

Abstract
The vagal afferent pathway is the major neural pathway by which information about ingested nutrients reaches the CNS and influences both GI function and feeding behavior. Vagal afferent neurons (VAN) express receptors for many of the regulatory peptides and molecules released from the intestinal wall, pancreas, and adipocytes that influence GI function, glucose homeostasis, and regulate food intake and body weight. As such, they play a critical role in both physiology and pathophysiology, such as obesity, where there is evidence that vagal afferent function is altered. This review will summarize recent findings on changes in vagal afferent function in response to ingestion of high fat diets and explore the hypothesis that changes in gut microbiota and integrity of the epithelium may not only be important in inducing these changes but may be the initial events that lead to dysregulation of food intake and body weight in response to high fat, high energy diets.
Keywords: TLR-4, leptin, CCK type 1 receptors, cocaine- and amphetamine-regulated peptide, EGR-1, phosphorylated STAT3, suppressor of cytokine signaling 3, Bacteroidales, Closatridiales, lipopolysaccharide
1.0 Introduction
The complex, hierarchical control of food intake and body weight involves central nervous system integration of information from different sources, including the peripheral nervous system and humoral signals from the gut, liver, and adipocytes. The gut is the initial interface in the body at which the quality and quantity of nutrients is accurately monitored; this information is key in the normal physiological functioning of the gastrointestinal tract and helps determines plasma glucose in the postprandial period. This information is also important in short term regulation of food intake; increasing evidence suggests that changes in the ability of the gut to accurately monitor luminal contents and/or to transmit this information to the central nervous system (CNS) is compromised in obesity.
It is generally accepted that enteroendocrine cells are the primary “gut sensors”; these cells are dispersed throughout the gastrointestinal mucosa and secrete hormones and amines in response to the three macronutrient types - carbohydrates, proteins and lipids [1]. The mechanisms for gut chemosensing and release of hormones and amines are varied and involve G protein coupled receptors, nutrient transporters and related proteins, and intracellular metabolism of nutrients. The precise mechanisms of gut chemosensing will not be considered further in this review; the reader should refer to several recent reviews [1–3]. Gut hormones can influence GI function and feeding behavior via true humoral pathways, acting on remote target tissue such as the endocrine and exocrine pancreas via the blood stream. However, other hormones and amines released from gut endocrine cells act via paracrine pathways, producing function changes via effects on local cells, including epithelial cells, immune cells and the nerve terminals of both intrinsic and extrinsic neurons.
One target of gut hormones is to activate vagal afferent neurons (VAN), whose terminals lie in the mucosa. This is the major neural pathway by which information about ingested nutrients reaches the CNS to influence both GI function and feeding behavior. VAN express receptors for many of the regulatory peptides and molecules released from the enteroendocrine cells in the gut. VAN express receptors for cholecystokinin (CCK), CCK type 1 receptors (CCK1R). The evidence for a functional role for these receptors in regulation of vago-vagal reflex control of gut function and in regulation of food intake is compelling.
2.0 CCK as master regulator of vagal afferent nerve function
Exogenous administration of CCK either in vivo in rats or in ex vivo preparations of the stomach or proximal intestine induces a rapid activation of VAN discharge via CCK1Rs [4–6]. CCK induces activation of second order neurons in the nucleus of the solitary tract (NTS), the brain region where vagal afferent neurons terminate, consistent with its observed effects on primary afferent neuronal discharge [7–9]. The action to depolarize VAN underlies the ability of CCK to produce reflex changes in GI function, such as inhibition of gastric motility, acid secretion and pancreatic secretion, all of which change rapidly after systemic administration of CCK [6, 10–11]. Moreover, the action of CCK on VAN is assumed to result in transmission of afferent information along the neuroaxis from the brainstem to the hypothalamus, resulting in inhibition of ongoing feeding, i.e. induction of satiation, comprising of a decrease in meal size and duration [11].
In humans, exogenous CCK or perfusion into the small intestine with protein or long chain triglycerides, nutrients known to release CCK from enteroendocrine cells, terminates feeding via a CCK1R pathway [12–13]. Experiments in rodents have shown the effects of lipid and CCK on food intake are mediated via a vagal afferent pathway [14–15]. Rats with a mutation in the CCK1R are hyperphagic and obese [16–17]. CCK1R null mice eat significantly longer and larger meals compared to wildtype mice, lending support to the hypothesis that the CCK1R plays an important role in the regulation of food intake [18–19].
