

Abstract
Elevated activation of the platelet-derived growth factor (PDGF) pathway, apoptosis evasion phenotype, and global DNA hypomethylation are hallmarks frequently observed in cancers, such as in low-grade glioma (LGG). However, the orchestration of these malignant functions is not fully elucidated in LGG. Our study reveals that the co-presence of these hallmarks in the same LGG is frequent and confers poor prognosis in patients with LGG. Our data also indicate that the apoptosis evasion phenotype of these cells harboring a hypomethylation-induced activation of the PDGF pathway is associated with a hypomethylation of the bcl-xl and bcl-w genes and the phosphorylation and/or downregulation of three major pro-apoptotic BH3-only proteins: PUMA, Bad, and Bim. Consistent with this, we demonstrate that the use of folate, a DNA-methylating agent, promotes the reprogramming of the sensitivity of glioma cells to ABT-737/etoposide-induced apoptosis and reduces the dose of ABT-737 required to promote etoposide-induced apoptosis. This work supports the idea that the inclusion of folate and/or ABT-737 could be a promising adjuvant in the design of anti-glioma therapeutic protocols in clinical studies.
Electronic supplementary material
The online version of this article (doi:10.1007/s13148-011-0035-5) contains supplementary material, which is available to authorized users.
Introduction
Acquired resistance to apoptosis or programmed cell death is one of the hallmarks of human cancer (Hanahan and Weinberg 2000). Defects in the apoptotic pathway and the disruption of the apoptotic program contribute to tumor initiation and progression, as well as to treatment resistance, since most current anti-cancer treatments including chemotherapy, radio- and immunotherapy act primarily by promoting cell death via the induction of apoptosis (Lowe and Lin 2000; Evan and Vousden 2001).
Studies over the last decade that aimed at identifying the underlying molecular mechanisms of apoptosis resistance have delineated multiple defects at various levels of the apoptosis signal transduction machinery. Among these multiple defects reported are changes in the expression of members of the Bcl-2 protein family that is a major cause for the resistance to apoptosis in tumor cells (Reed 2003). Thus, the presence of an “apoptosis evasion phenotype” in tumor cells is largely associated with the overexpression of certain anti-apoptotic proteins such as Bcl-2, Bcl-xl, and Bcl-w and/or is frequently correlated with the silencing, a low expression level, mutations, proteosomal degradation, and/or sequestration of certain pro-apoptotic proteins such as Bax, Bim, Bad, HRK, Bik, or Noxa (Martin et al. 2001). In glioma, these points have been illustrated and demonstrated by the fact that (1) high levels of Bcl-2 and/or Bcl-xl confer a resistance to radio- and/or chemotherapeutic drugs and promote the intracranial growth of glioma (Weller et al. 1995; Nagane et al. 1998; Del Bufalo et al. 2001; Bougras et al. 2004; Weiler et al. 2006), (2) Bax deficiency confers a high resistance to apoptosis induction (Cartron et al. 2003), (3) HRK is inactivated in astrocytic tumors, and this reduced HRK expression contributes to the loss of apoptotic control in high-grade tumors (Nakamura et al. 2005). Thus, the elucidation of these pathways over the past two decades has raised the possibility of developing and using therapies targeting the Bcl-2 protein family in order to induce apoptosis in tumor cells.
Among the multiple therapeutic strategies targeting the Bcl-2 protein family is the antagonism of the pro-survival function of anti-apoptotic proteins that seems to be the most attractive strategy. We and others have reported that HA14-1, the first small-molecule Bcl-2 inhibitor, has the capacity to overcome chemo- and radioresistance caused by Bcl-2 overexpression (Wang et al. 2000; Manero et al. 2006; Oliver et al. 2007). Several additional small-molecule inhibitors of anti-apoptotic proteins have been described including theaflavins and epigallechatechins, terphenyl derivates, NSC365400 (compound 6), gossypol derivates, GX015-070, and ABT-737 (Enyedy et al. 2001; Kutzki et al. 2002; Leone et al. 2003; Pellecchia and Reed 2004; Oltersdorf et al. 2005; Reed and Pellecchia 2005; Lessene et al. 2008). The latter molecule is the most potent and specific Bcl-2/Bcl-xl/Bcl-w inhibitor discovered to date. Mechanistic studies have revealed that ABT-737 is associated with the dissociation of interactions between pro-apoptotic and anti-apoptotic Bcl-2 family members, the change in conformation of Bax, cytochrome c release from mitochondria, and the activation of caspases (Oltersdorf et al. 2005; Kojima et al. 2006; Konopleva et al. 2006; van Delft et al. 2006).
Recently, we published that it is possible to abolish the “apopto-resistance” phenotype of glioma cells by reducing the expression of anti-apoptotic proteins such as Bcl-w via the folate-induced DNA (hyper)methylation of genes encoding these proteins (Hervouet et al. 2009). Based on these observations, we here complemented this point by (1) dissecting the mechanisms by which a folate treatment abrogates the “apopto-resistance” phenotype of glioma cells, (2) demonstrating that folate and ABT-737 can work together to reprogram the sensitivity of certain gliomas to the etoposide-induced apoptosis, (3) showing that the use of folate minimizes the dose of ABT-737 necessary to promote etoposide-induced apoptosis.
Results
Activation of the PDGF pathway correlates with a low degree of global DNA methylation and the presence of an apoptosis evasion phenotype in low-grade glioma
Low-grade glioma (LGG) tumors have an elevated platelet-derived growth factor (PDGF) signaling pathway, an “apoptosis evasion phenotype,” and a global DNA hypomethylation pattern. We initially searched for potential correlations between these three parameters in a collection of 65 LGG. For this purpose, we assessed the DNA methylation status by quantifying the number of the 5-methylcytosine (5mC) present on DNA by using an ELISA method. The phenotype of apoptosis evasion was estimated via the measure of intratumor apoptotic level through the activation of caspase-3/7 (referred as DEVDase activity) since caspase/DEVDase activity is the end step in apoptosis. The measurement of PDGF receptor beta (PDGFrβ) kinase activity was performed to assess the degree of activation of the PDGF pathway. All these parameters were plotted against each other, and a statistical analysis using Pearson’s correlation test indicated the presence of significant correlations between these parameters (Fig. 1a). This study showed that 31% (20/65; group A) of the LGG was characterized by the simultaneous presence of a high PDGFrβ activity (higher or equal to the median value), a low level of DNA methylation (lower than the median value), and a low DEVDase activity (lower than the median value), while 28% (18/65; group B) of the LGG was characterized by the opposite hallmarks (Fig. 1b).
Fig. 1.
