

Abstract
Protein Kinase D (PKD) is a subfamily of serine/threonine specific family of kinases, comprised of PKD1, PKD2 and PKD3 (PKCμ, PKD2 and PKCν in humans). It is known that PKCs activate PKD, but the relative expression of isoforms of PKD or the specific PKC isoform/s responsible for its activation in platelets is not known. This study is aimed at investigating the pathway involved in activation of PKD in platelets. We show that PKD2 is the major isoform of PKD that is expressed in human as well as murine platelets but not PKD1 or PKD3. PKD2 activation induced by AYPGKF was abolished with a Gq inhibitor YM-254890, but was not affected by Y-27632, a RhoA/p160ROCK inhibitor, indicating that PKD2 activation is Gq-, but not G12/13-mediated Rho-kinase dependent. Calcium-mediated signals are also required for activation of PKD2 as dimethyl BAPTA inhibited its phosphorylation. GF109203X, a pan PKC inhibitor abolished PKD2 phosphorylation but Go6976, a classical PKC inhibitor had no effect suggesting that novel PKC isoforms are involved in PKD2 activation. Importantly, Rottlerin, a non-selective PKCδ inhibitor, inhibited AYPGKF-induced PKD2 activation in human platelets. Similarly, AYPGKF- and Convulxin-induced PKD2 phosphorylation was dramatically inhibited in PKCδ-deficient platelets, but not in PKCθ– or PKCε–deficient murine platelets compared to that of wild type platelets. Hence, we conclude that PKD2 is a common signaling target downstream of various agonist receptors in platelets and Gq-mediated signals along with calcium and novel PKC isoforms, in particular, PKCδ activate PKD2 in platelets.
Keywords: Protein kinase C, Calcium, Protein kinase D, Protease activated receptor, Platelet, Gq
1. INTRODUCTION
Platelets maintain hemostasis preventing excessive bleeding or formation of unusual thrombus during injury [1]. Upon injury, collagen at the damaged site is exposed leading to initial tethering of platelets by GPIb-IX-V receptors and adhesion and activation through GPVI and α2β1 receptors [2, 3]. This step results in the recruitment of more platelets to the site of injury through secondary mediators released from activated platelets. Recruited platelets bind each other through αIIbβ3 receptors, change their shape, aggregate, undergo cytoskeletal rearrangements and secrete granular contents (ADP, serotonin) that can further activate platelets in a feedback fashion [4].
Platelets express G Protein-coupled receptors such as protease-activated receptors (PARs) and non-GPCRs such as GPVI [5] on their surface. Thrombin activates platelets through PARs and collagen activates platelets through GPVI receptors [2, 3]. PARs 1 and 4 are expressed on human platelets and PARs 2, 3 and 4 are expressed on murine platelets [6, 7]. PARs are activated by proteolytically unmasking the receptor’s N-terminal tethered ligand or they can also be activated in vitro by using peptides (PAR4 with AYPGKF and PAR1 with SFLLRN) that are similar to the revealed tethered ligands [8]. PARs couple to Gq and G12/13 pathways [9].
Platelet activation through either PARs or GPVI receptors involves activation of Protein Kinase C (PKC) isoforms, which are in turn involved in regulating platelet functional responses such as aggregation, secretion and thrombus formation [10]. PKCs are classified into three sub families [11] based on their distinct N-terminal regulatory domains: 1) Classical or Conventional class of PKC isoforms - PKCα, βI, βII and γ requiring calcium and DAG for their activation; 2) Novel class of PKC isoforms - PKC δ, θ, ε and η requiring only DAG for their activation; 3) Atypical class of PKC isoforms - PKC ι, λ and ζ requiring phospholipids for their activation [12]. Six isoforms of PKCs, PKCα, β, δ, θ, η, ε, and ζ were reported to be present in platelets [13, 14].
MARCKS [15], CDCrel-1 [16], Pleckstrin, Adducin [17] and Protein Kinase D (PKD) [18] are some of the substrates of PKC known so far but the precise role played by each substrate and the specific isoform/isoforms of PKC involved in the activation of these substrates are not known. PKD belongs to the serine/threonine family of kinases and can be activated directly by DAG or indirectly by PKC isoforms. Earlier they were considered as a part of the PKC family of kinases [19] but owing to their highest sequence homology to the catalytic domain of myosin light chain kinase and calcium-calmodulin kinase, they were given a separate status [20]. The PKD family comprises of PKD1 [21], PKD2 [22] and PKD3 [23] (PKCμ, PKD2 and PKCν in humans). Each isotype has a regulatory domain and a kinase domain. The regulatory domain inhibits the kinase domain until the enzyme is activated by phosphorylation [24] of two serine residues, 744 and 748 located within the kinase domain [25] upon which serine 916 in the C-terminal domain gets auto-phosphorylated [26]. The phosphorylation of PKD on serine residues 744/748 is taken as a read out for PKD activation [27]. However, Rybin et al [28] showed that phosphorylation on Ser 916 of PKD does not necessarily correspond to its activation and that this phosphorylation could be detected without any increase in its catalytic activity towards substrates. Structurally PKD has a N-terminal domain (rich in alanine and proline) followed by two cysteine-rich zinc finger domains, an acidic domain (rich in acidic amino acids), Pleckstrin homology domain and C-terminal catalytic domain. PKDs, unlike PKC isoforms do not have a C2 domain and the pseudo substrate site [21]. PKD is a protein of 918 amino acids with a molecular weight of around 115kDa. It is distributed primarily in the cytosol, and to some extent, in the nucleus, Golgi and mitochondria. PKD is recruited to the membrane, gets phosphorylated and then shuttles back to various sub cellular compartments to carry out its functions such as proliferation, differentiation, motility, invasion, protein transport, membrane trafficking, apoptosis and immune responses in B- and T-cells [29].
Although PKD has been shown to be present and phosphorylated in platelets [18], not much is known about the specific isoforms of PKD expressed in platelets or the specific PKC isoforms that activate PKD. In our current study, we have investigated the pathway involved in the phosphorylation of PKD in platelets downstream of PAR4. We found that PKD2, but not PKD1 or PKD3, is the predominant isoform of PKD expressed in human and murine platelets. We also found that PKD2 can be activated downstream of PAR4 through Gq-mediated signaling involving increases in intracellular calcium. We also demonstrate for the first time that novel PKC isoform PKCδ is required for the activation of PKD2 in platelets and hence PKD2 is the specific substrate for PKCδ but not PKCθ or PKCε.
