion
Nuclear receptors are attractive targets for the treatment of AD due to their ability to facilitate degradation of Aβ, affect microglial activation and suppress the inflammatory milieu of the brain. LXR agonists have proven difficult to move into clinical trials since long-term treatment results in hepatic steatosis. It is our view that PPARɣ activation remains a promising avenue for the treatment for AD, however, the poor BBB permeability of the currently available agonists and the negative outcome of the phase III clinical trials are likely to diminish interest in pursuing this target.
Keywords: Alzheimer’s disease, Liver X Receptors, Peroxisome Proliferator-Activated Receptors, Retinoid X Receptor, ApoE, Inflammation, Microglia
1.0 Introduction
Alzheimer’s disease (AD) is the leading cause of dementia in the elderly. This disease was first discovered by Dr. Alois Alzheimer in 1907, and is characterized by the deposition of extracellular amyloid plaques and intraneuronal neurofibrillary tangles. According to the World Health Organization there are currently 18 million people affected with AD worldwide and it is estimated that this number will double by the year 2050 [1]. The annual cost of treating AD in the United States is now $170 billion. AD is reaching epidemic proportions and treatments aimed at prevention or treating this disorder are urgently needed.
Alzheimer’s disease is a chronic neurodegenerative disease characterized by the progressive deposition of the amyloid beta (Aβ) in the parenchyma of the brain. A substantial body of literature supports the centrality Aβ homeostasis in disease progression. The Aβ peptide is generated by the sequential cleavage of the amyloid precursor protein (APP) by the beta and gamma secretases, resulting in the generation of peptides 40 or 42 amino acids in length [2] . These peptides are spontaneously deposited into both diffuse and dense-core plaques, principally in the cortex and hippocampus. Genetically inherited forms of AD arise from mutations in APP that are clustered in the vicinity of the beta and gamma secretase sites or within the gamma secretase itself, favoring Aβ generation [3]. Importantly, it was recently reported that sporadic, late onset AD is associated with an impaired ability to clear Aβ from the brain [4]. Thus, enhanced removal of Aβ from the diseased brain may alleviate symptoms associated with the disease. It has been postulated that soluble or small oligomeric forms of Aβ have deleterious effects in the brain, inducing impaired synaptic function and promoting neuronal degeneration [5].
The development of dense-core amyloid plaques is associated with a robust immune response mediated by microglial cells. Microglia are the principal immune effectors of the central nervous system and are derived embryonically from a myeloid lineage [6]. While this cell type was first discovered over a century ago, the complexity of microglial actions in inflammatory diseases of the CNS is just recently beginning to be understood in detail. In the AD brain, microglia are found to be intimately associated with amyloid plaques and display an “activated” morphology [7-9]. The plaque-associated microglia secrete a variety of cytotoxic species including the inflammatory cytokines , interferon gamma (INFɣ), tumor necrosis factor alpha (TNFα), interleukin 1β (IL-1β) and interleukin 6 (IL-6) and chemokines, most prominently CCL2 [10-12]. It is hypothesized that this inflammatory milieu contributes to a toxic environment in the brain and promotes the subsequent neuronal loss associated with AD.
2.0 Nuclear Receptors
Nuclear receptors (NRs) are ligand-activated transcription factors which provide a critical and direct linkage between the environment and the genome (Figure 1). In the past decade, drugs targeting the nuclear receptors Peroxisome proliferator-activated receptor ɣ (PPARɣ) and Liver X receptor (LXR) have shown to ameliorate pathogenesis in animal models of AD. These type II NRs form obligate heterodimers with the Retinoid X receptors (RXRs) to form a functional transcription factor. While the role of these receptors has been well characterized in the periphery, their actions in the brain have only recently been investigated. Recently , they have been shown to promote the degradation of the Aβ peptides in the brain by activating genes responsible for reverse cholesterol transport [13]. Activation of these receptors has been shown to suppress microglial-mediated inflammatory responses both in vitro and in vivo [11, 14]. Recent findings now suggest these receptors may play a key role in microglial activation by acting in concert to dampen the inflammatory response and promote tissue repair [15]. Agonists of RXRs are postulated to have similar effects on AD through their association and activation of both PPARɣ and LXR signaling pathways. While other nuclear receptors have also been shown to have effects on AD, the focus of this review will be on PPARs, LXRs and RXRs and their beneficial effects on the pathophysiology of AD.
Figure 1.
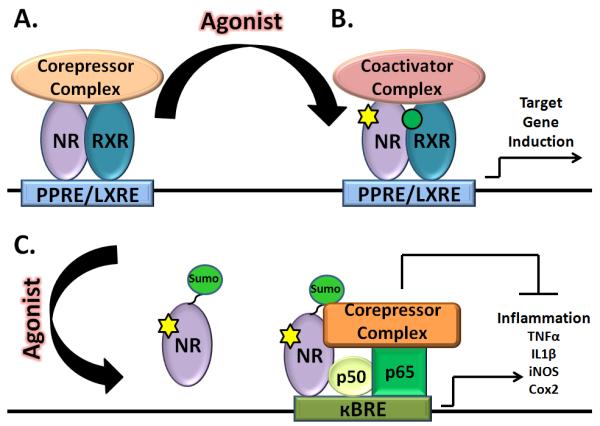
In the absence of ligand, type II nuclear receptors heterodimerize with RXR and bind response elements located on the promoters of target genes in association with a corepressor complex actively repressing the expression of target genes (A). In the presence of ligands, the receptor heterodimer undergoes a conformational change, resulting in the exchange of the corepressor complex for a coactivatior complex and allowing for gene transcription (B). Upon ligand binding, PPARɣ becomes sumoylated and recruited to the NFκB-corepressor complex. This interaction secures the corepressor complex on the κBRE promoter elements, preventing its stimulus-induced dismissal and maintains the NFκB inflammatory genes in a repressed state (C).
3.0 Gene Activation by Nuclear Receptors
Nuclear receptors are ligand-activated transcription factors that play diverse roles in a variety of signaling pathways involved in development and metabolism. All members of the nuclear receptor superfamily are structurally conserved and act to directly regulate gene expression through their association with sequence-specific elements within the promoter regions of their target genes. Type II nuclear receptors are comprised of three main domains, the amino (N)-terminal activation domain known as the activation function 1 domain (AF1), which is necessary for coactivator recruitment, the carboxy (C)-terminal ligand-binding domain (LBD) and the DNA-binding domain (DBD). The DBD is highly conserved, mediates binding to specific response elements (LXREs or PPREs) on the promoters of specific target genes [16].
Type II NRs, including the PPARs and LXRs, form obligate heterodimers with RXR. These heterodimers are retained in the nucleus regardless of their ligand binding status [17]. In the absence of ligand the coreceptor complex is constitutively associated with a nuclear corepressor complex consisting of nuclear receptor co-repressor (NCoR) or the silencing mediator of retinoic acid thyroid hormone (SMRT) and histone deactylases (Figure 1). Upon ligand binding a conformational change results in the release of the corepressor complex and the recruitment of a coactivator complex, comprised of CBP, p300 or others. The coactivator complex has intrinsic histone acetyltransferase activity allowing chromatin decondensation, thus promoting transcription of target genes (Figure 1).