More recently it has been recognized that CCK induces longer term changes in VAN function associated with the nutritional status of the animal; the changes involve expression of peptides and receptors involved in the regulation of food intake [6, 20]. Expression of mRNA and protein for orexigenic peptides, such as melanin-concentrating hormone (MCH), and their receptors, such as the MCH receptor (MCH1R) and cannabinoid receptor (CB1Rs) are increased in VAN in fasted compared to fed rats. At the same time, there is a decrease in expression of anorexigenic PYY3–36 receptor (Y2R) and the anorexigenic peptide, cocaine- and amphetamine-related transcript (CART) in fasted versus the fed state [21–23]. Administration of exogenous CCK to fasted rats or release of endogenous CCK by ad libitum feeding of a normal chow diet stimulates expression of CART and Y2R and inhibits expression of MCH, MCH1R and CB1Rs [24]. The reversal in the phenotype of VAN by CCK or feeding is mediated via a CCK1R pathway. Thus, CCK acts on VAN to decrease expression of peptides and receptors that stimulate appetite and feeding (MCH, MCHR1, CB1R), and to increase expression of peptides and receptors that inhibit feeding and decrease appetite (CART and Y2R).
Thus, VAN can exist in one of at least two states, expressing either a “hunger” or a “satiety” phenotype (Fig 1). The functional significance of the change in the VAN phenotype is not clear. However, it is possible that alterations in the expression of receptors and peptides play a role in setting the sensitivity of VAN to other hormones and neuromodulators, and in this way play an additional role in regulation of food intake and possibly body weight. Whether this altered function occurs at the level of VAN, via autocrine pathways for example, or at the site of termination of VAN in the brainstem is not known.
Figure 1.
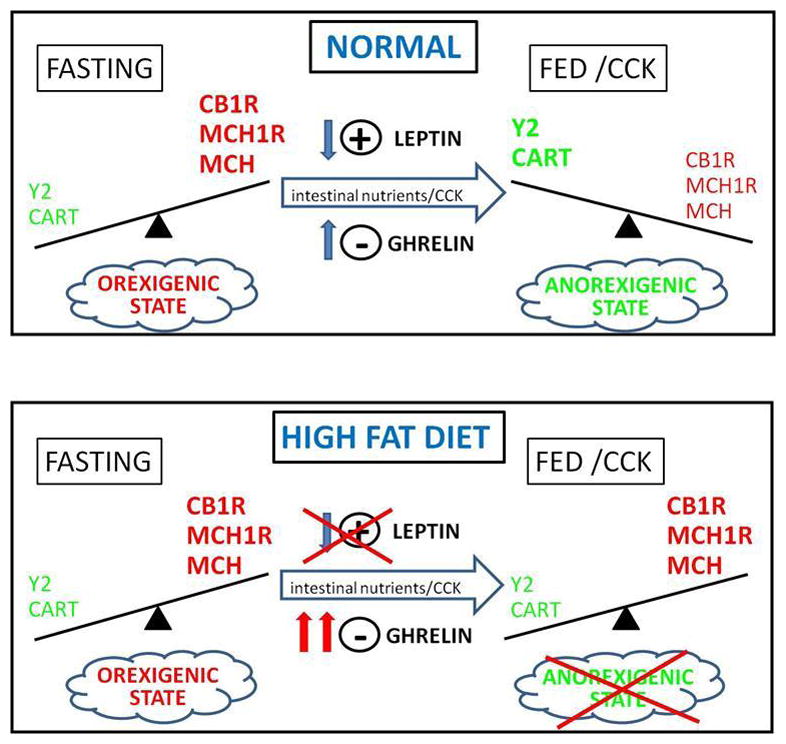
CCK or feeding decreases expression of CB1R, MCH1R and MCH and increases expression of CART and Y2R; in this way the phenotype of vagal afferent neurons switch to an “anorexigenic” state and the drive to eat will be decreased when food is in the intestine. This process is dependent on CCK, is synergized by leptin and inhibited by ghrelin. On e possible hypothesis for alteration in vagal afferent nerve activity in diet-induced obesity is that after long term ingestion of high fat diets, this phenotypic switch may be reduced or abolished, because of a decrease in leptin signaling and/or an increase in ghrelin receptor expression. Thus the neurons remain in an orexigenic state resulting in hyperphagia and obesity.
The cellular mechanism by which CCK alters the phenotype of VAN has been further investigated by the group of Dockray et al. Using VAN in short term culture, this group has shown activation of CCK1Rs induces a PKC-dependent pathway leading to CREB phosphorylation and expression of CART and Y2R. Transfection of VAN with a dominant negative mutant of CREB results in expression of MCH and not CART, suggesting that CREB plays a role in mediating both an increase in CART and a decrease in MCH expression in response to CCK [21, 25]. CCK also increases translocation of the immediate early gene EGR-1 from the cytoplasm to the nucleus; EGR-1 translocation is a required step for the CCK-induced increase in CART expression [26].
