

Abstract
Two orthogonal destabilizing domains have been developed based on mutants of human FKBP12 as well as bacterial DHFR and these engineered domains have been used to control protein concentration in a variety of contexts in vitro and in vivo. FKBP12 based destabilizing domains can not be rescued in the yeast Saccharomyces cerevisiae; ecDHFR based destabilizing domains are not degraded as efficiently in S. cerevisiae as in mammalian cells or Plasmodium, but provide a starting point for the development of domains with increased signal-to-noise in S. cerevisiae.
Interrogation of protein functions in specific contexts can be performed through systematic analysis upon its perturbation through gene deletion or overexpression, mRNA depletion or inhibition or activation with small molecules. Small molecules allow rapid, tunable and often reversible control over protein function, but discovery of small molecule ligands specific for a protein of interest is non-trivial and can be complicated by additional off-target effects. We previously developed a chemical-genetic approach that allows rapid, reversible and tunable expression of a protein of interest by genetic fusion to a ‘destabilizing domain’ (DD)1. Destabilizing domains are small protein domains that allow tunable control over protein function by acting as a dominant degron when fused to a protein of interest in the absence of a stabilizing ligand. Addition of a bio-orthogonal ligand allows stabilization of the domain and the protein of interest, allowing it to function normally. Thus far, we have discovered two ligand-protein pairs that have the desirable properties that allow them to be used as DDs: Shield-1 with mutant (human) FKBP12 (DDFKBP) and trimethoprim (TMP) with mutant (E. coli) DHFR (DDDHFR)1, 2. These DDs allow functional control over proteins of interest in a variety of contexts in mammalian cell culture, the apicomplexans Plasmodium and Toxoplasma, and in living mice1–6.
Because of its tractable genetics, it would be desirable to develop destabilizing domains that function in the yeast Saccharomyces cerevisiae. It can be propagated as haploid cells and DNA repair takes place primarily by homologous recombination; this combination allows for facile replacement of the endogenous gene with tagged versions. This strategy has been used to create several near genome-wide collections of S. cerevisiae where the endogenous gene has been knocked out, tagged with GFP or tagged with the TAP tag7–9. We aimed to use the DDs in yeast to control the concentration of endogenously tagged proteins, so we designed our yeast DD strategy to use C-terminal DDs in order to not disrupt the endogenous promoters of targeted genes.
We first examined the function of DDFKBP by integrating a yeast-enhanced GFP (yeGFP)-DDFKBP fusion at the ho locus in strains Sc252a, W303a or Y7092 (a derivative of S288c). Equivalent results were obtained from each strain (data not shown), and all data reported here are from Y7092. We examined several FKBP mutants, including the originally reported L106P mutant and the E31G/R71G/K105E mutant that was previously found to have superior characteristics to the L106P mutant as a C-terminal DD1, 10. In each case, we expected to observe low fluorescence in the absence of ligand, and higher fluorescence in the presence of Shield-1. The yeGFP-DDFKBP fusion proteins were not expressed highly and/or constitutively degraded (Fig 1a). Note that for the analysis of all flow cytometry data, a tight FSC-A vs. SSC-A gate was selected since yeast have considerably greater size variation than mammalian cells in culture and much of the variation in fluorescence in a yeast culture can be attributed to variations in size (Supp. Fig 1). Fluorescence of the yeGFP-DDFKBP expressing cells could not be rescued by the addition of 1μM Shield-1, 100nM rapamycin or 1μM FK506, as it could in mammalian cells under similar conditions. Because rapamycin and FK506 are known to penetrate yeast11, 12, it is unclear why the fluorescence of these constructs could not be rescued by the addition of these small-molecule ligands; however, this avenue was not pursued further.
Figure 1.