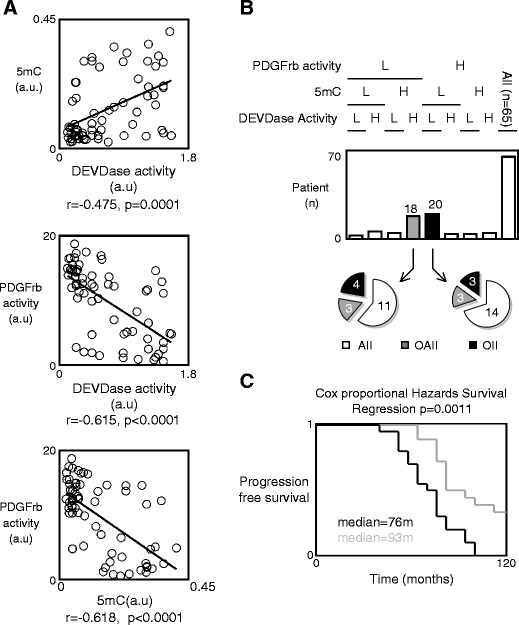
PDGF pathway activation correlates with the low degree of global DNA methylation and the presence of the apoptosis evasion phenotype in low-grade glioma (LGG). a Correlation between the degree of global DNA methylation (i.e., the number of 5-methylcytosine, 5mC) and the degree of apoptosis (i.e., the DEVDase activity), the degree of PDGFrβ activity and the degree of apoptosis (i.e., the DEVDase activity), and between the degree of PDGFrβ activity and the degree of global DNA methylation (i.e., the number of 5-methylcytosine, 5mC) in 65 LGG. 5mC was estimated by using the Methylamp Global DNA Methylation Quantification kit (Epigentek-Euromedex, France). PDGFrβ activity was assessed by using the HTScan® PDGF Receptor β Kinase Assay Kit (Ozyme, France). DEVDase activity was measured as previously described (Cartron et al. 2002). b Representation of the patient number whose LGG is characterized by the presence of a high PDGFrβ activity (higher or equal to the median value), a low level of DNA methylation (lower to the median value), and a low DEVDase activity (lower to the median value). AII grade II astrocytoma, OAII grade II oligoastrocytoma, OII grade II oligodendrocytoma. c Kalpan–Meier estimates of survival time for patients whose tumors had a high PDGFrβ activity (higher or equal to the median value), a low level of DNA methylation (lower to the median value), and a low DEVDase activity (lower to the median value) (in black) and those whose tumors did not (in gray)
We continued our study with these two groups. Firstly, we noted that the presence of these hallmarks was not associated with the histology of the LGG because a similar number of astrocytoma, oligodendrocytoma, or oligoastrocytoma were included in these two subgroups (Fig. 1b). Secondly, by estimating and comparing the survival curves for these two subgroups using Kaplan–Meier curve and the Cox proportional hazards survival regression analysis, we observed a statistical difference in the survival of these two groups of patients indicating that co-presence in LGG of a high PDGFrβ activity, a low level of DNA methylation, and a low DEVDase activity as a poor prognostic factor (Fig. 1c).
Collectively, these results suggest that the PDGF pathway plays an important role in apoptosis evasion in LGG, that this hallmark could be epigenetically regulated, and that the co-presence of a high PDGFrβ activity, a low level of DNA methylation, and a low DEVDase activity in LGG could be used as an alternative predictor of disease outcome and/or biomarker to select a subpopulation of patients.
The apoptosis evasion phenotype of Ntv-a/PDGF cells is generated by the disruption of the apoptotic program at the mitochondrial step symbolized by the loss of Bax activation.
To develop a therapeutic protocol based on a molecular rationale and able to promote apoptosis in tumor cells characterized by a low DNA methylation, a low intratumor apoptosis, and an elevated PDGF signaling pathway, we chose to use the Ntv-a/PDGF cells as these cells harbor these three signatures, as previously reported (Dai et al. 2001; Hervouet et al. 2009). In addition to using Ntv-a/LacZ cells as a control (non-tumor cells generated via a RCAS-LacZ infection), PDGFr activity was inhibited using a PDGFr tyrosine kinase inhibitor III or PDGFr inhibitor (PDGFri; Calbiochem #521232) or by promoting its silencing via the folate-induced methylation of the PDGFrβ gene (Fig. S1). Folate was also used to abolish the presence of DNA hypomethylation mechanisms of “apopto-resistance” in Ntv-a/PDGF cells.
Consistent with that published by our group recently, we confirmed that the PDGFri or the folate treatment of Ntv-a/PDGF cells increased the UV-induced apopto-sensitivity of these cells and limited the hypomethylation status of these cells compared to the Ntv-a/LacZ cells (Fig. 2a; Hervouet et al. 2009). In other terms, our data underlined the presence, in Ntv-a/PDGF cells, of molecular mechanisms inhibiting the apoptosis induced by UV and showed that these mechanisms were methylation-dependent and/or PDGF-dependent since folate (a DNA-methylating agent) or PDGFri treatments resensitized these Ntv-a/PDGF cells to apoptosis.
Fig. 2.
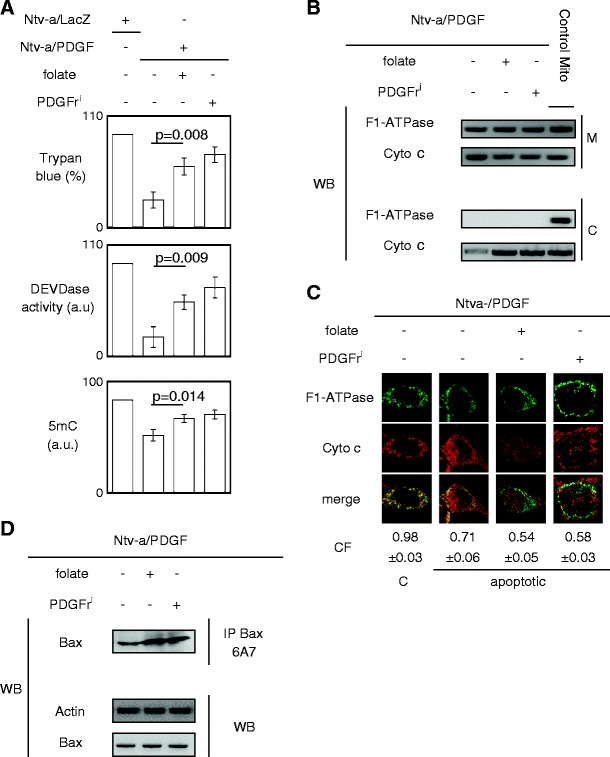
The apoptosis phenotype evasion is associated with the loss of Bax activation. a Effect of the folate or PDGFri (PDGFri 0.5 μM Calbiochem, France) treatment on the level of DNA methylation (i.e., on the 5methylcytosine number, 5mC) and on the UV-induced apoptosis. b Effect of the folate or PDGFri treatment on the cytochrome c release in cytosol (C ) from mitochondria (M) via subcellular fractionation experiment. F1-ATPase is used as control of mitochondrial protein. c Effect of the folate or PDGFri treatment on the cytochrome c release in cytosol (C) from mitochondria (M) via confocal microscopy experiment. F1-ATPase is used as control of mitochondrial protein. CF represents the correlation factor or co-localization of F1-ATPase (i.e., mitochondria) with cytochrome c. The pictures are representative of at least 100 different cells. d Effect of the folate or PDGFri treatment on degree of Bax activation via the Bax6A7-immunoprecipitation (IP) method. Western blot (WB) is performed to analyze the effect of the folate or PDGFri treatment on the Bax expression. Actin is used as loading control. All experiments were done after 7 days of treatment as previously described (Hervouet et al. 2009)
We then decided to identify the PDGF dependence and/or the methylation dependence of these molecular mechanisms conferring the phenotype of apoptosis evasion in LGG cells. For this purpose, we first investigated whether the apoptosis evasion of Ntv-a/PDGF cells was due to a negative regulation of the mitochondrial step in the apoptotic program by assessing the quantity of cytochrome c released from mitochondria after UV induction of apoptosis. The choice of UV to induce apoptosis is due to the fact that UV induces apoptosis by multiple processes including DNA damage, activation of the tumor suppressor gene p53, triggering of cell death receptors either directly by UV or by autocrine release of death ligands, mitochondrial damage, and cytochrome c release (Kulms and Schwarz 2000). Subcellular fractionation and densitometric analysis of Western blots revealed that the quantity of cytochrome c released from mitochondria in Ntv-a/PDGF cells was greatly inferior to that released from mitochondria in Ntv-a/PDGF cells treated with folate or PDGFri (Fig. 2b). Confocal microscopy and MetaMorph analyses confirmed this result (Fig. 2c).