Recent evidence suggests that CART can interact with CCK at the level of VAN and this action may play a role in CCK-induced inhibition of food intake [26]. In short term culture, CCK stimulates the secretion of CART from VAN; CCK-induced CART release is augmented by the presence of leptin and inhibited by ghrelin. CART is also found to increase expression of its own promoter suggesting CART can act on VAN via an autocrine loop. Blockade of CART expression or release inhibited the ability of CCK to increase nuclear pCREB and EGR-1. This data from isolated VAN suggests that CART may act synergistically with CCK to inhibit food intake. This hypothesis was tested by measuring food intake in rats after systemic administration of CART with or without a premeal to prime the release of endogenous CCK. After a short fast, prior administration of CART before refeeding augmented the endogenous CCK-induced decrease food intake, consistent with its ability to release CART from VAN as shown in short term culture. However, the site of action of CART is not clear; it could be at the level of the VAN or could be via its release within the NTS. Previous studies have shown that CART acts within the brain to inhibit food intake, although whether it acts at the level of the termination of VAN is not clear.
3.0 Chronic ingestion of a high fat diet on the CCK1R-vagal afferent pathway
In laboratory rodents, chronic ingestion of high fat (HF) diet leads to an increase in body weight. In humans, ingestion of high fat and high calorie diets is associated with an increase in adiposity, body weight and obesity. The increase in adiposity results in an increase in plasma levels of leptin, a hormone released from both the gastric epithelium and adipocytes. Chronic ingestion of high fat, high energy diets and an increase in plasma leptin are associated with leptin resistance in the hypothalamus [27–28]. Leptin resistance is characterized by the lack of downstream intracellular signaling in response to leptin. Peripheral leptin resistance refers to the process by which access of leptin derived from the periphery can no longer cross the blood-brain barrier and thus does not induce changes in hypothalamic neuronal activity as in a lean individual. However, the initial triggering event in this cycle is not clear. Leptin resistance in hypothalamic neurons or a decrease in its ability to cross the blood brain barrier only occurs when there is an increase in fat pad mass or function, and therefore hypothalamic leptin resistance does not necessarily explain the initial events that lead to dysregulation of food intake.
Several investigators have established that there is a decreased sensitivity of VAN to high fat feeding, although the mechanism by which this occurs is unclear [29]. Activation of neurons in the NTS measured by Fos protein immunoreactivity in response to either CCK or intestinal lipid is markedly decreased by chronic ingestion of high fat diets in rodents. Moreover, inhibition of food intake in response to either CCK or lipid is markedly reduced.
We investigated whether the diminished response to a HF diet and its effects on food intake were dependent on the caloric content of the diet or the fat content [30]. Rats were pair-fed isocaloric diets that were either high in fat (38% fat/kcal) or low in fat (10% fat/kcal; LF), both having the same energy density, and monitored meal patterns. Initially, meal size was reduced in rats ingesting the HF compared to the LF diet but overall daily food intake was the same due to a compensatory decrease in intermeal interval in the HF fed rats. However, after 8 weeks, there was a significant increase in individual meal size and daily food intake in rats ingesting the HF compared to LF diet and to the values obtained at 2 weeks. The change in meal patterning occurred in the absence of an increase in adiposity or weight gain. Moreover, there was significant decrease in activation of second order neurons in the NTS in response to intragastric gavage with lipid in the HF fed rats compared to LF fed controls. Thus, adaptation to HF diets does not depend upon an increase in adiposity but rather to the fat content of the ingested diet. This adaptation of the vagal afferent pathway may contribute to altered body weight regulation during ingestion of HF foods in humans.
The mechanism(s) leading to attenuation of vagal afferent function by HF diets and the contribution this plays to the development or maintenance of an obese phenotype is not clear. One possible hypothesis is that the attenuation is due to an altered balance in expression of anorexigenic and orexigenic peptides and receptors, leading to dysregulation of intestinal feedback control of GI function and food intake (Fig 1). Consumption of HF diets in freely feeding outbred Sprague Dawley rats for eight weeks causes animals prone to the obesigenic effects of the HF diets (diet-induced obesity; DIO) to become hyperphagic, increase in body weight and adiposity compared to rats resistant to the obesigenic effects of the diet (DR) or low fat (LF) fed controls [31–32]. We examined the gene expression of VAN in DIO compared to DR and LF rats. Expression of genes encoding for the orexigenic receptors, CB1R and GHSR1a increased in DIO compared to DR or LF fed rats. In addition, there was an increase in expression of the CCK1R but no change in leptin or Y2R expression. This data is particularly interesting as it shows that the change in receptor expression is not solely dependent on the ingestion of HF diet but is only evident in rats that actually become obese when ingesting the HF diet. Another possibility is the development of hormonal resistance in VAN as seen with leptin or insulin in diet-induced obesity.