RXRs have been classified as “promiscuous” due to their ability to form heterodimers with a variety of other type II NRs. RXR binding partners have been further characterized as either permissive or non-permissive binding partners. Permissive binding partners, such as LXRs and PPARs, can be activated by ligands specific to either receptor in the heterodimer (Figure 2). Heterodimers formed between RXR and non-permissive binding partners, such as the thyroid hormone receptor, can only be activated by ligands specific to the non-permissive binding partner [18].
Figure 2.

Activation of nuclear receptors PPARɣ and LXR induce the expression of genes necessary for insulin sensitization and lipid metabolism. Interestingly, activation of PPARɣ induces LXRα and LXRα induces expression of PPARɣ, driving a positive-feedback loop (A). RXR activation induces the activation of their permissive heterodimer binding partners, resulting the expression of both PPARɣ and LXRα target genes (B).
3.1 Peroxisome proliferator-activated receptors (PPARs)
PPARs have been shown to play essential roles in energy metabolism, adipocyte differentiation, insulin sensitization and tumor suppression. Their endogenouse ligands are dietary lipids and their metabolites [19]. They act as dominant regulators of lipid metabolism through their ability to transactivate genes encoding enzymes of lipid metabolism, providing a key linkage between the diet and the genome. This family is comprised of three isoforms, PPARα (NR1C1), PPARɣ (NR1C3) and PPARβ/δ (NR1C2). PPARα is known to regulate lipid oxidation but is not highly expressed in the brain [20]. PPARɣ is involved in lipid storage, insulin sensitivity and energy metabolism and has been shown to promote adipocyte differentiation [21]. PPARβ/δ is the most abundant isoform and plays a role in fatty acid oxidation [22]. Thiazolidinedione (TZD) agonists of PPARɣ, such as pioglitazone (Actos™) and rosiglitazone (Avandia™) are FDA approved for the treatment of type II diabetes [23]. PPARɣ has been most widely studied in the CNS and the effects of its agonists in the brain have been well documented [24]. All PPAR species have been shown to exert robust anti-inflammatory actions [25].
3.2 Liver X Receptors (LXRs)
LXR receptors are activated by oxysterols, most prominently hydroxylated forms of cholesterol, and play a critical role in the control of whole body cholesterol homeostasis, as well as exerting potent anti-inflammatory actions [26]. Two isoforms of LXR have been identified, LXRα (NR1H3) and LXRβ (NR1H2). LXRβ is ubiquitously expressed while LXRα is abundantly expressed in macrophages [27]. Due to their ability to regulate cholesterol metabolism, LXR agonists have emerged as possible therapeutic targets for atherosclerosis. However, their therapeutic utility is compromised owing to their induction of hepatic steatosis. LXRs have also been shown to play an important role in the CNS. LXRβ knockout mice display adult onset motor degeneration that is accompanied by axonal dystrophy, astrogliosis, and lipid accumulation [28]. Animals lacking both LXRα and β have a variety of abnormalities in the brain including the accumulation of lipid droplets, loss of neurons, astrocytic proliferation, closure of the ventricles and abnormalities in vasculature [29].
3.3 Retinoid X Receptors (RXRs)
RXRs play a central role in nuclear receptor biology due to their heterodimerization with many other type II nuclear receptor family members, including LXRs and PPARs (Figure 2). As a result they play a critical role in nuclear receptor-mediated gene expression [30, 31]. There are three RXR isotypes, RXRα (NR2B1), RXRβ (NR2B2) and RXRɣ (NR2B3). RXRα is most abundantly expressed in myeloid cells [32]. Ligands for RXRs include 9-cis retinoic acid, all-trans retinoic acid (ATRA), endogenous fatty acids and synthetic agonists, termed rexinoids [33, 34]. Currently, the rexinoid bexarotene (Targretin™) is prescribed for treatment of cutaneous T-cell lymphoma and displays a favorable side-effect profile. Recently, RXRɣ activation was shown to play a role in oligodendrocyte differentiation and to enhance remyelination [35].
4.0 Apolipoprotein E and Alzheimer’s Disease
The ApoE gene is the principal genetic risk factor for sporadic forms of AD. Humans express one of three ApoE alleles; ApoE2, ApoE3 and ApoE4, these isoforms differ only by two amino acids. The ApoE4 allele is most important genetic risk factor for AD, while the ApoE2 allele is thought to be protective. Forty percent of people afflicted with AD possess at least one copy of the ApoE4 allele which increases the risk for AD by 3-4 fold. Possession of two copies of the ApoE4 allele increases the risk factor for AD by 12-fold. The isoform-specific actions of ApoE on amyloid deposition have been demonstrated in APP mice expressing the human ApoE isoforms. Mice expressing the ApoE4 isoform exhibited higher levels of Aβ deposition in comparison to ApoE3 or ApoE2 expressing animals [36]. Interestingly, APP-expressing transgenic mice lacking murine apoe failed to develop compact, but not diffuse, plaques suggesting a role for this protein in the deposition of Aβ in the brain. It is of interest to note, however, that while no compact plaques were observed in the absence of ApoE, the total levels of Aβ within the brains of these animals was significantly elevated [37], arguing for a role in Aβ clearance.
ApoE acts to scaffold the formation of high density lipoproteins (HDL) that function to transport cholesterol and lipids throughout the body and in the brain. In the brain, ApoE is primarily synthesized and secreted by astrocytes although it is expressed at lower levels by microglia. The lipidation of ApoE is carried out primarily by the actions of the ATP-binding cassette transportor A1 (ABCA1) in the brain, which transfers phospholipids and cholesterol to ApoE, forming HDL particles. The size of the HDL particle is proportionate to its lipid content. The lipidation of ApoE governs its functions including its conformation, interactions with membrane receptors and its interaction with Aβ [38, 39]. ApoE4 is associated with smaller, poorly lipidated HDL particles [13]. Numerous studies have shown that both PPARɣ and LXRs induce the expression of ApoE and ABCA1 and it is through the expression of these proteins that they exert their effects on amyloid pathology.
5.0 Nuclear Receptors and Amyloid Clearance in Mouse Models of AD
Genetic studies have long suggested a relationship between dyslipidemia, high cholesterol levels and AD risk. Genes associated with cholesterol regulation such as APOE, ABCA1, LXRβ, ACAT and LRP1 have also been shown to share a linkage with AD [40, 41]. While genes of cholesterol metabolism, and most prominently the APOE4 allele, are known to play a crucial role in AD pathogenesis, the exact mechanisms through which they confer susceptibility to AD is just recently being understood. Jiang et al. were the first to demonstrate lipdated ApoE species act to promote Aβ proteolysis, providing a mechanistic linkage between the major genetic risk factor for AD and the normal clearance of Aβ from the brain. They were able to show that the lipidation of ApoE enhanced the degradation of soluble species of Aβ by neprilysin in the endolytic compartments of microglia as well as extra-cellularly through the actions of the insulin-degrading enzyme (IDE) [13]. Both ABCA1 and ApoE are important mediators of Aβ clearance since microglia lacking apoe or abca1 lost the capacity to degrade soluble Aβ. Importantly, this study utilized the LXR agonist, GW3965, to activate the LXRs and induce the expression of both ApoE and ABCA1. Significantly, a four month treatment of Tg2576 mice with GW3965 reduced plaque deposition by over 50% and improved contextual memory in these animals [13]. Importantly, the ability for microglial cells to degrade Aβ was ApoE isoform-dependent [13]. Indeed, macrophages expressing the ApoE2 allele were the most efficient at degrading Aβ, followed by ApoE3, then ApoE4; microglia lacking ApoE were the least efficient at degrading Aβ [13, 42]. LXR activation increased the ApoE particle size of all human ApoE isoforms, suggesting that activation of this pathways may enhance Aβ clearance regardless of the ApoE allele expressed [13]. Studies conducted by other laboratories have also shown LXR activation has beneficial effects on amyloid deposition and memory retention in mouse models of AD [43-45]. LXR activation was also shown to suppress amyloid deposition and improve behavior in APP23 mice induced by a high fat diet [46]. Consistent with this hypothesis, it was shown by Zelcer et al. that genetic inactivation of LXRα or LXRβ decreased levels of both ApoE and ABCA1 protein levels in APP/PS1 mice exacerbated plaque pathology[47]. A different result was reported by Vanmierlo et al., who found that LXR agonist treatment resulted in restoration of memory, but did not find a change in plaque burden [48]. Conversely, Terwel et al. found the LXR agonist treatment had modest effects on spatial learning, but robustly reduced plaque and Aβ levels [49].