Since the latter results indicated that Ntv-a/PDGF cells harbored a disruption of the apoptotic program at the mitochondrial step, we analyzed the level of activated Bax after UV treatment because the activation of Bax is a crucial event in mitochondrial step of apoptotic program leading to the release of apoptogenic proteins (such as the cytochrome c) from the mitochondria. We performed Bax6A7 immunoprecipitation since Bax activation is characterized by the exposure of the amino terminal epitope recognized by the monoclonal antibody 6A7 (Hsu and Youle 1997). After UV-induced apoptosis, the quantity of activated Bax (or Bax6A7 positive) was lower in Ntv-a/PDGF than in Ntv-a/PDGF cells treated with folate or PDGFri, while the levels of total Bax remained unchanged in these cells (Fig. 2d). Thus, these results demonstrate that the disruption of the apoptotic program at the mitochondrial step present in Ntv-a/PDGF cells was due to the absence of Bax activation.
The low level of Bax activation in Ntv-a/PDGF cells is associated with the low level of PUMA and Bim induced by PDGF-Akt/FoxO3a signaling and the presence of pBim via the activation of PDGF/Erk signaling.
We next attempted to identify the molecular causes inducing the loss of Bax activation in Ntv-a/PDGF cells. For this purpose, we determined whether the loss of Bax activation in Ntv-a/PDGF cells was associated with low expression pro-apoptotic BH3-only proteins. We focused our study on the level of expression of five pro-apoptotic BH3-only proteins: Bid, Bad, Bim, PUMA, and Noxa. Our experiments reveal no association between the level of Bax activation and the expression levels of Bad, Bid, and Noxa. However, we noted a concordance between the expression level of Bim (notably BimEL isoform) or PUMA and the level of Bax activation (Figs. 2d and 3a). This suggests that the loss of Bax activation in Ntv-a/PDGF cells was, in part, due to the low level of expression of PUMA and Bim, both BH3-only proteins that have the capacity to activate Bax.
Fig. 3.
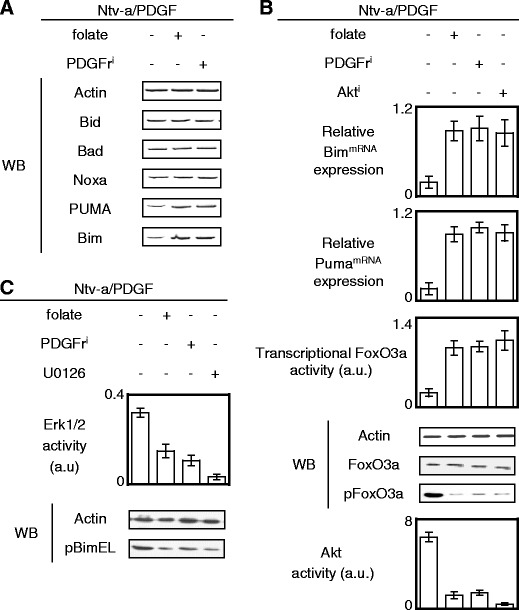
The apoptosis phenotype evasion is associated with the loss of Bax activation. a Effect of the folate or PDGFri treatment on the expression levels of BH3-only proteins. Actin is used as loading control. b Relative BimmRNA and PUMAmRNA expressions were estimated by qPCR. Transcriptional FoxO3a activity was estimated by using the pTL-Luc plasmid (Ozyme, France) i.e., a luciferase reporter vector. Western blot (WB) is used to monitor the effect of the folate or PDGFri treatment on the expression levels of FoxO3a and on its phosphorylation. Relative Akt activity was determined by using the PathScan® Phospho-Akt Sandwich ELISA Kit (Ozyme, France). c Effect of the folate, PDGFri, or U0126 treatment on the phosphorylation levels of Bim. Actin is used as loading control. U0126 (0.4 μM, Ozyme, France). Erk1/2 activity was monitored by ELISA method (Ozyme, France)
To identify the mechanism(s) leading to the low expression level of PUMA and Bim in Ntv-a/PDGF cells, we firstly quantified that the mRNAs coding for PUMA and Bim were overexpressed in Ntv-a/PDGF cells treated with PDGFri or folate (Fig. 3b). Since both PUMA and Bim are partially under the transcriptional control of FoxO3a, we analyzed the transcriptional activity of FoxO3a using the plasmid reporter method. As illustrated in Fig. 3b, both folate and PDGFri increased the FoxO3a transcriptional activity in Ntv-a/PDGF cells. These data suggest the presence of a FoxO3a inhibition mechanism in Ntv-a/PDGF cells. We also show that (1) the low transcriptional activity of FoxO3a in Ntv-a/PDGF cells was associated with its high phosphorylation in a context of high PDGF-induced Akt activity and (2) an Akt inhibitor treatment of Ntv-a/PDGF cells restored the high transcriptional activity of FoxO3a and a high expression of mRNAs coding for PUMA and Bim. Taken together, these results demonstrate that the low level of Bax activation in Ntv-a/PDGF cells is, in part, due to the low expression level of Bim and PUMA as a result of a negative regulation of FoxO3a initiated by PDGF and mediated by Akt.
Concerning Bim, we complemented our study by observing that the Ntv-a/PDGF cells harbored a high level of phospho-Bim (pBim), while the PDGFri and folate treatments decreased the presence of pBim in Ntv-a/PDGF cells (Fig. 3c). In addition, we noted that pBim was due to the PDGF–Erk pathway because the pBim level decreased when Ntv-a/PDGF cells were treated with PDGFri or U0126 (an Erk1/2 inhibitor). Thus, these observations reinforced and corroborated the relationship underlined above and existing between the low expression level of Bim and the low apoptosis level seen in Ntv-a/PDGF cells because literature reports that the Bim phosphorylation abrogates the pro-apoptotic function of this BH3-only protein via its degradation or via its incapacity to activate Bax (Harada et al. 2003; Luciano et al. 2003).
Overexpression of Bcl-w and Bcl-xl in Ntv-a/PDGF cells participates in the low level of Bax activation.