Preliminary data from our laboratory suggests that VAN become leptin resistant. Leptin potentiates the effects of CCK on VAN [11]. Leptin and CCK interact in VAN at the level of the transcription factor early growth response factor-1 (EGR-1); leptin-induced pSTAT3 upregulates expression of EGR-1 in VAN while CCK induces translocation of EGR-1 to the nucleus [26]. Thus an inability of VAN to respond to leptin would decrease the ability of CCK to activate VAN and to inhibit food intake. Preliminary data from our laboratory has shown that feeding has no effect on the CART/MCH expression in VAN from obese rats (de Lartigue, unpublished observations) suggesting that in diet-induced obesity, VAN get “stuck” in the hunger phenotype (Fig 1).
4.0 Intestinal Microflora
Considerable attention has focused recently on the role of the gut microbiota in obesity, prompted by a series of interesting studies from the Gordon group at Washington University, USA [33–35]. Initial observations in leptin-deficient ob/ob mice show a different proportion of the major phyla of cecal bacteria compared to lean WT mice; there was an increase in Firmicutes and a decrease in Bacteriodetes in the obese mice [36]. In humans, obesity is also associated with a decrease in Bacteriodetes and an overall decrease in bacterial diversity [37–38]. Germ-free mice have a reduced level of adiposity compared to conventionally raised mice, even when ingesting a low fat chow diet. Conventionalization of the germ free mice with the cecal microbiota from normal mice increased the level of adiposity, which was not dependent on an increase in chow intake or changes in energy expenditure [38]. Evidence was obtained to suggest that this might be due to an increase in energy extraction due to bacterial fermentation of polysaccharides and also to the ability of the gut microbiota to upregulate fasting induced adipose factor, a circulating lipoprotein lipase produced by the ileal epithelium, adipocytes and liver, which increases cellular uptake of fatty acids and triglyceride accumulation in adipocytes. These studies led to the concept of an “obese microbiota” [34].
Similar results linking gut microflora to body weight have been described in models of diet-induced obesity. In mice, ingestion of a high fat diet resulted in a bloom in a single clade of Firmicutes, the Mollicutes, and again this was transmissible to lean germ-free recipient mice [39]. Moreover, the absence of microbiota or its almost complete reduction with broad spectrum antibiotics prevents or reverses diet-induced obesity [40–41]. In a recent study on the effects of a high fat diet on the gut microbiota, Hildebrandt et al show that in switching lean mice to a HF diet, there was the expected decrease in Bacteriodetes and an increase in Firmicutes and Proteobacteria [42]. However, further studies have provided evidence that the alteration of the gut microbiota may not be enough to drive the obese phenotype; rather the phenotype depends on the response of the host to the obesigenic diet and/or the change in the microflora. In RELMβKO mice, a transgenic line relatively resistant to the obesigenic effects of a HF diet, the changes in the microbiota are similar regardless of whether the mouse displayed an obese or lean phenotype. This data suggests that it is the HF diet rather than obesity that accounts for the change in the microflora. It should be noted that these mice are only relatively resistant to the effects of a HF diet which confounds these findings. There is a complex interplay between the host and the microbiota, with the one shaping the response to the other. Gross perturbations, such as germ free animals or antibiotic treatment may not necessarily be good models to deduce much about interaction between the host, diet, microbiota and alteration of body weight regulation and glucose homeostasis.
We have recently published data to conclusively show that it is a HF diet rather than obesity that is correlated with changes in gut microbiota [43] We used the Sprague Dawley rat which is a non-congenic strain with individuals either prone or resistant to diet-induced obesity. We found a decrease in the total number of cecal bacteria and an increase in a clade of Firmicutes Clostridiales in all rats ingesting a HF diet regardless of whether they had the DIO or DR phenotype compared to LF fed controls. Taken together these studies suggest that the diet is driving changes in the gut microbiota. If this is the case, what is determining, in either rodent models or more importantly humans, whether they remain lean or increase weight?