2. MATERIALS AND METHODS
Approval for this study was obtained from the Institutional Review Board of Temple University (Philadelphia, PA).
2.1 Materials
Apyrase (type VII), bovine serum albumin (fraction V), acetylsalicylic acid and GR144053 were obtained from Sigma (St Louis, MO). PGE1, ADP, Go6976, LY294002, PP2 (4-amino-5- (4-chlorophenyl)-7-(t-butyl) pyrazole [3,4-d] pyrimidine) and Rottlerin were purchased from Enzo Life Sciences (Plymouth Meeting, PA). AYPGKF and SFLLRN were custom synthesized at Invitrogen (Carlsbad, CA). Convulxin was purchased from Centerchem, Inc. (Norwalk, CT). YM-254890 was a generous gift from Yamanouchi Pharmaceutical (Ibaraki, Japan). 5,5′-dimethyl-bis- (o-aminophenoxy) ethane-N, N, N′, N′-tetra acetic acid (dimethyl-BAPTA) was obtained from Molecular probes (Eugene, OR). Bisindolylmaleimide I (GF 109203X) and Y-27632 were from Calbiochem (San Diego, CA). Total PKD, Phospho Ser744/748 (recognizes equivalent serines on PKD2) and β-Actin antibodies were obtained from Cell Signaling Technologies (Beverly, MA). Total PKD (D-20), PKD3 (L-20) and Horseradish peroxidase-labeled secondary antibody were from Santa Cruz. Total PKD2 antibody was from Abcam. Chemiluminescent HRP-substrate was from Millipore (Billerica, MA). All the other reagents were of reagent grade, and de-ionized water was used throughout.
2.2 Animals
PKCδ−/− (C57BL/6 strain) mice were a generous gift from Dr Keiko Nakayama (Division of Developmental Genetics, Tohoku University Graduate School of Medicine). PKCθ −/− mice were obtained from The Jackson Laboratory (Bar Harbor, ME). PKCε −/− mice were a kind gift from Dr. Robert Messing (Gallo Center, San Francisco, CA). Wild type littermates were used as controls. The mice were used for physiologic measurements using the protocol approved by the Institutional Animal Care and Use Committee.
2.3 Human platelet preparation
All experiments using human subjects were performed in accordance with the Declaration of Helsinki. Whole blood was drawn from healthy human volunteers into tubes containing one-sixth volume of ACD (2.5 g of sodium citrate, 1.5 g of citric acid, 2 g of glucose in 100 ml of deionized water). Blood was centrifuged (Eppendorf 5810R centrifuge) at 230g for 20 min at room temperature to obtain platelet-rich plasma (PRP). PRP was incubated with 1mM aspirin for 30 min at 37°C. The PRP was then centrifuged for 10 min at 980g at room temperature to pellet the platelets. Platelets were resuspended in Tyrode’s buffer pH 7.4 (138mM NaCl, 2.7mM KCl, 1mM MgCl2, 3mM NaH2PO4, 5mM glucose and 10mM HEPES) containing 0.3U/mL apyrase. Platelets were counted using the Hemavet (Drew Scientific Inc., Dallas, TX) and concentration of cells was adjusted to 2 × 108 platelets/ml. Platelet samples used in all the experiments were treated with aspirin and apyrase to inhibit the feedback effects of thromboxane and ADP.
2.4 Murine platelet preparation
Blood was collected from ketamine-anesthetized mice by cardiac puncture into syringes containing 3.8% sodium citrate as anticoagulant. The whole blood was centrifuged (IEC Micromax Centrifuge, International Equipment Components, CA) at 100g for 10 minutes to isolate the PRP. Prostaglandin E1 (1 μM) was added to PRP. Platelets were centrifuged at 400g for 10 minutes, and the pellet was resuspended in Tyrode’s buffer (pH 7.4) containing 0.3U/mL apyrase.
2.5 Western Immunoblotting
Platelets were stimulated with agonists and the reaction was stopped by the addition of 6M perchloric acid. Samples were kept on ice and then centrifuged. Sample buffer (2M Tris, 10% by volume glycerol, 10% SDS, 0.5% bromophenol blue, 1mM Dithiothreitol (DTT)) was added to the pellet and boiled for 5 minutes. Proteins were separated by 8% SDS–polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were blocked by incubation with Tris-buffered saline (TBST: 20mM Tris, 140mM NaCl) containing 3% (wt/vol) bovine serum albumin (BSA) for 1 hour at room temperature. Membranes were incubated overnight at 4°C with the primary antibody in loading buffer (TBS with 3% BSA) with gentle agitation. PKD1, 2 or 3 isoform-selective antibodies were used at 1:500 dilutions and phospho-specific PKD antibody was used at 1:500 dilution. After three washes for 5 minutes each with Tris-buffered saline Tween-20 (TBS-T), the membranes were probed with HRP-conjugated rabbit/mouse secondary antibodies dissolved in TBS-T for 1 hour at room temperature. After additional washing steps, membranes were then incubated with the chemiluminescent HRP-substrate and immunoreactivity was detected using a Fuji Film Luminescent Image Analyzer (LAS-3000 CH, Tokyo, Japan).
2.6 Statistical analysis
Western blot data were compiled from at least three independent experimental results. The results were quantified and expressed as means ± SD. Statistical significance was tested by Student’s t test or ANOVA. P value < 0.05 was considered statistically significant and non-significance is indicated by NS.
3. RESULTS
3.1 PKD2 is the major isoform of PKD expressed in both human and murine platelets
To determine and confirm the presence of PKD in platelets, we used total antibodies against PKD1, PKD2 and PKD3. Washed platelet lysates were prepared from both human and murine platelets, subjected to western blotting and probed for total PKD1, PKD2 and PKD3. 293T cell lysate was used as control. Figure 1 shows that PKD2 is expressed in human and murine platelets, but not PKD1 or PKD3.
Fig 1. Expression profile of PKD isoforms in platelets.