We next analyzed whether the loss of Bax activation in Ntv-a/PDGF cells was also associated with the overexpression of anti-apoptotic proteins since these proteins block cell death signaling by antagonizing Bax activation. Western blot analyses of the expression levels of Mcl-1, Bcl-2, Bcl-w, and Bcl-xl proteins in Ntv-a/PDGF cells treated or not with PDGFri or folate revealed that these cells overexpressed Bcl-w and Bcl-xl (Fig. 4a). This observation was also validated at the transcriptome level (Fig. 4b).
Fig. 4.
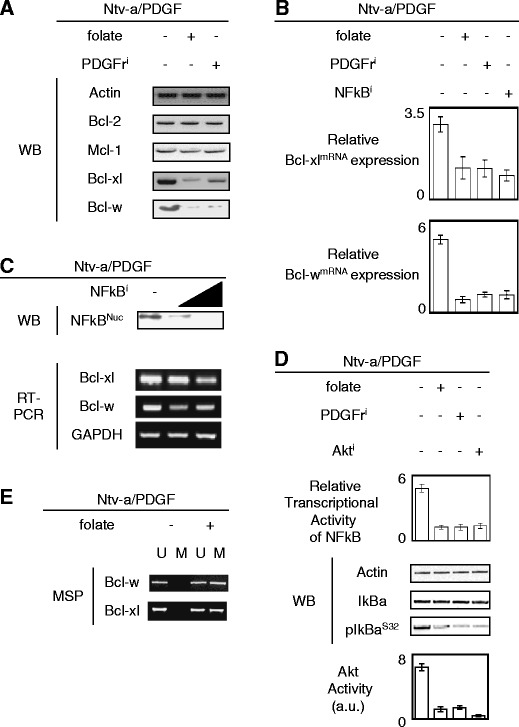
Overexpression of Bcl-w and Bcl-xl in Ntv-a/PDGF cells participates to the low level of Bax activation in Ntv-a/PDGF cells. a Effect of the folate or PDGFri treatment on the expression levels of anti-apoptotic proteins. Actin is used as loading control. b Relative BimmRNA and PUMAmRNA expressions were estimated by qPCR. Cells were treated with NFκBi: BAY11 (Calbiochem, France) at 100 μM for 42 h. c Effect of a range of the NFκBi (BAY11, 50, and 100 μM) treatment on the subcellular localization of NFκB and on the Bcl-w and Bcl-xl mRNA expression. Nuclear extraction was performed using the Nuclear Extract kit (Active Motif, France). GAPDHs are used as control in the RT-PCR analysis. d Correlation between the transcriptional activity of NFκB, the phosphorylation level of IκB, and the Akt activity in Ntv-a/PDGF cells treated or not with folate, PDGFri or Akti
We then asked whether the high expression of Bcl-wmRNA and Bcl-xlmRNA was dependent on the nuclear factor κB (NFκB) activity in Ntv-a/PDGF cells as NFκB has been shown to regulate the expression of these transcripts (Tran et al. 2005). The data presented in Fig. 4c using a NFκB inhibitor confirmed this observation. We also showed that the high transcriptional activity of NFκB in Ntv-a/PDGF was associated with the presence of high levels of phospho-IκBα and a high Akt activity, while the low transcriptional activity of NFκB was associated with the presence of low levels of phospho-IκBα and a low Akt activity in Ntv-a/PDGF cells treated with folate or PDGFri (Fig. 4d).
Based on the DNA-methylating function of folate, we hypothesized that the downregulation of both Bcl-wmRNA and Bcl-xlmRNA observed in Ntv-a/PDGF cells treated with folate could be due to the methylation of the promoters of both bcl-x and bcl-w. Consistent with the fact that NFκB regulates the transcription of both bcl-x and bcl-w genes, we designed primers to assess the methylation status of the NFκB boxes localized within the promoter regions of these two genes. MSP revealed that the CG dinucleotides included within the NFκB boxes of both the bcl-x and bcl-w genes were unmethylated in Ntv-a/PDGF cells, but methylated when these cells were treated with folate (Fig. 4e).
Thus, our data indicate that the high expression of Bcl-xl and Bcl-w seen in Ntv-a/PDGF cells is due to the PDGF-initiated Akt/IκBα-mediated activation of NFκB in a context of unmethylation of the DNA-binding domains of this transcription factor localized within the promoter regions of the bcl-x and bcl-w genes.
Functional hierarchy of the Bcl-2 family members in Ntv-a/PDGF cells
We next studied the relationship between the level of Bax activation and the interactions existing between the Bcl-2, Bcl-xl, and Bcl-w proteins and Bax, PUMA, and Bim proteins.
Changes in the expression of the anti-apoptotic proteins in these cells were quantified by immunoprecipitations performed on CHAPS cell extracts from Ntv-a/PDGF cells treated or not with folate or PDGFri. Under these conditions, the comparison of co-immunoprecipitated proteins from Ntv-a/PDGF cells treated or not with folate or PDGFri revealed that the Ntv-a/PDGF cells were characterized by the strong inhibition of Bax by Bcl-2, Bcl-xl, and Bcl-w, a high antagonism of PUMA and Bim by Bcl-2, Bcl-xl, and Bcl-w, and a low neutralization of Bcl-2, Bcl-xl, and Bcl-w by Bad (Fig. 5a). Thus, this result associates the apopto-resistance of Ntv-a/PDGF cells with the loss of Bax activation as a result of its inhibition by Bcl-2, Bcl-xl, and Bcl-w (Fig. 5a). Immunoprecipitation experiments also revealed a strong antagonism of PUMA and Bim by Bcl-2, Bcl-xl, and Bcl-w in Ntv-a/PDGF cells, and that this antagonism was reduced by PDGFri or folate treatments of Ntv-a/PDGF cells. All these results indicate that (1) the apoptosis evasion phenotype harbored by the Ntv-a/PDGF cells is, in part, due to the antagonism of pro-apoptotic proteins by the anti-apoptotic proteins and (2) the folate-induced apopto-sensitivity of Ntv-a/PDGF cells is associated with the dissociation of pro-apoptotic and anti-apoptotic protein interactions.
Fig. 5.
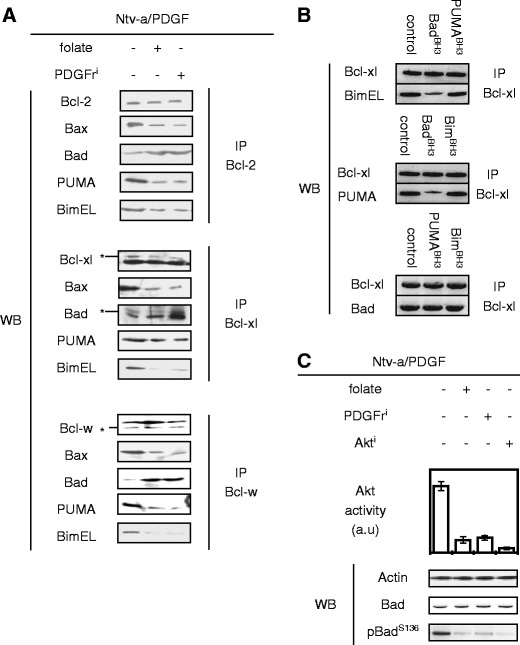
Hierarchical orchestration of the Bcl-2 family members in Ntv-a/PDGF cells. a Effect of the folate or PDGFri treatment on the interaction existing between anti-apoptotic proteins, Bax, and BH3-only proteins by using immunoprecipitation method (IP). b Effect of BH3 peptides (akin the BH3 domain of BH3-only proteins, 1 μM) on the interaction existing between Bcl-xl and BH3-only proteins by using immunoprecipitation method (IP). c Effect of the folate, PDGFri, or Akti treatment on the phosphorylation level of Bad in Ntv-a/PDGF cells
Taken into consideration the roles of activators and/or sensitizers/derepressors of BH3-only proteins and on their hierarchical regulation, our data seem to indicate that the weak neutralization of Bcl-2, Bcl-xl, and Bcl-w by Bad could be at the origin of the strong antagonism of Bax, PUMA, and Bim by these same anti-apoptotic proteins.