5. 0 Obesity as an inflammatory disease
It has been recognized for many years that obesity and type 2 diabetes (T2D) are inflammatory conditions and that “low grade, chronic” inflammation exists in obese patients and is associated with insulin and leptin resistance [44]. The source of the inflammation is regarded as the adipose tissue itself, known to produce inflammatory mediators that could then influence other tissues, such as muscle, liver, pancreas and the central nervous system. However, with evidence for a role of the gut microbiota, the source of the inflammation has been reevaluated and the possible role of the GI tract as a potential source of inflammation is being explored.
In humans it has been shown that ingestion of a high fat meal is associated with an increase in plasma levels of bacterial endotoxin, lipopolysaccharide (LPS) [45]. LPS is part of the outer membrane of gram negative bacteria and is released into the gut lumen when bacteria die. LPS can cross the intestinal epithelial barrier, possibly by phagocytosis by enterocytes but also via a leaky gut epithelium. Further, a link has been found between plasma levels of LPS and food intake in humans and it has been reported that there is a 2 fold increase in circulating levels of LPS in obese type 2 diabetic patients versus lean human subjects [46].
The group of Cani et al has investigated a role for LPS in inducing obesity and type 2 diabetes. They reported that in mice fed high fat diets there is a constant increase in circulating LPS and coined the term “metabolic endotoxemia” to describe the low chronic increase in LPS compared to the much larger, acute onset levels seen in sepsis [47]. Moreover, this group showed that mice with a deletion of CD14, the co-receptor for the LPS receptor TLR4, remained lean even when ingesting a HF diet. Similarly, TLR-4 KO mice are also relatively resistant to diet-induced obesity [48–49]. Obese ob/ob mice have [50] an increase in plasma levels of LPS and the use of antibiotics in these mice decreased the metabolic endotoxemia and improved measures of inflammation, such as macrophage infiltration of adipose tissue. This group also reported data to show that high fat fed mice increased intestinal permeability and ZO-1 expression. Treatment with a broad spectrum antibiotic decreased intestinal permeability and increased ZO-1 expression to levels seen in LF fed control mice. The data suggest that HF feeding induces changes in intestinal permeability via alteration of expression of tight junction proteins. Further, this group used implanation of osmotic minipumps to experimentally induce metabolic endotoxemia and found this treatment recapitulated many of the symptoms of T2D, such as impaired insulin sensitivity. Taken together, this data suggests that LPS may well be a link between the gut flora and metabolic disease.
However, these studies do not demonstrate the site of action of LPS, particularly for the initial hyperphagia (and change in energy expenditure) that must precede gain in body weight and adiposity. Our recent studies have focused on the role of the intestinal microbiota and LPS in altering VAN in diet-induced obese rats.
6. 0 Gut Inflammation
Several studies have now shown a strong link between intestinal inflammation and diet-induced obesity. DIO but not DR Sprague Dawley rats exhibit ileal inflammation (increase of myeloperoxidase, MPO, a commonly used marker of inflammation) together with activation of TLR4 (increase immunoreactivity for the activated receptor TLR4-MD2 complex) in ileal epithelial cells [43]. In DIO rats, there is an increase in plasma LPS, intestinal permeability, and altered tight junction proteins (increase phosphorylation of myosin light chain [MLC] and intracellular localization of occludin). As previously described, quantification of total bacterial-universal 16S rRNA gene copies showed that the HF diet decreased total bacterial density, and was associated with a relative increase in Clostridiales order in HF vs LF fed rats, but there was no difference between DIO and DR rats. However, we noted a marked and significant increase in Enterobacteriales order which only appeared in the obese microbiota. Evidence from inflammatory models suggests that this relative increase in Enterobacteriales order may be secondary to inflammation, rather than causal. Thus, consumption of a HF diet induces changes in the gut microbiota, but it is the development of inflammation, and the increase in plasma LPS in response to these changes, which is associated with the appearance of an obese phenotype.
A novel approach to elucidate the role between the gut microbiota, HF diet and gut inflammation was recently published by Ding et al [51]. Germ free or conventional mice were fed HF or LF diets and intestinal inflammation was assessed by measuring mRNA for TNFα or by use of NF-κBEGFP reporter mice. HF diet increased TNFα mRNA in the ileum and induced activation of NF-κB only in conventional mice. The authors concluded that the combination of HF diet and bacteria was necessary to induce intestinal inflammation and its development was strongly associated with weight gain, adiposity, and plasma levels of insulin and glucose. Thus intestinal inflammation may be on early event by which HF diets contributes or leads to obesity.