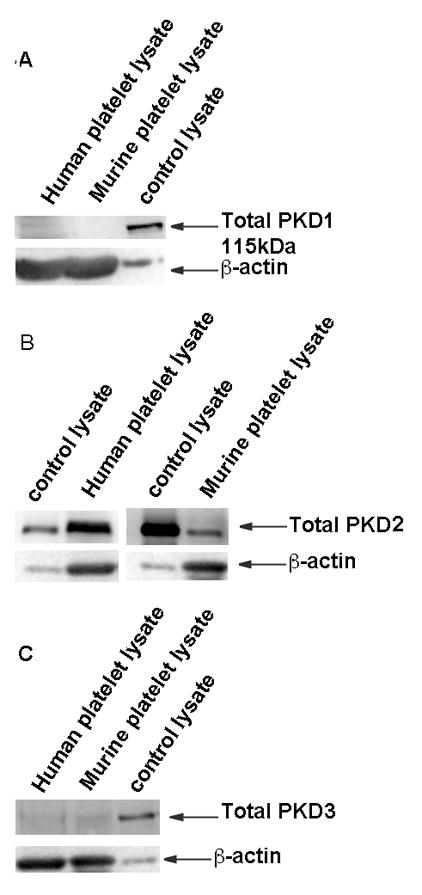
Washed platelets (2×108 cells/ml), prepared from human and murine blood, were lysed and proteins were precipitated with 6M perchloric acid. Samples were subjected to SDS-PAGE and analyzed for expression of PKD 1, 2, or 3 by western blotting by using total PKD antibodies. βActin was used as a loading control. The data are representative of at least three independent experiments. The samples from platelets were overloaded to detect PKD isoforms, relative to control, as reflected in the differences in β-actin bands.
3.2 PKD2 can be activated by either GPVI or PAR4 receptors
It was shown previously that thrombin and convulxin could activate PKD in platelets[18]. To determine whether individual PAR receptors could activate PKD2, we used peptides that can selectively stimulate PAR1 or PAR4. Washed human and murine platelets were stimulated with GPCR agonists SFLLRN (PAR1 agonist), AYPGKF (PAR4 agonist), ADP that acts on P2Y1 and P2Y12 receptors, a GPVI agonist, Convulxin. As shown, PKD2 was phosphorylated by SFLLRN, AYPGKF, and Convulxin but not by ADP stimulation (Figure 2A). Similarly PKD2 was activated downstream of PAR4 and GPVI receptors but not with ADP in murine platelets (Figure 2B). A time course experiment was also performed with 20 μM ADP in both human and murine platelets to investigate whether activation of PKD2 downstream of ADP is time-dependent. As shown in Figure 2C, PKD2 phosphorylation was not observed at any time point (up to 3 min) in either human or murine platelets indicating that PKD2 is not activated downstream of ADP. Stafford et al., [18] showed activation of PKD in platelets, however we were unable to detect phosphorylation of PKD2 [either phospho serine 744/748 or phospho serine 916 (Fig. 2C&D)] in platelets using the same experimental protocol.
Fig 2. PKD2 can be activated by either GPVI or GPCR receptors.

(A) Washed aspirin-treated human platelets (2×108 cells/ml) and (B) washed platelets from mice (2×108 cells/ml) were stimulated with 500 μM AYPGKF, 10 μM SFLLRN, 20 μM ADP, or 500 ng/ml Convulxin at 37°C under stirring conditions. The reaction was stopped after 1 minute by addition of 6 M perchloric acid. Time course studies were performed with 20 μM ADP in both (C) human and (D) murine samples and the reactions were stopped after respective time points. Samples were subjected to SDS-PAGE and analyzed for ser744/748 and ser916 phosphorylations of PKD2 by western blotting by using phospho-specific antibodies. β-Actin was used to ensure equal protein concentrations in all lanes. The data are representative of at least three independent experiments. US = unstimulated.
3.3 Concentration-response and kinetics of PKD2 phosphorylation
Concentration-response studies were performed to see the dependence of agonist on PKD2 phosphorylation. Washed platelets were stimulated with increasing concentrations of AYPGKF. As shown in Figure 3A and 3B, PKD2 phosphorylation increased in a concentration-dependent manner in both human and murine platelets. Next, we determined the kinetics of PKD2 phosphorylation with AYPGKF. Time-course studies showed that PKD2 was phosphorylated as early as 20 seconds and maximally phosphorylated at 1 minute in human platelets (Figure 3C). In murine platelets PKD2 was phosphorylated as early as 10 seconds and was maximally phosphorylated at 1 minute (Figure 3D). These studies indicate that PKD2 is activated at early time points during platelet activation.
Fig 3. Concentration-response and kinetics of PKD2 phosphorylation.

Washed platelets (2×108 cells/ml) from both (A) human and (B) mice were stimulated with increasing concentrations of AYPGKF at 37°C under stirring conditions. The reaction was stopped after 1 minute by addition of 6 M perchloric acid. Washed platelets from (C) human and (D) mice were stimulated with 500 μM AYPGKF at 37°C under stirring conditions. The reaction was stopped after different time points by addition of 6M perchloric acid. Samples were subjected to SDS-PAGE and analyzed for ser744/748 phosphorylations of PKD2 by western blotting by using phospho specific antibodies. β-Actin was used as a loading control. The data are representative of at least three independent experiments.
3.4 PKD2 phosphorylation is Gq-mediated downstream of PAR4 in platelets
We next sought to find out the signaling pathway involved in activating PKD2 downstream of PAR4. As PAR4 is coupled to both Gq and G12/13 pathways [9], we investigated the specific G protein pathway contributing to PKD2 phosphorylation in platelets. G12/13 activates Rho-kinase through which its actions are mediated [30]. We used a Rho-ROCK kinase inhibitor; Y-27632 and a Gq inhibitor, YM-254890 to evaluate the role of Gq and G12/13 pathways in PKD2 phosphorylation. Washed human platelets were pre-incubated with YM254890 or Y27632 followed by stimulation with AYPGKF under stirring conditions. As shown in Figure 4A, Rho-kinase inhibitor did not have any effect on the phosphorylation of PKD2 whereas Gq inhibitor abolished the phosphorylation of PKD2 indicating that PAR4-mediated PKD2 phosphorylation is Gq-dependent but not Rho-kinase dependent.
Fig 4. PKD2 phosphorylation is Gq-mediated downstream of PAR4 in platelets and requires intracellular increase in calcium.

Washed aspirin-treated human platelets (2×108 cells/ml) were incubated with (A) DMSO (vehicle control) or 50 nM YM-254890 or 10 μM Y-27632 for 5 minutes at 37°C and (B) with DMSO (vehicle control) or 10 μM Dimethyl BAPTA for 5 minutes at 37°C and stimulated with 500 μM AYPGKF at 37°C under stirring conditions. The reaction was stopped after 1 minute by addition of 6 M perchloric acid. Samples were subjected to SDS-PAGE and analyzed for ser744/748 phosphorylations of PKD2 by western blotting by using phospho specific antibodies. β-Actin was used as a loading control. The data are representative of at least three independent experiments. (C) Data obtained from three different sets of experiments were quantified and expressed as mean ± SD. The phosphorylation induced by AYPGKF stimulated samples without any inhibitors was considered 100%.