Based on the above data, we investigated whether Bad has the capacity to displace PUMA or Bim from anti-apoptotic proteins. For this purpose, we developed an acellular assay in which the capacity of a given BH3 domain to disrupt the co-immunoprecipitation of a given anti-apoptotic protein with a considered BH3-only protein was analyzed. Consistent with a previous report (Kim et al. 2006), we noted that BadBH3 displaced PUMA and Bim from anti-apoptotic proteins such as Bcl-xl, while PUMABH3 or BimBH3 was unable to displace Bad, Bim, or PUMA from Bcl-xl (Fig. 5b ). In addition to the fact that Bad has the capacity to displace Bax from anti-apoptotic proteins, these results strongly suggest that the strong inhibition or antagonism of Bax, Bim, and PUMA by Bcl-2, Bcl-xl, and Bcl-w is, in part, due to the weak neutralization of these anti-apoptotic proteins by Bad.
We next extended our study to the identification of factors promoting the weak neutralization of Bcl-2, Bcl-xl, and Bcl-w by Bad observed in Ntv-a/PDGF cells. For this purpose, we asked whether the process was associated with the presence of Akt-induced phospho-Bad (pBadS136) because this posttranslational modification is known to abrogate the capacity of Bad to bind the anti-apoptotic proteins and Akt is activated by the PDGF signaling pathway. Our analyses revealed that it was the case because the high levels of pBadS136 and Akt activity were associated with the weak neutralization of the anti-apoptotic proteins by Bad in Ntv-a/PDGF cells, while the inverse situation was observed when these cells were treated with PDGFri, Akt, and strikingly with folate (Fig. 5c).
Thus, these results identify the loss of Bad pro-apoptotic function as a molecular determinant governing the apoptosis evasion phenotype of Ntv-a/PDGF cells and suggest that the restoration of Bad pro-apoptotic function could abrogate the apoptosis evasion phenotype of Ntv-a/PDGF cells. To summarize, all our results support the idea that the apoptosis evasion phenotype of Ntv-a/PDGF cells is favored by the loss of pro-apoptotic function of Bad, PUMA, and Bim in a context of overexpression of anti-apoptotic proteins.
Effect of the folate and/or ABT-737 pretreatment on etoposide-induced cell death of PCTC-LGG
Our previous experiments demonstrate that a folate treatment abrogates the PDGF-induced apoptosis evasion phenotype of Ntv-a/PDGF cells by methylating certain genes (bcl-w and bcl-xl), and by reorchestrating the hierarchical interactions of the Bcl-2 family members, we next compared the effect of folate and/or ABT-737 on etoposide-induced apopto-sensibility of primary cultured tumor cells obtained from human LGG (PCTC-LGG). We used this approach because, like folate, ABT-737 (a chemotherapeutic agent that docks within the hydrophobic groove of anti-apoptotic proteins, thereby disabling their capacity to antagonize the actions of pro-apoptotic proteins) has a weak single-agent cell-killing activity but promotes apoptosis in combination with anti-cancer agents unable to initially elicit apoptosis in cancer cells (Oltersdorf et al. 2005; van Delft et al. 2006).
PCTC-LGG used were obtained from tumors characterized by the presence of a high PDGFrβ activity, a low level of 5-methylcytosine, and a low level of intratumor apoptosis intratumor of a high level, a high level of pBad, and the unmethylation of the bcl-w and bcl-xl genes, i.e., by molecular signatures characterizing the Ntv-a/PDGF cells. Thus, among the 20 LGG exhibiting a high PDGFrβ activity, a low level of 5-methylcytosine, and a low level of apoptosis intratumor of a high level, 60% (12/20) presented a high level of pBad and unmethylation of bcl-w gene simultaneously, and 2 different tumors of these 12 were used to obtain PCTC#LGG-1 and -2 (Fig. 6a).
Fig. 6.

Effect of the folate and/or ABT-737 pretreatment on the etoposide-induced cell death of PCTC-LGG. a Characterization of 20 LGG. 12/20 LGG present similar characteristics than the Ntv-a/PDGF cells, i.e., a high PDGFrβ activity (higher or equal to the median value), a low level of DNA methylation (lower to the median value), a low DEVDase activity (lower to the median value), a high level of phospho-Bad (higher or equal to the median value), and the unmethylation of the bcl-w and bcl-xl genes. b Two primary cultured tumor cells obtained from LGG (PCTC-LGG) were treated with folate, ABT-737, and/or epotoside (50 μg/ml), and cell death was estimated by using Trypan blue method
As expected, we initially noted that ABT-737 and folate have weak single-agent cell-killing activity because these components induced less than 20% of cell death (Fig. 6b). Secondly, our data indicated that ABT-737 and folate promoted massive cell death (equal or superior to 60%) in PCTC#LGG-1 when used in association with etoposide. In addition, the synergy of ABT-737 and etoposide is also supported by the fact that cell death increased when the ABT-737 concentration increased (p = 0.0424). Thirdly, we noted that the triple combination of folate/ABT-737 (0.1 μM)/etoposide was more effective than the double combination of folate/etoposide or ABT-737 (0.1 μM)/etoposide (p = 0.0334, p = 0.0031). Fourthly, our data indicated that the use of folate in combination with ABT-737 and etoposide permitted a reduction in the concentration of ABT-737 used to induce massive cell death in PCTC#LGG-1 because no significant difference was observed between the percentage of cell death obtained between PCTC#LGG-1 treated with folate/ABT-737 (0.1 μM)/etoposide and folate/ABT-737 (10 μM)/etoposide (p = 0.6803). Lastly, similar experiments done with PCTC#LGG-2 indicated that ABT-737/etoposide was weakly toxic in these cells in comparison with folate/etoposide (18%, 22%, and 67% cell death, p = 0.0016 and p = 0.0017). In other terms, folate amplified the etoposide-induced cell death in PCTC#LGG-2 cells but not ABT-737. However, we noted that ABT-737 amplified the folate/etoposide-induced death in PCTC#LGG-2 cells (p = 0.0397, p = 0.0376) and this independently of the dose of ABT-737 (p = 0.8601).
Thus, these results indicate that we can reverse the cell death resistance in LGG to anti-cancer agents by using folate and/or ABT-737. In addition, our work indicates that the use of folate in combination with ABT-737/etoposide can markedly enhanced the response of LGG cells to ABT-737/etoposide and may minimize the dose of ABT-737 in order to reduce the ABT-737-induced co-lateral damage, even if this drug is generally well tolerated (Oltersdorf et al. 2005; Konopleva et al. 2006).