The challenge now is to link gut inflammation with hyperphagia (Fig 2). Considerable evidence suggests that LPS activation of the TLR-4 can induce marked changes in the function of hypothalamic neurons [52]. However, the close proximity of VAN to the site of LPS release makes vagal afferent signaling a particularly interesting target for LPS. Clearly, the decrease in sensitivity of the vagal afferent pathway to CCK and intestinal lipid is associated with hyperphagia. It is interesting to note that VAN express TLR-4 receptors [53] and therefore could affect VAN signaling and function. TLR-4 receptors have also been localized to enteroendocrine cells and this may be an alternate or additional target of action [54]. Another challenge is to determine whether LPS is acting locally within the wall of the gut as it moves across leaky tight junctions in the more distal gut or whether it acts systemically to influence VAN function. Gut microbes are found throughout the length of the gut, although the highest numbers are in the large intestine and presumably the preponderance of LPS will come from the distal gut. How this alter vagal afferent neuronal function in the more proximal gut is not clear, but certainly would point to a systemic action of LPS.
Figure 2.

Proposed model by which ingestion of high fat diets may lead to hyperphagia and an increase in body weight. Ingestion of a high fat diet induces changes in the gut microbiota; in diet-induced obese prone rats this directly or indirectly leads to dysbiosis and gut inflammation, which in turn will dirupt tight junction function and increase gut permeability. An increase in gut permeability may allow more lipopolysacharide (LPS) to cross the gut epithelium leading to a low, but chronic increase in plasma levels of LPS (metabolic endotoxemia). The mechanism by which LPS induces hyperphagia is unclear but may involve effects on the gut-brain axis, including vagal afferent neurons.
7.0 Summary
There is evidence that a high fat diet will induce changes in the gut microbiota and, at least in rodent models, this dysbiosis will produce deleterious effects on gut inflammation and intestinal barrier function. This in turn can lead to a metabolic endotoxemia leading either directly or indirectly to changes in neuronal function including vagal afferent neurons. Diet-induced obesity is rats is associated with an absence of phenotypic change induced by feeding; the mechanism of which is unclear at this time but may involve leptin resistance.
A greater understanding of the plasticity of the vagal afferent pathway, particularly as it relates to changes in gut microflora and inflammation, will provide a better understanding of the effect of high fat diets on regulation of body weight.
Acknowledgments
Work funded in part by funds from NIHDDK and the California Dairy Research Foundation/Dairy Milk Initiative. We are grateful to the many individuals at UC Davis who have been part of discussions and collaborative work; Collin Ellis, Jack Rutledge, Gabriel Paulino, Jennifer Lee, and Sean Adams.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Raybould HE. Gut chemosensing: Interactions between gut endocrine cells and visceral afferents. Autonomic Neuroscience-Basic & Clinical. 2010;153(1–2):41–46. doi: 10.1016/j.autneu.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sternini C, Anselmi L, Rozengurt E. Enteroendocrine cells: a site of ‘taste’ in gastrointestinal chemosensing. Curr Opin Endocrinol Diabetes Obes. 2008;15(1):73–8. doi: 10.1097/MED.0b013e3282f43a73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raybould HE. Nutrient sensing in the gastrointestinal tract: possible role for nutrient transporters. J Physiol Biochem. 2008;64(4):349–56. doi: 10.1007/BF03174091. [DOI] [PubMed] [Google Scholar]
- 4.Blackshaw LA, Grundy D. Locally and reflexly mediated effects of cholecystokinin-octapeptide on the ferret stomach. J Auton Nerv Syst. 1991;36(2):129–37. doi: 10.1016/0165-1838(91)90109-g. [DOI] [PubMed] [Google Scholar]
- 5.Blackshaw LA, Grundy D. Effects of cholecystokinin (CCK-8) on two classes of gastroduodenal vagal afferent fibre. J Auton Nerv Syst. 1990;31(3):191–201. doi: 10.1016/0165-1838(90)90185-l. [DOI] [PubMed] [Google Scholar]
- 6.Dockray GJ. Cholecystokinin and gut-brain signalling. Regul Pept. 2009;155(1–3):6–10. doi: 10.1016/j.regpep.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 7.Chen DY, et al. The induction and suppression of c-fos expression in the rat brain by cholecystokinin and its antagonist L364,718. Neurosci Lett. 1993;149(1):91–4. doi: 10.1016/0304-3940(93)90355-o. [DOI] [PubMed] [Google Scholar]