3.5 PKD2 phosphorylation requires intracellular calcium increases
We next sought to find out the signaling molecules involved in the pathway of activation of PKD2. Downstream of Gq, Phospholipase Cβ is activated and cleaves PIP2 to IP3 and DAG. IP3 mobilizes calcium from intracellular reservoirs to the cytosol, which is necessary for subsequent activation of platelets [31]. Therefore, we investigated the requirement of calcium for PKD2 activation. Dimethyl BAPTA, a chelator of divalent ions was used to chelate calcium. Dimethyl BAPTA significantly inhibited the phosphorylation of PKD2 induced by AYPGKF (Figure 4B), suggesting a role for calcium in mediating PKD2 phosphorylation downstream of PAR4. Figure 4C shows the quantification of blots.
3.6 PKD2 phosphorylation does not require outside-in signaling
Inside-out signaling from agonist receptors results in the activation of fibrinogen receptor, αIIbβ3 integrin. Once the integrin is activated, it binds to fibrinogen and leads to activation of various signaling molecules downstream in a process termed outside-in signaling [32]. We investigated the requirement of outside-in signaling in phosphorylation of PKD2. Washed human platelets, pre-incubated with fibrinogen receptor antagonist GR144053, were stimulated with AYPGKF under stirring conditions. As shown in Figure 5A, inhibition of outside-in signaling had no effect on phosphorylation of PKD2 indicating that outside-in signaling downstream of PAR4 is not required to activate PKD2. These results are consistent with the time-course of PKD2 activation wherein integrin activation would not have occurred at the early time points of platelet stimulation.
Fig 5. Signaling pathways involved in PKD2 phosphorylation.

Washed and aspirin-treated human platelets (2×108 cells/ml) were incubated with (A) DMSO (vehicle control) or 200 nM GR144053 and (B) with DMSO (vehicle control) or 10 μM PP3 or 10 μM PP2 or 25 μM LY294002 for 5 minutes at 37°C or (D and E) with 100nM Go6976 for 10 minutes or 5μM GF109203X for 5 minutes in both human and murine platelets, followed by stimulation with 500 μM AYPGKF at 37°C under stirring conditions. The reaction was stopped after 1 minute by addition of 6M perchloric acid. The samples were analyzed for ser744/748 phosphorylation on PKD2 by western blotting by using phospho specific antibodies. β-Actin was used as a loading control. The data are representative of at least three independent experiments. (C) Data obtained from three different sets of experiments were quantified and expressed as mean ± SD. The phosphorylation induced by AYPGKF stimulated samples without any inhibitors was considered 100%.
3.7 Src-family kinases (SFK) and PI-3Kinases are not involved in PKD2 phosphorylation downstream of PAR4
As shown by Stafford et al [18], PKD2 has been shown to be phosphorylated downstream of GPVI in a SFK and PI-3 kinase-dependent manner. PAR-mediated signaling can cause Gi stimulation through P2Y12 receptor activation by secreted ADP, and subsequently activate PI-3kinases [33, 34]. To investigate the requirement of these signaling molecules in activation of PKD2 downstream of PAR4, we used a pan-SFK inhibitor (PP2), inactive PP2 analogue PP3, or a PI-3Kinase inhibitor (LY-294002), followed by stimulation with AYPGKF. As shown in Figure 5B, PKD2 phosphorylation was unaffected with SFK inhibitor or PI-3Kinase inhibitor suggesting that PKD2 phosphorylation is not dependent on SFK or PI-3Kinases downstream of PAR4. Figure 5C shows the quantification of blots.
Earlier work suggested that PKC isoforms are involved in phosphorylating PKD [18, 35]. To confirm the requirement of PKC isoforms, human and murine platelets were treated with GF109203X, a pan PKC inhibitor or Go6976, a classical PKC inhibitor [36] followed by stimulation with AYPGKF. As shown in Figure 5D and Figure 5E, GF109203X abolished AYPGKF-induced PKD2 phosphorylation but Go6976 had no effect on PKD2 phosphorylation in both human and murine platelets, suggesting that novel class of PKC isoforms are probably required for phosphorylation of PKD2 downstream of PAR4.
3.8 Novel PKC isoform δ but not θ or ε mediates PKD2 phosphorylation in platelets
It has been shown that novel PKC isoforms PKCε, PKCδ and PKCθ are associated with and are involved in the activation of PKD2 in various cell systems [37, 38]. To investigate the specific novel isoform(s) of PKC involved in phosphorylation of PKD2 in platelets, we used murine platelets lacking specific PKC isoforms. Washed platelets from wild type littermates and PKCδ–, θ– and ε– deficient mice were prepared followed by stimulation with 500 μM AYPGKF or 500 ng/ml Convulxin. AYPGKF- or Convulxin-induced phosphorylation of PKD2 was dramatically reduced in PKCδ–deficient platelets (Figure 6A & B). The residual phosphorylation observed in agonist-stimulated PKCδ knockout murine platelets was abolished with PKC inhibitor GF 109203X (Figure 6A & B), but was unaffected with Go6976 (data not shown). Total PKD2 protein levels were checked in wild type and PKCδ-null murine platelets to confirm that the reduced phosphorylation in PKCδ-deficient platelets is because of the signaling events from PKCδ but not due to the decreased ability of PKCδ-deficient platelets to make total PKD2. As shown in Figure 6C, there is no change in total PKD2 levels in both wild type and PKCδ-deficient platelets. PKD1 and PKD3 could not be detected in wild type or PKCδ-null murine platelets (data not shown). The phosphorylation on PKD2 was unaffected in PKCθ- (Figure 6D & E) and PKCε- (figure 6F & G) deficient platelets, downstream of PAR4 and GPVI receptors, compared to platelets from wild type littermates, indicating that PKC isoform δ plays a major role in mediating the activation of PKD2 in platelets.
Fig 6. Novel PKC isoform δ but not θ or ε mediates PKD2 phosphorylation downstream of PAR4.