Discussion
Recent advances in the identification of the molecular mechanisms that govern the chemo-resistance of cancer cells via the acquisition of apoptosis evasion phenotype have provided meaningful progress in the treatment of many human cancers. However, much more has to be done in order to design successful therapeutic protocols based on rational molecular targeting and the treatment personalization. Recently, we have begun to unveil that folate treatment has the capacity to increase apopto-sensitivity of Ntv-a/PDGF cells, i.e., in LGG cells induced by the constitutive activation of PDGF signaling pathway and characterized by a low level of DNA methylation and a phenotype of apopto-resistance (Hervouet et al. 2009). However, not much is known about how folate abrogates PDGF-induced resistance of anti-cancer treatments for the benefit of an apopto-sensitivity to these same anti-cancer treatments.
We have focused in this study on the role of folate in PDGF-induced dysfunctions in the apoptotic pathway responsible for resistance to anti-cancer drugs/treatments in low-grade glioma cells. Our data support the interest of this search because we found that the simultaneous presence of high PDGFr kinase activity, low level of DNA methylation, and low intratumor apoptosis constitutes a hallmark correlated with a poor survival prognosis in patients suffering from LGG. In addition, our results suggest that the presence of these three hallmarks could be used as a biomarker of disease outcome in LGG and/or for rational selection of patients susceptible to a personalized anti-cancer treatment including folate. However, the incorporation of folate into anti-glioma therapy must be supported by other experiments since folate as other epigenetic drugs may have a dual role in cancer prevention or progression because folate supplementation may decrease the risk of common cancers. Besides, numerous reports support this duality suggesting that high folate treatment may be associated with an increase in the risk of colorectal and prostate cancers in a randomized controlled trial and increase risk of breast cancer, while a low dose of folate may protect against colorectal cancer (Stolzenberg-Solomon et al. 2006; Van Guelpen et al. 2006; Cole et al. 2007; Lindzon et al. 2009; Stidley et al. 2010). Thus, since the importance of the dose schedule of the 5-aza-2′-deoxycytidine treatment in epigenetic therapy of cancer, the dose schedule of the folate treatment is certainly important (Lemaire et al. 2008).
Due to its dual role in PDGF signaling pathway and on the methylation status of genes coding for Bcl-w and Bcl-xl anti-apoptotic proteins, our data demonstrate that folate amplifies etoposide-induced apoptosis by dissociating Bax, Bim, and PUMA pro-apoptotic proteins from Bcl-2, Bcl-xl, and Bcl-w anti-apoptotic proteins for the benefit of Bad/Bcl-2, Bad/Bcl-xl, and Bad/Bcl-w interactions in a context of low expression of Bcl-xl and Bcl-w and of non-phosphorylation of Bad. Thus, these results support and complement the idea of an alternative model of hierarchical activation of pro-apoptotic functions of BH3-only proteins (Cartron et al. 2004; Certo et al. 2006; Kim et al. 2006; Gallenne et al. 2009). In other terms, we can speak of a model in which Bim and PUMA can activate directly Bax when they are no longer antagonized by Bcl-2, Bcl-xl, and Bcl-w anti-apoptotic proteins, i.e., when Bad (or other BH3-only proteins) engages and represses Bcl-2, Bcl-xl, and Bcl-w anti-apoptotic proteins. Consequently, the fact that the folate treatment “activates” the pro-apoptotic function of Bad via its non-phosphorylation indicates that this treatment acts, in part, likely by mimicking the effects of BH3 peptides or BH3 mimetic components. However, this activity is indirect since it is mediated by Bad, whereas that of BH3 mimetic components is direct since the majority of these small molecules have the capacity to bind anti-apoptotic proteins, leading to the disruption of interactions existing between anti- and pro-apoptotic proteins such as BH3-only proteins. Nevertheless, our data argue that folate and BH3 mimetic components such as ABT-737 have other common characteristics than the capacity to displace BH3-only proteins from its binding partners such as Bcl-2, Bcl-xl, and Bcl-w (Oltersdorf et al. 2005; Zhang et al. 2007b). Indeed, our data indicate that they have weak single-agent cell-killing activity, while they enhanced etoposide-induced cell death. In addition and despite the fact that these compounds share similar targets, it seems that folate and ABT-737 can work in synergy to amplify etoposide-induced cell death because the triple combination of folate/ABT-737/etoposide is more efficient than the double combination of folate/etoposide or ABT-737/etoposide. This point is also supported by the fact that the addition of ABT-737 to Ntv-a/PDGF cells treated with folate increases the release of Bim and PUMA from Bcl-xl (Fig. S2).
Due to the fact that interactions between pro- and anti-apoptotic proteins can be directly or indirectly disrupted by ABT-737 and folate treatment, it appears that the combination of folate and ABT-737 markedly amplifies etoposide-induced cell death and permits a reduction in ABT-737 doses. This point is crucial for the future use of ABT-737 in preclinical and/or clinical application because there are reports that ABT-737 can reduce lymphocyte counts and can cause dose-dependent acute thrombocytopenia by reducing the number of circulating platelets, whose turnover is regulated by apoptosis, even if ABT-737 seems to be well tolerated in animal model, or by normal hematopoietic and bone marrow cells (Oltersdorf et al. 2005; Konopleva et al. 2006; Mason et al. 2007; Zhang et al. 2007a).
Finally, the capacity of folate to promote ABT-737/etoposide-induced cell death in cells initially resistant to ABT-737/etoposide-induced cell death (such as PCTC#LGG-2) would suggest that among the potential pre-tropic effects of folate, some of them have the capacity to restore the caspase activation because ABT-737-induced apoptosis is caspase-dependent (van Delft et al. 2006). Besides, this point is consistent with what we have recently reported and the fact that folate treatment has the capacity to methylate the promoter region of survivin gene, i.e., a gene coding for an anti-apoptotic protein able to block apoptosis by inhibiting the caspases (Shin et al. 2001; Hervouet et al. 2009).
By dissecting a mechanism generating the phenotype of apopto-resistance in a context of malignant transformation induced by (or associated with) the activation of PDGF signaling pathway, our data open a new door to a better management of LGG by proposing new therapies including folate and ABT-737 as “amplificators” of response to usual chemotherapeutic drugs, and developed in association with the detection of biomarkers providing a better rationalization and personalization of anti-glioma treatments.
Materials and methods
Analyses of DNA methylation
DNA was extracted by using the QiaAmp DNA mini Kit (Qiagen, France). Four micrograms of genomic DNA was treated with the EZ DNA methylation Gold kit (Zymo Research/Proteigene, France) according to the manufacturer’s instructions. Five microliters of bisulfite DNA was used for methylation-specific PCR analysis.
qPCR
RNA was isolated from cells by using the RNeasy Mini Kit (Qiagen, France). Reactions were realized in triplicate by using the Multiplex Quantitative PCR system (Mx40000), qPCR Core Reagent kit (Stratagene). PDGFrβ: TGCCTCAGCCAAATGTCACCTGC and TCACCACCTCGTATTCC; PUMA: GAAGAGCAAATGAGCCAAACG and GGAGCAACCGGCAAACG; Bim: TGGCAAAGCAACCTTCTGATG and GCAGGCTGCAATTGTCTACCT. Bcl-xl: GATGGAGGAACCAGGTTGTG and CCCCGGAAGGTCTTTTGTAT. Relative expression was calculated using the 2–Ct method.