- 8.Day HE, et al. Evidence that cholecystokinin induces immediate early gene expression in the brainstem, hypothalamus and amygdala of the rat by a CCKA receptor mechanism. Neuropharmacology. 1994;33(6):719–27. doi: 10.1016/0028-3908(94)90111-2. [DOI] [PubMed] [Google Scholar]
- 9.Monnikes H, Lauer G, Arnold R. Peripheral administration of cholecystokinin activates c-fos expression in the locus coeruleus/subcoeruleus nucleus, dorsal vagal complex and paraventricular nucleus via capsaicin-sensitive vagal afferents and CCK-A receptors in the rat. Brain Res. 1997;770(1–2):277–88. doi: 10.1016/s0006-8993(97)00865-2. [DOI] [PubMed] [Google Scholar]
- 10.Raybould HE. Mechanisms of CCK signaling from gut to brain. Curr Opin Pharmacol. 2007;7(6):570–4. doi: 10.1016/j.coph.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peters JH, Simasko SM, Ritter RC. Modulation of vagal afferent excitation and reduction of food intake by leptin and cholecystokinin. Physiol Behav. 2006;89(4):477–85. doi: 10.1016/j.physbeh.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 12.Gutzwiller JP, et al. Interaction between CCK and a preload on reduction of food intake is mediated by CCK-A receptors in humans. Am J Physiol Regul Integr Comp Physiol. 2000;279(1):R189–95. doi: 10.1152/ajpregu.2000.279.1.R189. [DOI] [PubMed] [Google Scholar]
- 13.Matzinger D, et al. The role of long chain fatty acids in regulating food intake and cholecystokinin release in humans. Gut. 2000;46(5):688–693. doi: 10.1136/gut.46.5.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beglinger C, Degen L. Fat in the intestine as a regulator of appetite--role of CCK. Physiol Behav. 2004;83(4):617–21. doi: 10.1016/j.physbeh.2004.07.031. [DOI] [PubMed] [Google Scholar]
- 15.Raybould HE. Nutrient tasting and signaling mechanisms in the gut. I. Sensing of lipid by the intestinal mucosa. Am J Physiol. 1999;277(4 Pt 1):G751–5. doi: 10.1152/ajpgi.1999.277.4.G751. [DOI] [PubMed] [Google Scholar]
- 16.Moran TH, et al. Disordered food intake and obesity in rats lacking cholecystokinin A receptors. Am J Physiol. 1998;274(3 Pt 2):R618–25. doi: 10.1152/ajpregu.1998.274.3.R618. [DOI] [PubMed] [Google Scholar]
- 17.Moran TH. Unraveling the obesity of OLETF rats. Physiol Behav. 2008;94(1):71–8. doi: 10.1016/j.physbeh.2007.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whited KL, et al. Targeted disruption of the murine CCK1 receptor gene reduces intestinal lipid-induced feedback inhibition of gastric function. Am J Physiol Gastrointest Liver Physiol. 2006;291(1):G156–62. doi: 10.1152/ajpgi.00569.2005. [DOI] [PubMed] [Google Scholar]
- 19.Bi S, et al. Differential body weight and feeding responses to high-fat diets in rats and mice lacking cholecystokinin 1 receptors. Am J Physiol Regul Integr Comp Physiol. 2007;293(1):R55–63. doi: 10.1152/ajpregu.00002.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dockray GJ. The versatility of the vagus. Physiol Behav. 2009;97(5):531–6. doi: 10.1016/j.physbeh.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 21.de Lartigue G, et al. Cocaine- and amphetamine-regulated transcript: stimulation of expression in rat vagal afferent neurons by cholecystokinin and suppression by ghrelin. J Neurosci. 2007;27(11):2876–82. doi: 10.1523/JNEUROSCI.5508-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burdyga G, et al. Expression of cannabinoid CB1 receptors by vagal afferent neurons is inhibited by cholecystokinin. J Neurosci. 2004;24(11):2708–15. doi: 10.1523/JNEUROSCI.5404-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burdyga G, et al. Feeding-dependent depression of melanin-concentrating hormone and melanin-concentrating hormone receptor-1 expression in vagal afferent neurones. Neuroscience. 2006;137(4):1405–15. doi: 10.1016/j.neuroscience.2005.10.057. [DOI] [PubMed] [Google Scholar]
- 24.Burdyga G, et al. Cholecystokinin regulates expression of Y2 receptors in vagal afferent neurons serving the stomach. J Neurosci. 2008;28(45):11583–92. doi: 10.1523/JNEUROSCI.2493-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Lartigue G, et al. Cocaine- and Amphetamine-Regulated Transcript Mediates the Actions of Cholecystokinin on Rat Vagal Afferent Neurons. Gastroenterology. 2010;138(4):1479–1490. doi: 10.1053/j.gastro.2009.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Lartigue G, et al. EGR1 Is a Target for Cooperative Interactions between Cholecystokinin and Leptin, and Inhibition by Ghrelin, in Vagal Afferent Neurons. Endocrinology. 2010;151(8):3589–3599. doi: 10.1210/en.2010-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Myers MG, Cowley MA, Munzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol. 2008;70:537–56. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- 28.Morris DL, Rui L. Recent advances in understanding leptin signaling and leptin resistance. Am J Physiol Endocrinol Metab. 2009;297(6):E1247–59. doi: 10.1152/ajpendo.00274.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Covasa M. Deficits in gastrointestinal responses controlling food intake and body weight. Am J Physiol Regul Integr Comp Physiol. 2010;299(6):R1423–39. doi: 10.1152/ajpregu.00126.2010. [DOI] [PubMed] [Google Scholar]