Washed platelets (2×108 cells/ml) from wild type or PKCδ Knockout mice, were stimulated with (A) 500μM AYPGKF with or without 5 μM GF 109203X or (B) with 500ng/ml Convulxin with or without 5 μM GFX at 37°C under stirring conditions. (C) Samples from wild type and PKCδ-deficient murine platelets were probed for total PKD2. PKCθ kinockout (D and E) and PKCε knockout murine platelets (F and G) were stimulated with 500μM AYPGKF or 500 ng/ml Convulxin. The samples were analyzed for ser744/748 phosphorylation on PKD2 by western blotting by using phospho-specific antibodies. The knockout murine platelets of individual PKCs were confirmed by checking for total PKC levels in respective knockouts. β-Actin was used as a loading control. The data are representative of at least three independent experiments.
In order to evaluate whether PKCδ phosphorylates PKD2 in human platelets, we used a non-selective PKCδ inhibitor, Rottlerin in human platelets. A dose response study (Figure 7A) in PKCδ deficient murine platelets showed that PKD2 phosphorylation was abolished with 150 μM and 250 μM concentrations of AYPGKF and was inhibited with 500 μM AYPGKF. Similarly, human platelets pre-incubated with Rottlerin showed reduced phosphorylation of PKD2 when stimulated with 150 μM and 250 μM AYPGKF compared to stimulation with 500 μM AYPGKF (Figure 7B). Hence we conclude that PKCδ is the major isoform of PKC that mediates activation of PKD2 in both human and murine platelets at low concentrations of agonist. Other PKC isoforms like PKCη also might contribute to PKD2 phosphorylation at higher agonist concentrations. The results are summarized in Figure 8.
Fig 7. PKCδ mediates phosphorylation of PKD2 in human and murine platelets.
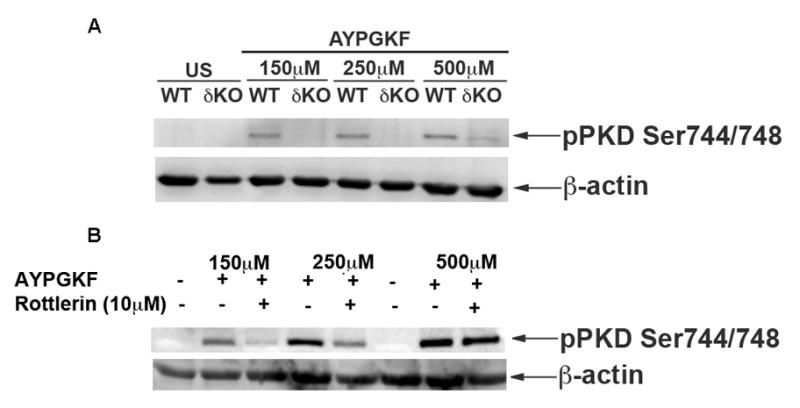
(A)Washed murine platelets from PKCδ-deficient mice were stimulated with 150, 250 and 500μM of AYPGKF and were subjected to western blotting. (B) Washed human platelets were pre-incubated with 10μM Rottlerin for 15 minutes and were stimulated with 150, 250 and 500μM of AYPGKF and were subjected to western blotting. The blots were probed for phospho ser744/748 antibody. β-actin was used as a loading control.
Fig 8. Overview of PKD2 activation.

PAR4 (Protease activated receptor4) is coupled to both Gq and G12/13. The Gq pathway involves phospholipase C-β activation that converts Phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol triphosphate (IP3) and diacyl glycerol (DAG), which subsequently cause intraplatelet calcium mobilization and PKC activation respectively. The G12/13 pathway involves Rho/Rho kinase activation and actin remodeling causing platelet shape change. PKD2 phosphorylation on serines 744/748 downstream of PAR4 in platelets requires calcium and/or PKCs particularly novel class of PKC isoform, PKCδ.
3. DISCUSSION
Different isoforms of Protein kinase C have been implicated in regulating different platelet functional responses [10, 14, 39–42]. As individual PKC isoforms have isoform- specific functions, it is important to identify the specific substrates which could help us understand the functions mediated by individual PKCs. Identifying PKC specific substrates also help us design and evaluate isoform specific inhibitors which could be used to treat various cardiovascular and thrombosis related disorders. PKD is one such substrate activated downstream of PKC in platelets but the relative expression of isoforms of PKD expressed and the specific PKC isoform involved in PKD activation is not known. In this study, we show that PKD2 is the major isoform of PKD expressed in human and murine platelets. We also show that PKD2 is activated by a PAR4 agonist, but not by ADP. It has been shown previously by Stafford et al [18] previously showed that ADP can activate PKD, but we did not observe PKD activation with ADP, despite using the same experimental conditions. Inhibitors for signaling intermediates downstream of G12/13 and Gi (Rho-kinase and PI3Kinase respectively) had no effects on PAR4-induced PKD2 activation. In contrast, Gq inhibitors blocked PAR4-mediated PKD2 activation. Hence we conclude that Gq, but not G12/13 or Gi mediates PKD2 activation downstream of PAR4. Calcium plays an important role in mediating platelet functional responses downstream of Gq and is also required for activation of PKD2 as inhibition of calcium inhibited PKD2 phosphorylation. The inhibitors used in the study were tested for their efficacy on their intended targets as shown previously in our lab [43–47] and were found effective (data not shown).
Three alternative pathways can activate PKD: 1) caspases cleave PKD between the acidic domain and activation domain and activate it upon genotoxic damage; 2.) Gβγ subunits in the Golgi can activate PKD by binding to the Pleckstrin homology domain, and 3.) PLC-DAG-PKC phosphorylation upon receptor stimulation can activate PKD [48]. PKD activity has been shown to be modulated by various PKCs in different cell types [49]. PKD has been shown to be associated with PKCη in COS-7 cells [50] and PKCη–activated PKD can modulate ERK and JNK signaling pathways [35]. It has been shown that PKCδ and PKCε associate with PKD in smooth muscle cells but PKD activity is regulated only by PKCδ [38]. However, PKCε has been shown to modulate the activity of PKD in HEK293 cells [51]. Although PKCθ has not been shown to be associated with PKD, it regulates PKD activity in COS-7 cells and lymphocytes [37]. Recent studies with PKCε showed that although this isoform is expressed in murine platelets, it had minimal functional role in platelets [52]. PKCθ null murine platelets show reduced PAR-mediated platelet functional responses [39, 53], but our studies show that PKD2 phosphorylation is not affected in PKCθ-null murine platelets. Hence the positive regulatory role of PKCθ in PAR-mediated functional responses may not be occurring through PKD2.