Western blot and immunoprecipitation
In brief, proteins were size fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Proteins were transferred onto nitrocellulose or PVDF membrane. Saturation and blotting were realized by using SNAP i.d™ Protein Detection System (Millipore, France). The detection of proteins was performed using ECL™ (GE-Amersham Biosciences, France) chemiluminescence reagents. Bands were quantified using Quantity One® quantification software (BioRad, France). Immunoprecipitations were realized by using the Catch and Release® v2.0 Reversible Immunoprecipitation System (Millipore, France). Antibodies used are listed in Table S1.
Transcriptional activity
We used Fugene6 reagent (Roche, France) to transfect the cells with 200 ng of each pTransLuc (Ozyme, France), with 10 ng of CMV-Renilla luciferase. Day 0 was obtained 24 h after transfections. At each time point, cells were harvested, and protein extracts were prepared for dual luciferase assay as described by the manufacturer (Promega, France), and luciferase levels were normalized to Renilla luciferase activity.
Statistical analysis
According to the internal committee of bioethics, the overall survival time was measured from the date of surgical resection to the last follow-up visit or death. Patients with tumor recurrence were censored. Data in bar graphs are expressed as the mean ± SD. Each result is representative of, at least, three independent experiments.
Ntv-a/LacZ and Ntv-a/PDGF cells
These cells come from the laboratory directed by Dr. Eric C. Holland (Memorial Sloan Kettering Cancer Center, New York, USA; Dai et al. 2001). Ntv-a cells were cultivated with Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum and antibiotics. Folate treatment was performed by adding 4 μg/ml of folic acid into the DMEM medium. Thus, the concentration of folic acid was multiplied by two when cells were treated with folate treatment since DMEM contains 4 μg/ml of folic acid according to the manufacturer’s information.
Electronic supplementary materials
Below is the link to the electronic supplementary material.
Effect of the folate or PDGFri treatment on the PDGFrβ activity, the expression of the PDGFrβmRNA, and PDGFrβ and on the methylation status of the −681/-583 region of the PDGFrβ gene. (PDF 65 kb)
Impact of the folate or folate/ABT-737 treatment on the Bcl-xl/PUMA and Bcl-xl/Bim interactions. (PDF 48 kb)
List of antibodies used (PDF 60 kb)
Acknowledgments
We thank the Neurosurgery Department of the Hôpital G and R Laennec, CHU Nantes, and the Oncology Department of the Centre René Gauducheau, Nantes-Atlantique for the tumor samples. We thank microPICell (Plateau technique d’imagerie cellulaire—Centre de Recherche en Cancérologie Nantes-Angers, INSERMU892-IRF26) for its assistance for the confocal microscopy. We thank Dr. Lisa Oliver for the critical reading of this manuscript and her comments.
Conflict of interest
The authors declare that they have no financial relationship with the organization that sponsored the research.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- Dnmt
DNA methyltransferase
- LGG
Low-grade glioma
- 5mC
5-Methylcytosine
References
- Bougras G, Cartron P, Gautier F, Martin S, LeCabellec M, et al. Opposite role of Bax and BCL-2 in the anti-tumoral responses of the immune system. BMC Cancer. 2004;4(1):54. doi: 10.1186/1471-2407-4-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartron PF, Oliver L, Martin S, Moreau C, LeCabellec MT, et al. The expression of a new variant of the pro-apoptotic molecule Bax, Baxpsi, is correlated with an increased survival of glioblastoma multiforme patients. Hum Mol Genet. 2002;11(6):675–687. doi: 10.1093/hmg/11.6.675. [DOI] [PubMed] [Google Scholar]
- Cartron PF, Juin P, Oliver L, Martin S, Meflah K, et al. Nonredundant role of Bax and Bak in Bid-mediated apoptosis. Mol Cell Biol. 2003;23(13):4701–4712. doi: 10.1128/MCB.23.13.4701-4712.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartron P, Gallenne T, Bougras G, Gautier F, Manero F, et al. The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Mol Cell. 2004;16(5):807–818. doi: 10.1016/j.molcel.2004.10.028. [DOI] [PubMed] [Google Scholar]
- Certo M, Del Gaizo MV, Nishino M, Wei G, Korsmeyer S, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9(5):351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Cole B, Baron J, Sandler R, Haile R, Ahnen D, et al. Folic acid for the prevention of colorectal adenomas: a randomized clinical trial. JAMA. 2007;297(21):2351–2359. doi: 10.1001/jama.297.21.2351. [DOI] [PubMed] [Google Scholar]
- Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, et al. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15(15):1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bufalo D, Trisciuoglio D, Biroccio A, Marcocci L, Buglioni S, et al. Bcl-2 overexpression decreases BCNU sensitivity of a human glioblastoma line through enhancement of catalase activity. J Cell Biochem. 2001;83(3):473–483. doi: 10.1002/jcb.1245. [DOI] [PubMed] [Google Scholar]
- Enyedy I, Ling Y, Nacro K, Tomita Y, Wu X, et al. Discovery of small-molecule inhibitors of Bcl-2 through structure-based computer screening. J Med Chem. 2001;44(25):4313–4324. doi: 10.1021/jm010016f. [DOI] [PubMed] [Google Scholar]
- Evan G, Vousden K. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411(6835):342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- Gallenne T, Gautier F, Oliver L, Hervouet E, Noël B, et al. Bax activation by the BH3-only protein Puma promotes cell dependence on antiapoptotic Bcl-2 family members. J Cell Biol. 2009;185(2):279–290. doi: 10.1083/jcb.200809153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg R. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Harada H, Quearry B, Ruiz-Vela A, Korsmeyer S. Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc Natl Acad Sci USA. 2003;101(43):15313–15317. doi: 10.1073/pnas.0406837101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervouet E, Debien E, Charbord J, Menanteau J, Vallette FM, et al. Folate supplementation: a tool to limit the aggressiveness of gliomas via the re-methylation of DNA repeat element and genes governing apoptosis and proliferation. Clin Cancer Res. 2009;15(10):3519–3529. doi: 10.1158/1078-0432.CCR-08-2062. [DOI] [PubMed] [Google Scholar]
- Hsu Y, Youle R. Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem. 1997;272(21):13829–13834. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- Kim H, Rafiuddin-Shah M, Tu H, Jeffers J, Zambetti G, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8(12):1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- Kojima K, Konopleva M, Samudio I, Schober W, Bornmann W, et al. Concomitant inhibition of MDM2 and Bcl-2 protein function synergistically induce mitochondrial apoptosis in AML. Cell Cycle. 2006;5(23):2778–2786. doi: 10.4161/cc.5.23.3520. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo P, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10(5):375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Kulms D, Schwarz T. Molecular mechanisms of UV-induced apoptosis. Photodermatol Photoimmunol Photomed. 2000;16(5):195–201. doi: 10.1034/j.1600-0781.2000.160501.x. [DOI] [PubMed] [Google Scholar]