The importance of ABCA1 function in Aβ clearance was confirmed in three independent studies where the abca1 gene was inactivated in four different APP-expressing transgenic mouse models. The loss of abca1 resulted in not only the reduction of ApoE levels, but also a paradoxical increase in Aβ deposition in the brain parenchyma of these animals owing to enhanced deposition of poorly lipidated ApoE in the brain [50-52]. Conversely, overexpression of ABCA1 in a mouse model of AD, was shown to decrease both soluble and fibrillar pools of Aβ in 12 month old mice and reduce plaque burden [53].
The vital role of ABCA1 in LXR-mediated amyloid clearance was demonstrated in a recent study by Donkin et al. In this study, AD transgenic mice that expressed or lacked abca1 were treated with the LXR agonist GW3965. APP/PS1 mice treated with high or low doses of GW3965 showed elevated brain levels of ABCA1 and ApoE, decreased amyloid burden and significant improvement in memory. However, APP/PS1 mice lacking abca1 displayed little change in brain ApoE levels, no reduction in amyloid load or behavioral improvements. These studies, taken together, suggest that LXRs are excellent therapeutic targets for AD and that both ABCA1 and ApoE are necessary for the beneficial effects of LXR agonists [54].
PPARɣ was recognized as a therapeutic target for AD about a decade ago, owing not only to its actions on inflammation, but also its effects on insulin sensitization and energy metabolism [24, 55, 56]. Similar to LXRs, PPARɣ activation can also induce the expression of both ABCA1 and ApoE. Additionally, PPARɣ can also induce the expression of LXRα creating a metabolically linked cycle. This critical feedback loop was first described by Chawla et al. in macrophages and is critical for PPARɣ mediated degradation of Aβ [57].
The synthetic thiazolidinedione (TZD) PPARɣ agonists, are widely prescribed for the treatment of type II diabetes mellitus, and have also been shown to be efficacious in a number of CNS disease models [21]. Currently, two TZD agonists, Actos™ (pioglitazone) and Avandia™ (rosiglitazone), are FDA approved for the treatment of diabetes. However, the use of these drugs for CNS-targeted disease treatments is compromised due to their poor blood brain barrier (BBB) penetrance. The permeability of pioglitazone across the BBB is poor, and rosiglitazone is even less permeable and subject to P-glycoprotein-mediated efflux from the brain [58]. However, a number of studies employing AD mouse models have been carried out and have demonstrated the utility of these synthetic agonists in AD disease pathogenesis.
In a study carried out by Yan et al., 12 month old Tg2576 animals were treated orally with 20 mg/kg/day of pioglitazone for 4 months. These animals did not exhibit a change in plaque pathology, however, showed a trend towards a decrease in soluble Aβ42 levels [59]. Treatment of 16 month old J20 animals with 20 mg/kg/day of pioglitazone yielded no effects on Aβ burden or AD associated behavioral deficits in this model [60]. When a higher dose of pioglitazone (7 days/40mg/kg/day) was employed in 10 month old transgenic mice overexpressing the APP V717I mutation, a 20-25% decrease in plaque burden was observed with significant reduction in Aβ42 levels within the brains of these animals [61]. This was the first study that provided conclusive evidence for the utility of PPARɣ agonists in an animal model of AD.
Pederson and colleagues examined the effects of rosiglitazone and found that activation of PPARɣ ameliorated behavioral deficits in the Tg2576 AD mouse model. However, these animals displayed no changes in plaque pathology, but had reduced brain Aβ42 levels. Since the BBB permeability of rosiglitazone is poor, the authors postulated that its effects were due to the peripheral actions of this drug and concluded the improvements in behavior were a result of suppression of plasma glucocorticoid levels [62]. A more recent study carried out by Toledo et al. observed the effects of long term rosiglitazone treatments in APP/PS1 animals. These animals were treated with a low dose of rosiglitazone (3mg/kg/day) for 12 weeks and evaluated for plaque deposition and behavior. These animals displayed an approximate 50% decrease in amyloid deposition, a decrease in Aβ oligomers, preservation of pre- and post-synaptic proteins and the attenuation of cognitive deficits in the Morris water maze. The authors argue that the effects of rosiglitazone were due to the activation of the wnt signaling cascade which they show by an increase in β-catenin expression and a decrease in GSK-3β levels [63]. In a study by Escribano et al., 9 month old J20 animals were treated with 5mg/kg/day of rosiglitazone for a period of 4 months [64]. After four weeks of treatment, the animals showed significant improvements in the object recognition task. After four months of treatment, the 13 month old J20 animals showed a 50% reduction in levels of Aβ40 and Aβ42. The authors also showed a decrease in levels of Aβ*56. While the authors did not detect an increase in ApoE levels in the treated animals they did observe a modest increase in ABCA1 levels and argue that the enhanced Aβ clearance could be attributed to an increase in lipidation of ApoE by ABCA1 [64]. Another study reported a reversal of associative learning and memory deficits in 9 month old Tg2576 animals after 1 month of rosiglitazone treatments (30mg/kg/day). However, this effect was not observed in 5 or 13 month old Tg2576 animals [65].
Recent studies in our laboratories have indicated that PPARɣ activation leads to an increase in both ApoE and ABCA1 levels in astrocytes and microglial cells. Inhibition of PPARɣ action by a receptor antagonist blocked all effects of pioglitazone on Aβ degradation in vitro. Both ApoE and LXRs are necessary for the actions of this nuclear receptor, because cells lacking ApoE or wildtype cells treated with an LXR antagonist lose their ability to degrade Aβ in response to pioglitazone. Furthermore, the treatment of APP/PS1 mouse model of AD with pioglitazone (80/mg/kg/day) for 9 days lowered plaque burden by approximately 50% and reversed behavioral deficits in contextual fear conditioning assay. Significantly, the levels of ABCA1 and ApoE were elevated in the brains of these animals [66]. While it is unclear what the dominant mode of action for PPARɣ activation is in ameliorating AD pathologies, it is now evident that ABCA1 and ApoE play critical roles on Aβ homeostasis and this pathway is positively regulated by PPARɣ’s actions on LXRs (Table 1).