Our results show that PAR4-mediated PKD2 activation requires PKCδ but not PKCθ or PKCε in platelets. Total PKD2 levels showed no change in wild type and PKCδ-deficient murine platelets, indicating that the decrease in activation is not due to a decrease in total levels of protein. Furthermore, using Go6976 in PKCδ-deficient murine platelets showed no additional inhibition of PKD2 phosphorylation (data not shown), indicating that classical PKC isoforms play little to no role in phosphorylating PKD2. Furthermore, ADP, which does not activate PKCδ [14] also did not cause phosphorylation of PKD2 in platelets. Similarly, Rottlerin, a non-selective PKCδ inhibitor, inhibited PKD2 phosphorylation in human platelets. Hence, we suggest that PKCδ plays a major role in mediating the phosphorylation of PKD2 in platelets downstream of PAR4. Our studies do not agree with the recent work of Konopatskaya et al [54], who concluded that PKD2 phosphorylation is mediated by classical PKC isoforms but not by novel PKC isoforms. Although they used PKCδ null murine platelets in their study, they did not evaluate the phosphorylation status of Ser744/748 residues in these murine platelets, which is well correlated with its activity. However, their functional data with Ser744/748 knock-in mice is consistent with our predicted functional role of PKCδ/PKD2 nexus.
In platelets, however, there is still some PKD2 phosphorylation seen with high concentrations of agonist (AYPGKF) indicating that the residual phosphorylation could be mediated through other PKC isoforms either through compensatory mechanisms or through PKCη. Unfortunately there are no PKC isoform- specific inhibitors to test this hypothesis. Interestingly, PKCδ is a novel class of PKC isoform that does not require calcium for its activation, but PKD2 seems to require calcium and PKCδ for its activation. PKC- and calcium-mediated pathways activate PKD2 independently. In addition, Src family kinases are shown to phosphorylate PKCδ on tyrosine 311 and hence are important in TxA2 generation. Inhibition of Src family kinases (Figure 5B) did not have any effect on PKD2 phosphorylation indicating that PKCδ tyrosine phosphorylation does not have a role in mediating PKD2 phosphorylation. Hence phosphorylation on threonine 505, an activation marker for PKCδ, probably is sufficient to cause PKD2 activation.
As specific PKD2 inhibitors or mice deficient in PKD isoforms are unavailable, evaluating the role of PKD2 in platelet functional responses is difficult. PKCδ regulates platelet responses such as aggregation and dense granule secretion [10]. This could be mediated by phosphorylating proteins involved in secretion [55]. As PKD2 is activated specifically downstream of PKCδ, the actions mediated by PKCδ could be attributed to the actions mediated by PKD2, but further experimentation is required to establish this.
In summary, we conclude that PKD2 is the major isoform of PKD expressed in both human and murine platelets and can be activated downstream of PAR4 in a Gq-dependent manner. Novel class PKC isoforms are required for phosphorylation of PKD2, and specifically PKC isoform δ is required for its activation. Finally we predict that some of the actions mediated by PKCδ including platelet dense granule secretion, might be mediated by PKD2.
Acknowledgments
We thank Dr Keiko Nakayama (Division of Developmental Genetics, Tohoku University Graduate School of Medicine) for the PKCδ-null mice and Dr. Robert Messing (Gallo Center, San Francisco, CA) for the PKCε-null mice. We also thank Ms. Monica Wright for maintenance, breeding and genotyping the mice. Research Grants HL60683, HL81322, and HL 93231 from the National Institutes of Health supported this work.
ABBREVIATIONS
- ADP
Adenosine diphosphate
- PKC
Protein kinase C
- PKD2
Protein kinase D
- PAR-4
Protease activated receptor-4
- P2Y12
platelet ADP receptor coupled to inhibition of adenylyl cyclase
- DAG
Diacyl glycerol
- MARCKS
Myristoylated alanine-rich C-kinase substrate
- CDCrel1
cell division cycle related-1
- PI 3-kinase
Phosphoinositide 3-kinase
- SFK
Src family kinases
- GPVI
Glycoprotein
- PIP2
Phosphotidylinositol 4,5-bisphosphate
- BAPTA
1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Packham MA. Role of platelets in thrombosis and hemostasis. Can J Physiol Pharmacol. 1994;72:278–84. doi: 10.1139/y94-043. [DOI] [PubMed] [Google Scholar]
- 2.Watson SP, Gibbins J. Collagen receptor signalling in platelets: extending the role of the ITAM. Immunol Today. 1998;19:260–4. doi: 10.1016/s0167-5699(98)01267-5. [DOI] [PubMed] [Google Scholar]
- 3.Watson S, Berlanga O, Best D, Frampton J. Update on collagen receptor interactions in platelets: is the two-state model still valid? Platelets. 2000;11:252–8. doi: 10.1080/09537100050129260. [DOI] [PubMed] [Google Scholar]
- 4.Brass LF. More pieces of the platelet activation puzzle slide into place. J Clin Invest. 1999;104:1663–5. doi: 10.1172/JCI8944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clemetson JM, Polgar J, Magnenat E, Wells TN, Clemetson KJ. The platelet collagen receptor glycoprotein VI is a member of the immunoglobulin superfamily closely related to FcalphaR and the natural killer receptors. J Biol Chem. 1999;274:29019–24. doi: 10.1074/jbc.274.41.29019. [DOI] [PubMed] [Google Scholar]
- 6.Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci U S A. 1994;91:9208–12. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, et al. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–4. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- 8.Hollenberg MD. Protease-activated receptors: PAR4 and counting: how long is the course? Trends Pharmacol Sci. 1999;20:271–3. doi: 10.1016/s0165-6147(99)01333-4. [DOI] [PubMed] [Google Scholar]