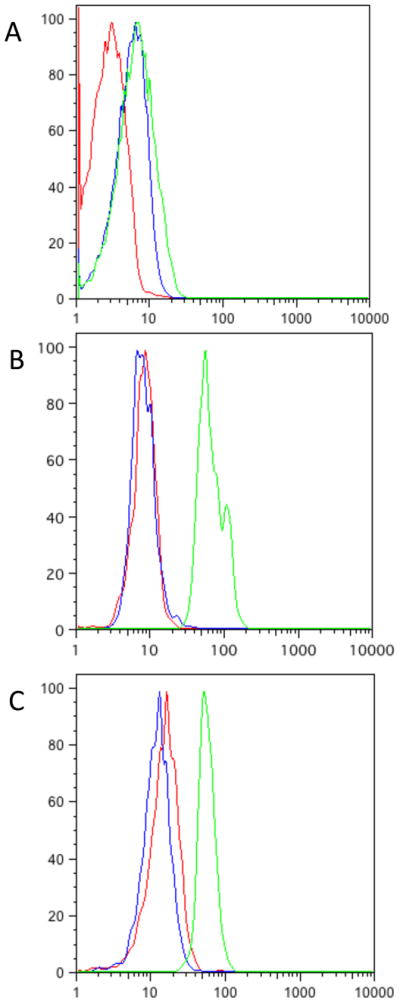
a) Flow cytometry data from Y7092 expressing yeGFP-DDFKBP; untransformed Y7092 (red), YRR030 (Y7092 ho::PSOD1:yeGFP-DDFKBP::KANMX4 (blue), YRR030 + 5μM Shield-1 (green), YRR030 +100nM rapamycin (orange). YRR030 +1μM FK506 (green) is shown in the right panel, for clarity. b) Flow cytometry data from Y7092 expressing yeGFP-DDDHFR; untransformed Y7092 (red), YRR009 (Y7092 ho::PSOD1:yeGFP-DDDHFR::NATMX4)(blue), YRR009 + 100μM TMP (green). c) Flow cytometry data from NIH 3T3 cells; untransduced (red), transduced with YFP-DDDHFR (blue), transduced with YFP-DDDHFR +10μM TMP (green).
The activity of DDs based on E. coli DHFR was similarly interrogated. Very low background levels of the fusion protein in the absence of stabilizing ligand are a desirable property of DDs as low levels of expressed proteins may be sufficient to mask interesting phenotypes. Yeast strains expressing yeGFP-DDDHFR (R12Y/Y100I mutant) integrated at ho are highly fluorescent upon the addition of 10μM TMP. Cultures in exponential growth phase grown in the absence of TMP however display significant background fluorescence (Fig 1b). The observed background fluorescence and signal-to-noise above background in the presence of 10μM TMP in yeast compared poorly to that observed in (mouse) NIH 3T3 cells (Fig 1c).
Because yeast can be cultured under a variety of conditions, including aerobic or fermentative growth, carbon source, temperature and culture density, we investigated whether any of these variables would affect the performance of the DDDHFR in S. cerevisiae. It is likely that the DDs are functioning as misfolded proteins recognized by the cellular quality control machinery where the folding defect can be corrected by the addition of the ligand. Since the folding of proteins, especially marginally stable ones, can be greatly influenced by temperature, we suspected that the poorer performance of the DDs in yeast could be attributed in part to their growth conditions at 30C instead of 37C, as for the other organisms tested. Yeast grown at 37C, however, showed almost identical background fluorescence, but failed to stabilize to as high a maximum fluorescence in the presence of TMP as those grown at 30C (Fig 2a). Cultures grown to near saturation (A600nm~20), however, have much lower background fluorescence than those in exponential phase, barely higher than autofluorescence of non-transformed cells (Fig 3b). Culturing in fermentative media (glucose, YPD), or forced respiration (ethanol+glycerol, YPEG) produced cells with similar levels of background fluorescence in the absence of TMP and maximum fluorescence in the presence of 100μM TMP (Fig 2c).
Figure 2.
a) Flow cytometry data from Y7092 grown at 37C (red), YRR009 grown at 37C (blue) and YRR009 +100μM TMP at 37C (green). b) Flow cytometry data from yeast grown to near saturation, Y7092 (red), YRR009 (blue) YRR009 +100μM TMP (green). c) same as in b), but grown in YPEG.
Figure 3.

Cell-cycle analysis by flow cytometry; a) W303a cells were grown to overnight in 2x YPD, diluted to a starting concentration of A600=0.05–0.1 and grown for two generations prior to addition of various concentrations of alpha factor 2hrs prior to fixation and staining (as indicated). b) W303a derivatives treated as in a), as indicated.
We then investigated whether the DDDHFR could be used to exert phenotypic control in S. cerevisiae. We tested two genes with easily discernable phenotypes when inactivated. Yeast mating occurs through classical GPCR-coupled MAPK signalling resulting in pheromone mediated cell-cycle arrest of haploid cells in G1. FAR1 encodes a cyclin dependent kinase inhibitor that is activated by the mating pheromone. Deletion of FAR1 produces cells that no longer arrest in response to mating pheromone13. We integrated DDDHFR at the 3′ end of FAR1 of W303a; correct integration was confirmed by PCR using primers upstream and downstream of the site of integration. We expected that cells would arrest with 1S DNA content as measured by flow cytometry when treated with alpha factor (mating pheromone) under permissive conditions (Fig 3a), but Far1p-DDDHFR strains should fail to arrest in the absence of TMP. However, Far1p-DDDHFR strains still arrested with 1S DNA content when treated with 2μM alpha factor (Fig 3b).