- 30.Paulino G, et al. Adaptation of lipid-induced satiation is not dependent on caloric density in rats. Physiol Behav. 2008;93(4–5):930–6. doi: 10.1016/j.physbeh.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paulino G, et al. Increased expression of receptors for orexigenic factors in nodose ganglion of diet-induced obese rats. Am J Physiol Endocrinol Metab. 2009;296(4):E898–903. doi: 10.1152/ajpendo.90796.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levin BE, et al. Selective breeding for diet-induced obesity and resistance in Sprague-Dawley rats. Am J Physiol. 1997;273(2 Pt 2):R725–30. doi: 10.1152/ajpregu.1997.273.2.R725. [DOI] [PubMed] [Google Scholar]
- 33.Tilg H, Moschen AR, Kaser A. Obesity and the microbiota. Gastroenterology. 2009;136(5):1476–83. doi: 10.1053/j.gastro.2009.03.030. [DOI] [PubMed] [Google Scholar]
- 34.Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol. 2009;587(Pt 17):4153–8. doi: 10.1113/jphysiol.2009.174136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ley RE. Obesity and the human microbiome. Curr Opin Gastroenterol. 2010;26(1):5–11. doi: 10.1097/MOG.0b013e328333d751. [DOI] [PubMed] [Google Scholar]
- 36.Ley RE, et al. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102(31):11070–5. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ley RE, et al. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–3. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 38.Turnbaugh PJ, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–31. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 39.Turnbaugh PJ, et al. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3(4):213–23. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Backhed F, et al. Mechanisms underlying the resistance to diet-induced obesity in germ–free mice. Proc Natl Acad Sci U S A. 2007;104(3):979–84. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Membrez M, et al. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. FASEB J. 2008;22(7):2416–26. doi: 10.1096/fj.07-102723. [DOI] [PubMed] [Google Scholar]
- 42.Hildebrandt MA, et al. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology. 2009;137(5):1716–24. e1–2. doi: 10.1053/j.gastro.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de La Serre CB, et al. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2010;299(2):G440–G448. doi: 10.1152/ajpgi.00098.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–7. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 45.Erridge C, et al. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr. 2007;86(5):1286–92. doi: 10.1093/ajcn/86.5.1286. [DOI] [PubMed] [Google Scholar]
- 46.Cani PD, Delzenne NM. The role of the gut microbiota in energy metabolism and metabolic disease. Curr Pharm Des. 2009;15(13):1546–58. doi: 10.2174/138161209788168164. [DOI] [PubMed] [Google Scholar]
- 47.Cani PD, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–72. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 48.Tsukumo DM, et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56(8):1986–98. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- 49.Davis JE, et al. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity (Silver Spring) 2008;16(6):1248–55. doi: 10.1038/oby.2008.210. [DOI] [PubMed] [Google Scholar]
- 50.Cani PD, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–81. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 51.Ding S, et al. High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One. 2010;5(8):e12191. doi: 10.1371/journal.pone.0012191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brown R, Imran SA, Wilkinson M. Lipopolysaccharide (LPS) stimulates adipokine and socs3 gene expression in mouse brain and pituitary gland in vivo, and in N-1 hypothalamic neurons in vitro. J Neuroimmunol. 2009;209(1–2):96–103. doi: 10.1016/j.jneuroim.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 53.Hosoi T, et al. Novel pathway for LPS-induced afferent vagus nerve activation: possible role of nodose ganglion. Auton Neurosci. 2005;120(1–2):104–7. doi: 10.1016/j.autneu.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 54.Palazzo M, et al. Activation of enteroendocrine cells via TLRs induces hormone, chemokine, and defensin secretion. J Immunol. 2007;178(7):4296–303. doi: 10.4049/jimmunol.178.7.4296. [DOI] [PubMed] [Google Scholar]