Table 1.
Effects of PPARγ agonists in AD mouse models and their effects on plaque burden and memory retention.
| Author | Drug | Dose | Mouse Model |
Treatment Age |
Treatment Duration |
Amyloid Burden | Memory Improvements |
|---|---|---|---|---|---|---|---|
| Yan | Pioglitazone | 20 mg/kg | Tg2576 | 11 months | 4 months | ↓ soluble Aβ42 | Not Assessed |
| Heneka | Pioglitazone | 40 mg/kg | VAPP717I | 10 months | 7 days | ↓ soluble Aβ42 | Not Assessed |
| Pederson | Rosiglitazone | 30 mg/kg | Tg2576 | 13 months | 7 months | ↓ soluble Aβ42 | Yes |
| Toledo | Rosiglitazone | 3 mg/kg | APP/PS1 | 4 months | 12 weeks | ↓ 50% plaque load | Yes |
| Escribiano | Rosiglitazone | 5 mg/kg | J20 | 9 months | 10 weeks | ↓ 50% plaque load | Yes |
| Dinely | Rosiglitazone | 30 mg/kg | Tg2576 | 5, 9, 13 months | 1 month | Not Assessed | 9 months-Yes |
| Nicolakakis | Pioglitazone | 20 mg/kg | J20 | 16 month | 6-8 weeks | No Change | No |
| Mandrekar- Colucci (unpublished) |
Pioglitazone | 80 mg/kg | APP/PS1 | 6, 12 months | 9 days | 6 month- ↓ 50% plaque load 12 month- ↓ 40% plaque load |
12 months- Yes |
Importantly, PPARɣ activation by pioglitazone has also been shown to rescue cerebrovascular function in an aged AD mouse model. A study conducted on 14 month old APP V717I animals treated with 20mg/kg/day of pioglitazone for 6-8 weeks showed that PPARɣ activation rescued deficits in neurometabolic coupling and cholinergic denervation. These authors observed no effects of pioglitazone on plaque load in these animals, but this could be attributed to the low drug dose utilized in this study [60].
Owing to the promising effects on AD mouse models, a small clinical trial was conducted to determine the efficacy of pioglitazone in patients exhibiting signs of mild to moderate AD. In this study, pioglitazone treatment was shown to improve memory and cognition in these patients [67, 68]. Recently, the effect of rosiglitazone in treating AD was examined in larger clinical trials. A phase II clinical trial, where patients treated with rosiglitazone for 6 months, showed improvements in attention and memory retention, but only in patients that did not have an ApoE4 allele. However, much larger phase III trials failed to show any efficacy in mild to moderate AD patients [69, 70]. It is important to note, that rosiglitazone is known to have very poor BBB permeability and was administered at approximately ten percent the dosage known to be efficacious in rodent models of AD. Additionally, rosiglitazone is a substrate for p-glycoprotein, promoting its efflux from the brain into peripheral circulation [58]. Thus, due to the design of these trials it is unclear whether existing PPARɣ agonists will be beneficial in treatment of AD.
PPARδ activation has also been shown to promote reverse cholesterol transport. The PPARδ agonist, GW501516, induced expression of ABCA1 and apolipoprotein A1, a peripheral lipid transporter, in macrophages [71]. In a study reported by Kalinin et al., treatment of 5xFAD mice with the PPARδ agonist, GW742, resulted in decreased plaque burden and an increase in the expression of two Aβ proteases, neprilysin and IDE [72]. PPARδ activation has also been shown to have robust anti-inflammatory actions [73]. These studies suggest that PPARδ activation may reduce amyloid burden in a similar manner to both LXR and PPARs.
RXR activation by numerous ligands has shown to increase levels of both ApoE and ABCA1 in vitro [74-76]. Due to its central role in nuclear receptor signaling and its interactions with both PPAR and LXR, it is hypothesized that RXR activation would simultaneously activate both LXR and PPAR signaling pathways, making it an excellent candidate for AD therapeutics (Figure 2). A study by Ding et al., described the effects of all-trans retinoic acid (ATRA) in a mouse model of AD. ATRA not only activates RXR but also activates the Retinoic Acid Receptor (RAR). Treatment of 5 month old APP/PS1 mice for 8 weeks with ATRA (20mg/kg/day) resulted in significant decreases in Aβ deposition and tau phosphorylation in these mice. Additionally, it attenuated memory deficits seen in the Morris water maze [77]. However, the ability of ATRA to activate both receptors does not allow for any conclusions to be drawn about its mechanism of action.
Bexarotene is a highly specific RXR agonist and is currently FDA approved with a favorable side-effect profile. Studies in our laboratory have shown that treatment of APP/PS1 animals with bexarotene for only 3 days results in a dramatic induction of ApoE and ABCA1 and the rapid reversal of AD-associated pathological hallmarks including, reduction in amyloid deposition and deficits in behavior as well as neural networks. More recently, a naturally occurring RXR agonist, honokiol, has been identified. This agonist is capable of activating RXR/LXR heterodimers and has been shown to induce the expression of ABCA1 and ApoE and should be tested in AD models [75, 78].
6.0 Nuclear Receptors and Inflammation
The appearance of amyloid deposition in the AD brain coincides with a dramatic phenotypic activation of microglial cells in the surrounding area. These cells are stably associated with the plaque and extend their processes deep within the plaque core. Microglial cells are able to detect Aβ through a multi-component cell surface receptor complex composed of CD36 (a B class scavenger receptor), the α1β6 integrin, the integrin associated protein, CD47, and the scavenger receptor A [79]. More recently, Toll Like Receptor 2 (TLR2), 4 (TLR4) and 6 (TLR6) and their coreceptors CD14 and CD36 have been shown to participate in this complex [80]. Activation of this cell surface receptor complex initiates intracellular signaling cascades that lead to NFκB-mediated proinflammatory gene transcription. This results in the production of pro-inflammatory cytokines, chemokines and the production of reactive nitrogen and oxygen species. In vitro, fibrillar Aβ stimulation stimulates phagocytosis, however, in vivo this effect is not observed and the plaque-associated microglial cells are described to be phagocytically inactive [81].
The deposition of fibrillar Aβ in the AD brain results in the recruitment of microglia to the plaques owing to their expression of CCL2, which acts to attract microglia [82]. CCL2 is a cytokine that is secreted by microglia and astrocytes, its expression is increased by cells surrounding amyloid deposits in the AD brain. Its receptor, CCR2, is also expressed my microglia and it is thought that that the upregulation of CCL2 in the vicinity of plaques results in the recruitment of microglia from adjacent regions to these deposits [83, 84]. The number of microglial cells surrounding a plaque is proportional to the dimensions of the deposits. This seems to be an ideal situation since microglia are competent phagocytes and are found in abundance surrounding amyloid deposits in the AD brain. It would be assumed that they would clear the deposits and maintain brain Aβ homeostasis. However, this is not the case, the deposition of plaques and the recruitment of microglia to these deposits does not result in their removal, but leads to the sustained activation of microglia and chronic production of inflammatory agents. While in normal circumstances inflammation plays a beneficial role in pathogen removal, in AD, the persistence of inflammation is detrimental to the surrounding tissue [14].