- 9.Offermanns S, Laugwitz KL, Spicher K, Schultz G. G proteins of the G12 family are activated via thromboxane A2 and thrombin receptors in human platelets. Proc Natl Acad Sci U S A. 1994;91:504–8. doi: 10.1073/pnas.91.2.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chari R, Getz T, Nagy B, Jr, Bhavaraju K, Mao Y, Bynagari YS, et al. Protein kinase C[delta] differentially regulates platelet functional responses. Arterioscler Thromb Vasc Biol. 2009;29:699–705. doi: 10.1161/ATVBAHA.109.184010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newton AC. Regulation of protein kinase C. Curr Opin Cell Biol. 1997;9:161–7. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- 12.Violin JD, Newton AC. Pathway illuminated: visualizing protein kinase C signaling. IUBMB Life. 2003;55:653–60. doi: 10.1080/152165401310001642216. [DOI] [PubMed] [Google Scholar]
- 13.Crosby D, Poole AW. Physical and functional interaction between protein kinase C delta and Fyn tyrosine kinase in human platelets. J Biol Chem. 2003;278:24533–41. doi: 10.1074/jbc.M301847200. [DOI] [PubMed] [Google Scholar]
- 14.Murugappan S, Tuluc F, Dorsam RT, Shankar H, Kunapuli SP. Differential role of protein kinase C delta isoform in agonist-induced dense granule secretion in human platelets. J Biol Chem. 2004;279:2360–7. doi: 10.1074/jbc.M306960200. [DOI] [PubMed] [Google Scholar]
- 15.Elzagallaai A, Rose SD, Brandan NC, Trifaro JM. Myristoylated alanine-rich C kinase substrate phosphorylation is involved in thrombin-induced serotonin release from platelets. Br J Haematol. 2001;112:593–602. doi: 10.1046/j.1365-2141.2001.02642.x. [DOI] [PubMed] [Google Scholar]
- 16.Dent J, Kato K, Peng XR, Martinez C, Cattaneo M, Poujol C, et al. A prototypic platelet septin and its participation in secretion. Proc Natl Acad Sci U S A. 2002;99:3064–9. doi: 10.1073/pnas.052715199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilligan DM, Sarid R, Weese J. Adducin in platelets: activation-induced phosphorylation by PKC and proteolysis by calpain. Blood. 2002;99:2418–26. doi: 10.1182/blood.v99.7.2418. [DOI] [PubMed] [Google Scholar]
- 18.Stafford MJ, Watson SP, Pears CJ. PKD: a new protein kinase C-dependent pathway in platelets. Blood. 2003;101:1392–9. doi: 10.1182/blood-2002-08-2384. [DOI] [PubMed] [Google Scholar]
- 19.Johannes FJ, Prestle J, Eis S, Oberhagemann P, Pfizenmaier K. PKCu is a novel, atypical member of the protein kinase C family. J Biol Chem. 1994;269:6140–8. [PubMed] [Google Scholar]
- 20.Rozengurt E, Rey O, Waldron RT. Protein kinase D signaling. J Biol Chem. 2005;280:13205–8. doi: 10.1074/jbc.R500002200. [DOI] [PubMed] [Google Scholar]
- 21.Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E. Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc Natl Acad Sci U S A. 1994;91:8572–6. doi: 10.1073/pnas.91.18.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sturany S, Van Lint J, Muller F, Wilda M, Hameister H, Hocker M, et al. Molecular cloning and characterization of the human protein kinase D2. A novel member of the protein kinase D family of serine threonine kinases. J Biol Chem. 2001;276:3310–8. doi: 10.1074/jbc.M008719200. [DOI] [PubMed] [Google Scholar]
- 23.Hayashi A, Seki N, Hattori A, Kozuma S, Saito T. PKCnu, a new member of the protein kinase C family, composes a fourth subfamily with PKCmu. Biochim Biophys Acta. 1999;1450:99–106. doi: 10.1016/s0167-4889(99)00040-3. [DOI] [PubMed] [Google Scholar]
- 24.Waldron RT, Iglesias T, Rozengurt E. Phosphorylation-dependent protein kinase D activation. Electrophoresis. 1999;20:382–90. doi: 10.1002/(SICI)1522-2683(19990201)20:2<382::AID-ELPS382>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 25.Waldron RT, Rey O, Iglesias T, Tugal T, Cantrell D, Rozengurt E. Activation loop Ser744 and Ser748 in protein kinase D are transphosphorylated in vivo. J Biol Chem. 2001;276:32606–15. doi: 10.1074/jbc.M101648200. [DOI] [PubMed] [Google Scholar]
- 26.Matthews SA, Rozengurt E, Cantrell D. Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/Protein kinase Cmu. J Biol Chem. 1999;274:26543–9. doi: 10.1074/jbc.274.37.26543. [DOI] [PubMed] [Google Scholar]
- 27.Iglesias T, Waldron RT, Rozengurt E. Identification of in vivo phosphorylation sites required for protein kinase D activation. J Biol Chem. 1998;273:27662–7. doi: 10.1074/jbc.273.42.27662. [DOI] [PubMed] [Google Scholar]
- 28.Rybin VO, Guo J, Steinberg SF. Protein kinase D1 autophosphorylation via distinct mechanisms at Ser744/Ser748 and Ser916. J Biol Chem. 2009;284:2332–43. doi: 10.1074/jbc.M806381200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matthews SA, Navarro MN, Sinclair LV, Emslie EA, Feijoo-Carnero MC, Cantrell D. Unique functions for protein kinase D1 and protein kinase D2 in mammalian cells. Biochem J. doi: 10.1042/BJ20101188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klages B, Brandt U, Simon MI, Schultz G, Offermanns S. Activation of G12/G13 results in shape change and Rho/Rho-kinase-mediated myosin light chain phosphorylation in mouse platelets. J Cell Biol. 1999;144:745–54. doi: 10.1083/jcb.144.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferris CD, Huganir RL, Supattapone S, Snyder SH. Purified inositol 1,4,5-trisphosphate receptor mediates calcium flux in reconstituted lipid vesicles. Nature. 1989;342:87–9. doi: 10.1038/342087a0. [DOI] [PubMed] [Google Scholar]
- 32.Shattil SJ. Signaling through platelet integrin alpha IIb beta 3: inside-out, outside-in, and sideways. Thromb Haemost. 1999;82:318–25. [PubMed] [Google Scholar]
- 33.Selheim F, Idsoe R, Fukami MH, Holmsen H, Vassbotn FS. Formation of PI 3-kinase products in platelets by thrombin, but not collagen, is dependent on synergistic autocrine stimulation, particularly through secreted ADP. Biochem Biophys Res Commun. 1999;263:780–5. doi: 10.1006/bbrc.1999.1450. [DOI] [PubMed] [Google Scholar]