- Kutzki O, Park H, Ernst J, Orner B, Yin H, et al. Development of a potent Bcl-x(L) antagonist based on alpha-helix mimicry. J Am Chem Soc. 2002;124(40):11838–11839. doi: 10.1021/ja026861k. [DOI] [PubMed] [Google Scholar]
- Lemaire M, Chabot G, Raynal N, Momparler L, Hurtubise A, et al. Importance of dose-schedule of 5-aza-2′-deoxycytidine for epigenetic therapy of cancer. BMC Cancer. 2008;8:128. doi: 10.1186/1471-2407-8-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone M, Zhai D, Sareth S, Kitada S, Reed J, et al. Cancer prevention by tea polyphenols is linked to their direct inhibition of antiapoptotic Bcl-2-family proteins. Cancer Res. 2003;63(23):8118–8121. [PubMed] [Google Scholar]
- Lessene G, Czabotar P, Colman P. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7(12):989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- Lindzon G, Medline A, Sohn K, Depeint F, Croxford R, et al. Effect of folic acid supplementation on the progression of colorectal aberrant crypt foci. Carcinogenesis. 2009;30(9):1536–1543. doi: 10.1093/carcin/bgp152. [DOI] [PubMed] [Google Scholar]
- Lowe S, Lin A. Apoptosis in cancer. Carcinogenesis. 2000;21(3):485–495. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, et al. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22(43):6785–6793. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- Manero F, Gautier F, Gallenne T, Cauquil N, Grée D, et al. The small organic compound HA14-1 prevents Bcl-2 interaction with Bax to sensitize malignant glioma cells to induction of cell death. Cancer Res. 2006;66(5):2757–2764. doi: 10.1158/0008-5472.CAN-05-2097. [DOI] [PubMed] [Google Scholar]
- Martin S, Toquet C, Oliver L, Cartron P, Perrin P, et al. Expression of bcl-2, bax and bcl-xl in human gliomas: a re-appraisal. J Neurooncol. 2001;52(2):129–139. doi: 10.1023/A:1010689121904. [DOI] [PubMed] [Google Scholar]
- Mason K, Carpinelli M, Fletcher J, Collinge J, Hilton A, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128(6):1035–1036. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- Nagane M, Levitzki A, Gazit A, Cavenee W, Huang H. Drug resistance of human glioblastoma cells conferred by a tumor-specific mutant epidermal growth factor receptor through modulation of Bcl-XL and caspase-3-like proteases. Proc Natl Acad Sci USA. 1998;95(10):5724–5729. doi: 10.1073/pnas.95.10.5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Ishida E, Shimada K, Nakase H, Sakaki T, et al. Frequent HRK inactivation associated with low apoptotic index in secondary glioblastomas. Acta Neuropathol. 2005;110(4):402–410. doi: 10.1007/s00401-005-1065-x. [DOI] [PubMed] [Google Scholar]
- Oliver L, Mahé B, Gréé R, Vallette F, Juin P. HA14-1, a small molecule inhibitor of Bcl-2, bypasses chemoresistance in leukaemia cells. Leuk Res. 2007;31(6):859–863. doi: 10.1016/j.leukres.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore S, Shoemaker A, Armstrong R, Augeri D, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Pellecchia M, Reed J. Inhibition of anti-apoptotic Bcl-2 family proteins by natural polyphenols: new avenues for cancer chemoprevention and chemotherapy. Curr Pharm Des. 2004;10(12):1387–1398. doi: 10.2174/1381612043384880. [DOI] [PubMed] [Google Scholar]
- Reed J. Apoptosis-targeted therapies for cancer. Cancer Cell. 2003;3(1):17–22. doi: 10.1016/S1535-6108(02)00241-6. [DOI] [PubMed] [Google Scholar]
- Reed J, Pellecchia M. Apoptosis-based therapies for hematologic malignancies. Blood. 2005;106(2):408–418. doi: 10.1182/blood-2004-07-2761. [DOI] [PubMed] [Google Scholar]
- Shin S, Sung B, Cho Y, Kim H, Ha N, et al. An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and −7. Biochemistry. 2001;40(4):1117–1123. doi: 10.1021/bi001603q. [DOI] [PubMed] [Google Scholar]
- Stidley C, Picchi M, Leng S, Willink R, Crowell R, et al. Multivitamins, folate, and green vegetables protect against gene promoter methylation in the aerodigestive tract of smokers. Cancer Res. 2010;70(2):568–574. doi: 10.1158/0008-5472.CAN-09-3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolzenberg-Solomon R, Chang S, Leitzmann M, Johnson K, Johnson C, et al. Folate intake, alcohol use, and postmenopausal breast cancer risk in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Am J Clin Nutr. 2006;83(4):895–904. doi: 10.1093/ajcn/83.4.895. [DOI] [PubMed] [Google Scholar]
- Tran N, McDonough W, Savitch B, Sawyer T, Winkles J, et al. The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling system regulates glioma cell survival via NFkappaB pathway activation and BCL-XL/BCL-W expression. J Biol Chem. 2005;280(5):3483–3492. doi: 10.1074/jbc.M409906200. [DOI] [PubMed] [Google Scholar]
- van Delft M, Wei A, Mason K, Vandenberg C, Chen L, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10(5):389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Guelpen B, Hultdin J, Johansson I, Hallmans G, Stenling R, et al. Low folate levels may protect against colorectal cancer. Gut. 2006;55(10):1461–1466. doi: 10.1136/gut.2005.085480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Liu D, Zhang Z, Shan S, Han X, et al. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc Natl Acad Sci USA. 2000;97(13):7124–7129. doi: 10.1073/pnas.97.13.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler M, Bähr O, Hohlweg U, Naumann U, Rieger J, et al. BCL-xL: time-dependent dissociation between modulation of apoptosis and invasiveness in human malignant glioma cells. Cell Death Differ. 2006;13(7):1156–1169. doi: 10.1038/sj.cdd.4401786. [DOI] [PubMed] [Google Scholar]
- Weller M, Malipiero U, Aguzz IA, Reed J, Fontana A. Protooncogene bcl-2 gene transfer abrogates Fas/APO-1 antibody-mediated apoptosis of human malignant glioma cells and confers resistance to chemotherapeutic drugs and therapeutic irradiation. J Clin Invest. 1995;95(6):2633–2643. doi: 10.1172/JCI117965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Nimmer P, Tahir S, Chen J, Fryer R, et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14(4):703–715. doi: 10.1038/sj.cdd.4402072. [DOI] [PubMed] [Google Scholar]
- Zhang L, Ming L, Yu J. BH3 mimetics to improve cancer therapy; mechanisms and examples. Drug Resist Updat. 2007;10(6):207–217. doi: 10.1016/j.drup.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of the folate or PDGFri treatment on the PDGFrβ activity, the expression of the PDGFrβmRNA, and PDGFrβ and on the methylation status of the −681/-583 region of the PDGFrβ gene. (PDF 65 kb)
Impact of the folate or folate/ABT-737 treatment on the Bcl-xl/PUMA and Bcl-xl/Bim interactions. (PDF 48 kb)
List of antibodies used (PDF 60 kb)