There is an extensive literature documenting the effects of chronic inflammation in AD pathogenesis. It is postulated to exacerbate the primary disease process, facilitating the deposition of Aβ, neuronal loss and contributing to cognitive deficits [11, 14]. Notably, studies aimed at suppressing the inflammatory response in AD have been shown to have a beneficial effect on disease progression. Animal models lacking the TNF type 1 receptor or the INFɣ receptor type 1 exhibit a significant decrease in microglial activation, Aβ deposition as well as reversal of Aβ associated cognitive deficits [85, 86]. Epidemiological studies have also reported that chronic use of nonsteroidal anti-inflammatory drugs (NSAIDs) is associated with a dramatic reduction in the incidence of AD [87]. Activation of LXR, PPAR and RXR have all been shown to exert robust anti-inflammatory actions [15, 88-90]. Indeed, treatment of AD mouse models with LXR or PPAR agonists has resulted in the suppression of microglial activation [44, 45, 47, 59, 63]. The anti-inflammatory effects of these receptors are due, in part, to their functional inactivation of the NFκB promoters. A number of different mechanisms have been proposed to explain how nuclear receptors repress inflammatory genes, including cross-coupling, coactivator squelching and corepressor interference [91-93]. The most compelling of these mechanisms, corepressor interference, was first described by Glass and colleagues in 2005. Normally, inflammatory genes are repressed through the association of the co-repressors NCoR and SMRT acting on kBRE elements. They were able to show that ligand dependent SUMOylation of PPARɣ directs the nuclear receptor to NFκB positioned on the promoters of inflammatory genes, thereby stabilizing co-repressor complexes and inhibiting inflammatory gene expression [92]. Both PPARs and LXRs have been shown to become SUMOylated after ligand binding [16] (Figure 1). The ability of RXRs to be SUMOylated is still unknown. Owing to their ability to broadly inhibit inflammatory gene expression and promote Aβ degradation, LXRs, PPARs, and perhaps RXRs, serve as an attractive therapeutic target for AD.
7.0 Nuclear Receptors and Macrophage and Microglial Activation Status
While comprising only 5% of the glial population in the cortex, microglia are the principal contributors of the chronic inflammation seen in the AD brain. One of the enigmas in AD pathogenesis is the inability of microglial cells to clear amyolid deposits even though they are found associated with these structures and are competent phagocytes. Genetic studies have revealed that the deposition of Aβ in the brain parenchyma is the initiating factor in disease pathogenesis. In sporadic forms of AD, there is a disruption of Aβ homeostasis, leading to the progressive accumulation and deposition of these peptides in the brain. This can occur via two mechanism, enhanced production or decreased clearance of Aβ. A recent study conducted by Bateman et al. has demonstrated that in human cases of AD, Aβ is generated at comparable levels between AD patients and control subjects, however, clearance of the peptide is impaired in AD patients [94]. Indeed, in AD patients Aβ clearance rates are reduced by approximately 30%, accounting for the accumulation of Aβ in the diseased brain [4]. Several studies have shown that microglia are unable to digest fibrillar deposits of Aβ [95-97]. However, immunotherapy with anti-Aβ antibodies demonstrated that these cells retain the capacity to phagocytically take up and remove amyloid deposits when appropriately stimulated [98]. It is therefore thought that one mechanism to alleviate AD related pathophysiology is to stimulate microglial cells to re-engage their phagocytic machinery and enhance amyloid clearance.
It has recently been appreciated that microglial cells can exhibit a variety of activation states and this finding may explain sustained microglial activation and the effects they exert in the AD brain. Originally, it was thought that microglia represented a homogenous population of cells in the brain and exhibited two phenotypic polarities; a resting, quiescent state or an activated, pro-inflammatory state. However, recently their biology has been re-evaluated, due in part to the ability to image these cells in vivo with 2-photon microscopy. It has been shown that microglial cells are quite active in their “resting” state and survey the whole brain every few hours by extending and retracting their processes [99]. It has also been recognized that they represent a phenotypically diverse group of cells that vary in their response to different local and environmental cues, as evidenced by their differential expression of phenotypic markers and secretion of distinct array of chemokines and cytokines. A new classification system has been suggested to more easily identify and categorize the different macrophage/microglial activation states. The “classical” pro-inflammatory activated macrophage/microglia are now referred to as being in an M1 state, while the “alternative”, anti-inflammatory activation states are referred to as M2 [100]. It is important to note that M1 and M2 activation states are reflective of a continuum of phenotypic states of these cells and their peripheral counterparts, the macrophage, and this nomenclature does not capture the complexity of their phenotypic diversity.
The M1 activation state is associated with the secretion of the typical Th1 pro-inflammatory cytokines, which are found in abundance in the AD brain. While classical activation is a host defense mechanism, it is necessary to down-regulate this response and initiate tissue repair to attain homeostasis in the CNS after pathogen infections or injury. The M2 state, termed “alternative activation” is characterized by the production of Th2 anti-inflammatory cytokines including IL-10, TGF-β, IL-4 and IL-13. It is generally associated with the suppression of Th1 pro-inflammatory cytokines and the expression of genes that play a role in tissue repair such as, YM1, FIZZ1, Arginase 1 (Arg1), and the mannose receptor [101]. The M2 state itself has been further subdivided into M2a, M2b and M2c phenotypes. IL-4 and IL-13 polarize macrophages to an M2a anti-inflammatory state, M2b macrophages are a mix between M1 and M2 states because they express high levels of pro-inflammatory cytokines as well as high levels of the anti-inflammatory cytokine IL-10. The M2c macrophages are thought to be in an “acquired deactivation” state induced by IL-10, TGF-β, glucocorticoids or contact with apoptotic cells, and are associated with a suppression of the innate immune response. The M2c state is also associated with an increased phagocytic capacity. Although macrophage activation states have been extensively characterized in the periphery it is unknown to what extent microglia can acquire these various states in the brain. A study by Colton et al. demonstrated that AD brains as well as mouse models show a switch in microglial activation status in response to disease progression. In this study it was shown that samples from human AD brains as well as two aged mouse models of AD showed increased mRNA levels of the M2 markers, Arg1 and Ym1, when compared to age matched controls [101]. It is thought that the environment of the AD brain alters the microglial response to fibrillar forms of Aβ and a question of importance now is how microglial polarization not only affects the inflammatory milieu of the brain but how it affects Aβ clearance.
Importantly, in vitro studies have shown that M2 polarization of microglia using M-CSF or IL-4 promote Aβ phagocytosis [102, 103]. Systemic administration of M-CSF also cleared amyloid deposition in a mouse model of AD, although how this is linked to the acquisition of a M2 status in microglia is unclear [104]. Activation of LXRs, PPARs and RXRs results in the polarization of microglia to M2 states due to their suppression of NFκB gene induction, but also through transactivation of genes that induce the M2 states. Recently, it has been shown that activation of PPARɣ promotes the acquisition of an M2 phenotype in peripheral macrophages. Activation of PPARɣ or PPARδ has been shown to induce Arg1 and IL-4 expression [105, 106]. Indeed, Odegaard et al. discovered a putative PPRE in the distal enhancer of the Arg1 gene [107]. Studies have shown disruption of PPARɣ or PPARδ in myeloid cells results in impaired maturation of alternative macrophage activation [107-109]. PPARδ and LXRs have been shown to also play a role in M2c polarization, since ligand activation of these receptors promotes phagocytosis of apoptotic cell bodies [110, 111]. Similarly, stimulation of microglia with the LXR agonist, GW3965, acts simultaneously to suppress inflammation and promote fibrillar Aβ stimulated phagocytosis [47].