- 34.Kim S, Foster C, Lecchi A, Quinton TM, Prosser DM, Jin J, et al. Protease-activated receptors 1 and 4 do not stimulate G(i) signaling pathways in the absence of secreted ADP and cause human platelet aggregation independently of G(i) signaling. Blood. 2002;99:3629–36. doi: 10.1182/blood.v99.10.3629. [DOI] [PubMed] [Google Scholar]
- 35.Brandlin I, Hubner S, Eiseler T, Martinez-Moya M, Horschinek A, Hausser A, et al. Protein kinase C (PKC)eta-mediated PKC mu activation modulates ERK and JNK signal pathways. J Biol Chem. 2002;277:6490–6. doi: 10.1074/jbc.M106083200. [DOI] [PubMed] [Google Scholar]
- 36.Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, et al. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem. 1993;268:9194–7. [PubMed] [Google Scholar]
- 37.Yuan J, Bae D, Cantrell D, Nel AE, Rozengurt E. Protein kinase D is a downstream target of protein kinase Ctheta. Biochem Biophys Res Commun. 2002;291:444–52. doi: 10.1006/bbrc.2002.6469. [DOI] [PubMed] [Google Scholar]
- 38.Tan M, Xu X, Ohba M, Ogawa W, Cui MZ. Thrombin rapidly induces protein kinase D phosphorylation, and protein kinase C delta mediates the activation. J Biol Chem. 2003;278:2824–8. doi: 10.1074/jbc.M211523200. [DOI] [PubMed] [Google Scholar]
- 39.Nagy B, Jr, Bhavaraju K, Getz T, Bynagari YS, Kim S, Kunapuli SP. Impaired activation of platelets lacking protein kinase C-theta isoform. Blood. 2009;113:2557–67. doi: 10.1182/blood-2008-07-169268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bynagari YS, Nagy B, Jr, Tuluc F, Bhavaraju K, Kim S, Vijayan KV, et al. Mechanism of activation and functional role of protein kinase Ceta in human platelets. J Biol Chem. 2009;284:13413–21. doi: 10.1074/jbc.M808970200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hall KJ, Harper MT, Gilio K, Cosemans JM, Heemskerk JW, Poole AW. Genetic analysis of the role of protein kinase Ctheta in platelet function and thrombus formation. PLoS One. 2008;3:e3277. doi: 10.1371/journal.pone.0003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soriani A, Moran B, de Virgilio M, Kawakami T, Altman A, Lowell C, et al. A role for PKCtheta in outside-in alpha(IIb)beta3 signaling. J Thromb Haemost. 2006;4:648–55. doi: 10.1111/j.1538-7836.2006.01806.x. [DOI] [PubMed] [Google Scholar]
- 43.Kim S, Jin J, Kunapuli SP. Akt activation in platelets depends on Gi signaling pathways. J Biol Chem. 2004;279:4186–95. doi: 10.1074/jbc.M306162200. [DOI] [PubMed] [Google Scholar]
- 44.Kim S, Jin J, Kunapuli SP. Relative contribution of G protein-coupled pathways to protease-activated receptor-mediated Akt phosphorylation in platelets. Blood. 2006;107:947–54. doi: 10.1182/blood-2005-07-3040. [DOI] [PubMed] [Google Scholar]
- 45.Kim S, Mangin P, Dangelmaier C, Lillian R, Jackson SP, Daniel JL, et al. Role of phosphoinositide 3-kinase beta in glycoprotein VI-mediated Akt activation in platelets. J Biol Chem. 2009;284:33763–72. doi: 10.1074/jbc.M109.048553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murugappan S, Chari R, Palli VM, Jin J, Kunapuli SP. Differential regulation of threonine and tyrosine phosphorylations on protein kinase Cdelta by G-protein-mediated pathways in platelets. Biochem J. 2009;417:113–20. doi: 10.1042/BJ20080235. [DOI] [PubMed] [Google Scholar]
- 47.Murugappan S, Shankar H, Bhamidipati S, Dorsam RT, Jin J, Kunapuli SP. Molecular mechanism and functional implications of thrombin-mediated tyrosine phosphorylation of PKC{delta} in platelets. Blood. 2005;106:550–7. doi: 10.1182/blood-2004-12-4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Lint J, Rykx A, Maeda Y, Vantus T, Sturany S, Malhotra V, et al. Protein kinase D: an intracellular traffic regulator on the move. Trends Cell Biol. 2002;12:193–200. doi: 10.1016/s0962-8924(02)02262-6. [DOI] [PubMed] [Google Scholar]
- 49.Zugaza JL, Sinnett-Smith J, Van Lint J, Rozengurt E. Protein kinase D (PKD) activation in intact cells through a protein kinase C-dependent signal transduction pathway. Embo J. 1996;15:6220–30. [PMC free article] [PubMed] [Google Scholar]
- 50.Waldron RT, Iglesias T, Rozengurt E. The pleckstrin homology domain of protein kinase D interacts preferentially with the eta isoform of protein kinase C. J Biol Chem. 1999;274:9224–30. doi: 10.1074/jbc.274.14.9224. [DOI] [PubMed] [Google Scholar]
- 51.Brandlin I, Eiseler T, Salowsky R, Johannes FJ. Protein kinase C(mu) regulation of the JNK pathway is triggered via phosphoinositide-dependent kinase 1 and protein kinase C(epsilon) J Biol Chem. 2002;277:45451–7. doi: 10.1074/jbc.M205299200. [DOI] [PubMed] [Google Scholar]
- 52.Pears CJ, Thornber K, Auger JM, Hughes CE, Grygielska B, Protty MB, et al. Differential roles of the PKC novel isoforms, PKCdelta and PKCepsilon, in mouse and human platelets. PLoS One. 2008;3:e3793. doi: 10.1371/journal.pone.0003793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cohen S, Braiman A, Shubinsky G, Ohayon A, Altman A, Isakov N. PKCtheta is required for hemostasis and positive regulation of thrombin-induced platelet aggregation and alpha-granule secretion. Biochem Biophys Res Commun. 2009;385:22–7. doi: 10.1016/j.bbrc.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 54.Konopatskaya O, Matthews SA, Harper MT, Gilio K, Cosemans JM, Williams CM, et al. Protein kinase C mediates platelet secretion and thrombus formation through protein kinase D2. Blood. doi: 10.1182/blood-2010-10-312199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Polgar J, Lane WS, Chung SH, Houng AK, Reed GL. Phosphorylation of SNAP-23 in activated human platelets. J Biol Chem. 2003;278:44369–76. doi: 10.1074/jbc.M307864200. [DOI] [PubMed] [Google Scholar]