Conclusions
There in now an extensive body of evidence demonstrating the efficacy of nuclear receptors agonists in ameliorating AD-related pathology in animal models. Targeting the family of receptors described in this review has shown to ameliorate plaque burden, reverse memory related deficits and reduce inflammation in animal models, all critical components of AD disease pathogenesis. Thus, we believe that nuclear receptors represent new and promising therapeutic targets for AD treatment.
Expert Opinion
Currently, there are no approved drugs available that target the underlying causes of Alzheimer’s disease. Nuclear receptors are attractive targets for the treatment of AD due to their ability to facilitate degradation of Aβ, affect microglial activation and suppress the inflammatory milieu of the brain. LXR agonists have proven difficult to move into clinical trials since long-term treatment results in hepatic steatosis. It is our view that PPARɣ activation remains a promising avenue for the treatment for AD, however, the poor BBB permeability of the currently available agonists and the negative outcome of the phase III clinical trials are likely to diminish interest in pursuing this target.
It is our opinion that RXR agonists are the most promising agents for AD treatment. Bexarotene, currently an FDA-approved drug, has an excellent side-effect profile allowing for its rapid translation into clinical trials. This drug not only targets both LXR and PPAR pathways but also has been shown to readily pass through the BBB making it an ideal candidate for AD treatment.
Highlights.
Nuclear receptors provide a promising target for the treatment of Alzheimer’s disease.
The capacity of nuclear receptors to ameliorate AD-related pathophysiology is reliant on their ability to increase expression of ABCA1 and ApoE, promoting the creation of highly lipidated HDL particles that are able to efficiently degrade soluble species of Aβ.
Nuclear receptors may further enhance amyloid clearance by controlling microglial activation allowing them to re-engage the phagocytic machinery and remove fibrillar amyloid deposits from the brain parenchyma.
Footnotes
Declaration of interest The authors declare no conflict of interest and have received no payment in preparation of this manuscript.
REFERENCES
- 1.2010 Alzheimer’s disease facts and figures. Alzheimers Dement. 2010;6(2):158–94. doi: 10.1016/j.jalz.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Sisodia SS, Price DL. Role of the beta-amyloid protein in Alzheimer’s disease. FASEB J. 1995;9(5):366–70. doi: 10.1096/fasebj.9.5.7896005. [DOI] [PubMed] [Google Scholar]
- 3.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120(4):545–55. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 4.Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330(6012):1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lacor PN, Buniel MC, Furlow PW, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27(4):796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–5. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akiyama H, Mori H, Saido T, et al. Occurrence of the diffuse amyloid beta-protein (Abeta) deposits with numerous Abeta-containing glial cells in the cerebral cortex of patients with Alzheimer’s disease. Glia. 1999;25(4):324–31. doi: 10.1002/(sici)1098-1136(19990215)25:4<324::aid-glia2>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 8.Mackenzie IR, Hao C, Munoz DG. Role of microglia in senile plaque formation. Neurobiol Aging. 1995;16(5):797–804. doi: 10.1016/0197-4580(95)00092-s. [DOI] [PubMed] [Google Scholar]
- 9.Stalder M, Phinney A, Probst A, et al. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999;154(6):1673–84. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21(3):383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mandrekar-Colucci S, Landreth GE. Microglia and inflammation in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2010;9(2):156–67. doi: 10.2174/187152710791012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Khoury J, Luster AD. Mechanisms of microglia accumulation in Alzheimer’s disease: therapeutic implications. Trends Pharmacol Sci. 2008;29(12):626–32. doi: 10.1016/j.tips.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Q, Lee CY, Mandrekar S, et al. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58(5):681–93. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer’s disease. Neurobiol Dis. 2010;7(3):503–9. doi: 10.1016/j.nbd.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong C, Tontonoz P. Coordination of inflammation and metabolism by PPAR and LXR nuclear receptors. Curr Opin Genet Dev. 2008;18(5):461–7. doi: 10.1016/j.gde.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10(5):365–76. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- 17.Klinge CM, Bodenner DL, Desai D, et al. Binding of type II nuclear receptors and estrogen receptor to full and half-site estrogen response elements in vitro. Nucleic Acids Res. 1997;25(10):1903–12. doi: 10.1093/nar/25.10.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desvergne B, Michalik L, Wahli W. Transcriptional regulation of metabolism. Physiol Rev. 2006;86(2):465–514. doi: 10.1152/physrev.00025.2005. [DOI] [PubMed] [Google Scholar]
- 19.Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci U S A. 1997;94(9):4312–7. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kliewer SA, Forman BM, Blumberg B, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci U S A. 1994;91(15):7355–9. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405(6785):421–4. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- 22.Barish GD, Narkar VA, Evans RM. PPAR delta: a dagger in the heart of the metabolic syndrome. J Clin Invest. 2006;116(3):590–7. doi: 10.1172/JCI27955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lehmann JM, Moore LB, Smith-Oliver TA, et al. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270(22):12953–6. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 24.Landreth G, Jiang Q, Mandrekar S, et al. PPARgamma agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics. 2008;5(3):481–9. doi: 10.1016/j.nurt.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chawla A. Control of macrophage activation and function by PPARs. Circ Res. 2010;106(10):1559–69. doi: 10.1161/CIRCRESAHA.110.216523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann JM, Kliewer SA, Moore LB, et al. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J Biol Chem. 1997;272(6):3137–40. doi: 10.1074/jbc.272.6.3137. [DOI] [PubMed] [Google Scholar]
- 27.Calkin AC, Tontonoz P. Liver x receptor signaling pathways and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30(8):1513–8. doi: 10.1161/ATVBAHA.109.191197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersson S, Gustafsson N, Warner M, et al. Inactivation of liver X receptor beta leads to adult-onset motor neuron degeneration in male mice. Proc Natl Acad Sci U S A. 2005;102(10):3857–62. doi: 10.1073/pnas.0500634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L, Schuster GU, Hultenby K, et al. Liver X receptors in the central nervous system: from lipid homeostasis to neuronal degeneration. Proc Natl Acad Sci U S A. 2002;99(21):13878–83. doi: 10.1073/pnas.172510899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83(6):841–50. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 31.Szanto A, Narkar V, Shen Q, et al. Retinoid X receptors: X-ploring their (patho)physiological functions. Cell Death Differ. 2004;11(Suppl 2):S126–43. doi: 10.1038/sj.cdd.4401533. [DOI] [PubMed] [Google Scholar]
- 32.Ricote M, Snyder CS, Leung HY, et al. Normal hematopoiesis after conditional targeting of RXRalpha in murine hematopoietic stem/progenitor cells. J Leukoc Biol. 2006;80(4):850–61. doi: 10.1189/jlb.0206097. [DOI] [PubMed] [Google Scholar]
- 33.Heyman RA, Mangelsdorf DJ, Dyck JA, et al. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell. 1992;68(2):397–406. doi: 10.1016/0092-8674(92)90479-v. [DOI] [PubMed] [Google Scholar]
- 34.Lala DS, Mukherjee R, Schulman IG, et al. Activation of specific RXR heterodimers by an antagonist of RXR homodimers. Nature. 1996;383(6599):450–3. doi: 10.1038/383450a0. [DOI] [PubMed] [Google Scholar]
- 35.Huang JK, Jarjour AA, Oumesmar BN, et al. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat Neurosci. 2011;14(1):45–53. doi: 10.1038/nn.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fagan AM, Watson M, Parsadanian M, et al. Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2002;9(3):305–18. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- 37.Bales KR, Verina T, Cummins DJ, et al. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 1999;96(26):15233–8. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LaDu MJ, Pederson TM, Frail DE, et al. Purification of apolipoprotein E attenuates isoform-specific binding to beta-amyloid. J Biol Chem. 1995;270(16):9039–42. doi: 10.1074/jbc.270.16.9039. [DOI] [PubMed] [Google Scholar]
- 39.Fisher CA, Ryan RO. Lipid binding-induced conformational changes in the N-terminal domain of human apolipoprotein E. J Lipid Res. 1999;40(1):93–9. [PubMed] [Google Scholar]
- 40.Wollmer MA, Streffer JR, Lutjohann D, et al. ABCA1 modulates CSF cholesterol levels and influences the age at onset of Alzheimer’s disease. Neurobiol Aging. 2003;24(3):421–6. doi: 10.1016/s0197-4580(02)00094-5. [DOI] [PubMed] [Google Scholar]
- 41.Puglielli L, Tanzi RE, Kovacs DM. Alzheimer’s disease: the cholesterol connection. Nat Neurosci. 2003;6(4):345–51. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- 42.Zhao L, Lin S, Bales KR, et al. Macrophage-mediated degradation of beta-amyloid via an apolipoprotein E isoform-dependent mechanism. J Neurosci. 2009;29(11):3603–12. doi: 10.1523/JNEUROSCI.5302-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burns MP, Vardanian L, Pajoohesh-Ganji A, et al. The effects of ABCA1 on cholesterol efflux and Abeta levels in vitro and in vivo. J Neurochem. 2006;98(3):792–800. doi: 10.1111/j.1471-4159.2006.03925.x. [DOI] [PubMed] [Google Scholar]
- 44.Koldamova RP, Lefterov IM, Staufenbiel M, et al. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer’s disease. J Biol Chem. 2005;280(6):4079–88. doi: 10.1074/jbc.M411420200. [DOI] [PubMed] [Google Scholar]
- 45.Riddell DR, Zhou H, Comery TA, et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol Cell Neurosci. 2007;34(4):621–8. doi: 10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 46.Fitz NF, Cronican A, Pham T, et al. Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J Neurosci. 2010;30(20):6862–72. doi: 10.1523/JNEUROSCI.1051-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zelcer N, Khanlou N, Clare R, et al. Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc Natl Acad Sci U S A. 2007;104(25):10601–6. doi: 10.1073/pnas.0701096104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vanmierlo T, Rutten K, Dederen J, et al. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 49.Terwel D, Steffensen KR, Verghese PB, et al. Critical Role of Astroglial Apolipoprotein E and Liver X Receptor-{alpha} Expression for Microglial A{beta} Phagocytosis. J Neurosci. 2011;31(19):7049–59. doi: 10.1523/JNEUROSCI.6546-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wahrle SE, Jiang H, Parsadanian M, et al. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem. 2005;280(52):43236–42. doi: 10.1074/jbc.M508780200. [DOI] [PubMed] [Google Scholar]
- 51.Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005;280(52):43224–35. doi: 10.1074/jbc.M504513200. [DOI] [PubMed] [Google Scholar]
- 52.Hirsch-Reinshagen V, Maia LF, Burgess BL, et al. The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J Biol Chem. 2005;280(52):43243–56. doi: 10.1074/jbc.M508781200. [DOI] [PubMed] [Google Scholar]
- 53.Wahrle SE, Jiang H, Parsadanian M, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118(2):671–82. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Donkin JJ, Stukas S, Hirsch-Reinshagen V, et al. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2010;285(44):34144–54. doi: 10.1074/jbc.M110.108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Landreth GE, Heneka MT. Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer’s disease. Neurobiol Aging. 2001;22(6):937–44. doi: 10.1016/s0197-4580(01)00296-2. [DOI] [PubMed] [Google Scholar]
- 56.Heneka MT, Landreth GE. PPARs in the brain. Biochim Biophys Acta. 2007;1771(8):1031–45. doi: 10.1016/j.bbalip.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 57.Chawla A, Boisvert WA, Lee CH, et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7(1):161–71. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- 58.Hemauer SJ, Patrikeeva SL, Nanovskaya TN, et al. Role of human placental apical membrane transporters in the efflux of glyburide, rosiglitazone, and metformin. Am J Obstet Gynecol. 2010;202(4):383, e1–7. doi: 10.1016/j.ajog.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yan Q, Zhang J, Liu H, et al. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer’s disease. J Neurosci. 2003;23(20):7504–9. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nicolakakis N, Aboulkassim T, Ongali B, et al. Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J Neurosci. 2008;28(37):9287–96. doi: 10.1523/JNEUROSCI.3348-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heneka MT, Sastre M, Dumitrescu-Ozimek L, et al. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain. 2005;128(Pt 6):1442–53. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- 62.Pedersen WA, Flynn ER. Insulin resistance contributes to aberrant stress responses in the Tg2576 mouse model of Alzheimer’s disease. Neurobiol Dis. 2004;17(3):500–6. doi: 10.1016/j.nbd.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 63.Toledo EM, Inestrosa NC. Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer’s disease. Mol Psychiatry. 2010;15(3):272–85. 228. doi: 10.1038/mp.2009.72. [DOI] [PubMed] [Google Scholar]
- 64.Escribano L, Simon AM, Gimeno E, et al. Rosiglitazone rescues memory impairment in Alzheimer’s transgenic mice: mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology. 2010;35(7):1593–604. doi: 10.1038/npp.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rodriguez-Rivera J, Denner L, Dineley KT. Rosiglitazone reversal of Tg2576 cognitive deficits is independent of peripheral gluco-regulatory status. Behav Brain Res. 2011;216(1):255–61. doi: 10.1016/j.bbr.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mandrekar-Colucci S, Karlo C, Landreth GE. PPARψ activation rapidly ameliorates amyloid pathology and restores cognition in a mouse model of Alzheimer’s disease. In Preparation. 2011 [Google Scholar]
- 67.Sato T, Hanyu H, Hirao K, et al. Efficacy of PPAR-gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 68.Hanyu H, Sato T, Kiuchi A, et al. Pioglitazone improved cognition in a pilot study on patients with Alzheimer’s disease and mild cognitive impairment with diabetes mellitus. J Am Geriatr Soc. 2009;57(1):177–9. doi: 10.1111/j.1532-5415.2009.02067.x. [DOI] [PubMed] [Google Scholar]
- 69.Risner ME, Saunders AM, Altman JF, et al. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 2006;6(4):246–54. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- 70.Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord. 2010;30(2):131–46. doi: 10.1159/000318845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oliver WR, Jr., Shenk JL, Snaith MR, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci U S A. 2001;98(9):5306–11. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kalinin S, Richardson JC, Feinstein DL. A PPardelta Agonist Reduces Amyloid Burden and Brain Inflammation in a Transgenic Mouse Model of Alzheimer’s Disease. Curr Alzheimer Res. 2009 doi: 10.2174/156720509789207949. [DOI] [PubMed] [Google Scholar]
- 73.Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28(12):551–8. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 74.Nishimaki-Mogami T, Tamehiro N, Sato Y, et al. The RXR agonists PA024 and HX630 have different abilities to activate LXR/RXR and to induce ABCA1 expression in macrophage cell lines. Biochem Pharmacol. 2008;76(8):1006–13. doi: 10.1016/j.bcp.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 75.Jung CG, Horike H, Cha BY, et al. Honokiol increases ABCA1 expression level by activating retinoid X receptor beta. Biol Pharm Bull. 2010;33(7):1105–11. doi: 10.1248/bpb.33.1105. [DOI] [PubMed] [Google Scholar]
- 76.Costet P, Lalanne F, Gerbod-Giannone MC, et al. Retinoic acid receptor-mediated induction of ABCA1 in macrophages. Mol Cell Biol. 2003;23(21):7756–66. doi: 10.1128/MCB.23.21.7756-7766.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ding Y, Qiao A, Wang Z, et al. Retinoic acid attenuates beta-amyloid deposition and rescues memory deficits in an Alzheimer’s disease transgenic mouse model. J Neurosci. 2008;28(45):11622–34. doi: 10.1523/JNEUROSCI.3153-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kotani H, Tanabe H, Mizukami H, et al. Identification of a naturally occurring rexinoid, honokiol, that activates the retinoid X receptor. J Nat Prod. 2010;73(8):1332–6. doi: 10.1021/np100120c. [DOI] [PubMed] [Google Scholar]
- 79.Bamberger ME, Harris ME, McDonald DR, et al. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23(7):2665–74. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reed-Geaghan EG, Savage JC, Hise AG, et al. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci. 2009;29(38):11982–92. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koenigsknecht J, Landreth G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J Neurosci. 2004;24(44):9838–46. doi: 10.1523/JNEUROSCI.2557-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Glabinski AR, Balasingam V, Tani M, et al. Chemokine monocyte chemoattractant protein-1 is expressed by astrocytes after mechanical injury to the brain. J Immunol. 1996;156(11):4363–8. [PubMed] [Google Scholar]
- 83.El Khoury J, Toft M, Hickman SE, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13(4):432–8. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 84.Naert G, Rivest S. CC Chemokine Receptor 2 Deficiency Aggravates Cognitive Impairments and Amyloid Pathology in a Transgenic Mouse Model of Alzheimer’s Disease. J Neurosci. 2011;31(16):6208–20. doi: 10.1523/JNEUROSCI.0299-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yamamoto M, Kiyota T, Horiba M, et al. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170(2):680–92. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.He P, Zhong Z, Lindholm K, et al. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J Cell Biol. 2007;178(5):829–41. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vlad SC, Miller DR, Kowall NW, et al. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008;70(19):1672–7. doi: 10.1212/01.wnl.0000311269.57716.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Combs CK, Bates P, Karlo JC, et al. Regulation of beta-amyloid stimulated proinflammatory responses by peroxisome proliferator-activated receptor alpha. Neurochem Int. 2001;39(5-6):449–57. doi: 10.1016/s0197-0186(01)00052-3. [DOI] [PubMed] [Google Scholar]
- 89.Nunez V, Alameda D, Rico D, et al. Retinoid X receptor alpha controls innate inflammatory responses through the up-regulation of chemokine expression. Proc Natl Acad Sci U S A. 2010;107(23):10626–31. doi: 10.1073/pnas.0913545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hoi CP, Ho YP, Baum L, et al. Neuroprotective effect of honokiol and magnolol, compounds from Magnolia officinalis, on beta-amyloid-induced toxicity in PC12 cells. Phytother Res. 2010;24(10):1538–42. doi: 10.1002/ptr.3178. [DOI] [PubMed] [Google Scholar]
- 91.Li M, Pascual G, Glass CK. Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Mol Cell Biol. 2000;20(13):4699–707. doi: 10.1128/mcb.20.13.4699-4707.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pascual G, Fong AL, Ogawa S, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437(7059):759–63. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Subbaramaiah K, Lin DT, Hart JC, et al. Peroxisome proliferator-activated receptor gamma ligands suppress the transcriptional activation of cyclooxygenase-2. Evidence for involvement of activator protein-1 and CREB-binding protein/p300. J Biol Chem. 2001;276(15):12440–8. doi: 10.1074/jbc.M007237200. [DOI] [PubMed] [Google Scholar]
- 94.Bateman RJ, Munsell LY, Morris JC, et al. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006;12(7):856–61. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bolmont T, Haiss F, Eicke D, et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28(16):4283–92. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chung H, Brazil MI, Soe TT, et al. Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer’s amyloid beta-peptide by microglial cells. J Biol Chem. 1999;274(45):32301–8. doi: 10.1074/jbc.274.45.32301. [DOI] [PubMed] [Google Scholar]
- 97.Paresce DM, Chung H, Maxfield FR. Slow degradation of aggregates of the Alzheimer’s disease amyloid beta-protein by microglial cells. J Biol Chem. 1997;272(46):29390–7. doi: 10.1074/jbc.272.46.29390. [DOI] [PubMed] [Google Scholar]
- 98.Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 99.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–8. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 100.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 101.Colton CA, Mott RT, Sharpe H, et al. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25(36):8240–9. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Majumdar A, Chung H, Dolios G, et al. Degradation of fibrillar forms of Alzheimer’s amyloid beta-peptide by macrophages. Neurobiol Aging. 2008;29(5):707–15. doi: 10.1016/j.neurobiolaging.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Boissonneault V, Filali M, Lessard M, et al. Powerful beneficial effects of macrophage colony-stimulating factor on beta-amyloid deposition and cognitive impairment in Alzheimer’s disease. Brain. 2009;132(Pt 4):1078–92. doi: 10.1093/brain/awn331. [DOI] [PubMed] [Google Scholar]
- 105.Loane DJ, Deighan BF, Clarke RM, et al. Interleukin-4 mediates the neuroprotective effects of rosiglitazone in the aged brain. Neurobiol Aging. 2009;30(6):920–31. doi: 10.1016/j.neurobiolaging.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 106.Gallardo-Soler A, Gomez-Nieto C, Campo ML, et al. Arginase I induction by modified lipoproteins in macrophages: a peroxisome proliferator-activated receptor-gamma/delta-mediated effect that links lipid metabolism and immunity. Mol Endocrinol. 2008;22(6):1394–402. doi: 10.1210/me.2007-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447(7148):1116–20. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Odegaard JI, Ricardo-Gonzalez RR, Eagle A. Red, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7(6):496–507. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kang K, Reilly SM, Karabacak V, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008;7(6):485–95. doi: 10.1016/j.cmet.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.N AG, Bensinger SJ, Hong C, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31(2):245–58. doi: 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mukundan L, Odegaard JI, Morel CR, et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med. 2009;15(11):1266–72. doi: 10.1038/nm.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]