CDC28 encodes the only cyclin dependent kinase (CDK) in S. cerevisiae. CDC28 is an essential gene and conditional cdc28 mutants do not progress through the cell cycle and are unviable14. Dohmen and Varshasky could conditionally regulate Cdc28p (the protein product of CDC28), and cell viability, through an N-terminal fusion with a temperature sensitive allele of mouse DHFR when shifted from 24C to 37C15. We integrated the DDDHFR at the 3′ locus of CDC28 in strain W303a; correct integration was confirmed by PCR using primers upstream and downstream of the site of integration. Cells were first grown in YPD overnight at 30C in the presence of 10μM TMP, washed twice in YPD and transferred to fresh media containing either 10μM TMP or in the absence of TMP. There was no obvious growth defect in the Cdc28p-DDDHFR strain in the absence of TMP, suggesting that residual Cdc28p-DDDHFR is sufficient to support robust growth (data not shown). Since alpha factor and loss of Cdc28p should both arrest cells in G1, we tested whether low concentrations of alpha-factor would in combination with lowered concentrations of Cdc28p in the Cdcd28p-DDDHFR strain would be sufficient for arresting cells in G1. While we did observe an enrichment of cells with G1 DNA content in this strain, we did not observe any additional cell-cycle arrest that could be attributed to loss of function in CDC28 (Fig 3b).
We have tested the two destabilizing domains developed in our lab for their activity in the yeast S. cerevisiae. We found that the FKBP based DD is unstable under all conditions tested, whereas the E. coli DHFR based DD is degradable and stabilizable in the presence of TMP, but with weak performance in signal-to-noise and residual protein in the absence of drug. We are currently attempting to improve the performance of the E. coli DHFR based DDs in yeast by screening for additional mutants with improved characteristics.
Materials and Methods
Trimethoprim (Sigma) was dissolved to a stock concentration of 100mM in DMSO. Shield-1, rapamycin and FK506 were dissolved at a stock concentration of 10mM in absolute ethanol.
All sequences were obtained from the Saccharomyces Genome Database (SGD). A plasmid was constructed based on pBluscript II SK+ for high efficiency integration into the ho locus. These plasmids contain the PSOD1-yeGFP-(DHFR or FKBP)-TADH1-(KANMX4 or NATMX4) cassette flanked by 500bp of homology to ho. 600bp upstream of the SOD1 start was used as the promoter since its transcription is invariant to carbon source and is highly expressed. Yeast-enhanced GFP, FKBP12 and bacterial DHFR were as previously reported and separated by a six amino acid (-GRGEGQ-) linker.
Yeast strains were constructed and cultured using standard techniques. Flow cytometry was carried out using a BD FACSCalibur instrument where at least 25000 events were collected. Analysis was carried out using FlowJo 9.1 (TreeStar), with tight FSC-SSC gating representing ~2–5% of total events represented in the reported data (see Supplementary Fig. 1).
For cell cycle arrest experiments, cultures were grown overnight at 30C in 2x YPD to a density of A600 <1. These were then washed once in PBS and resuspended at a density of A600~0.05 in 2x YPD and grown for a further three hours. Alpha factor (Zymo Research) was added to the indicated concentration for two hours prior to harvesting. Cells from a ~5ml culture were washed twice in PBS, followed by resuspending in 300μL of water, followed by dropwise addition of 700μL of absolute ethanol while vortexing. Cells were washed with PBS and resuspended in PBS containing 1mg/ml RNAse A and incubated at 50C for at least two hours. Cells were washed in PBS and digested with 0.1mg/ml Proteinase K for 5 minutes at 50C. Cells were washed again PBS and incubated with 0.25mg/ml propidium iodide at room temperature for at least one hour. Cells were briefly vortexed prior to analysis using a BD FACSCalibur instrument where at least 10000 events were collected.
Genotypes of strains reported in this study:
| YRR009 | ho::PSOD1:yeGFP-DDDHFR::NATMX4 | Y7092 (mat alpha can1delta::STE2pr-Sp_his5 lyp1delta his3delta1 leu2delta0 ura3delta0 met15delta0) |
| YRR030 | ho::PSOD1:yeGFP-DDFKBP::KANMX4 | Y7092 |
| YRR031 | FAR1-DDDHFR::KANMX4 | W303a (MATa ADE2 his3 trp1 leu2 ura3) |
| YRR032 | CDC28-DDDHFR::KANMX4 | W303a |
Supplementary Material
Acknowledgments
We would like to thank Ling Chen for advice and help with cloning, and Joe Horecka and Angela Chu (Stanford), Charlie Boone (University of Toronto) and Wendell Lim (UCSF) for plasmids, strains, and advice. This work is supported through NIH (GM068589) and the Ellison Medical Foundation. R.R. is supported by a post-doctoral fellowship from CIHR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, Wandless TJ. Cell. 2006;126:995. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iwamoto M, Bjorklund T, Lundberg C, Kirik D, Wandless TJ. Chem Biol. 2010;17:981. doi: 10.1016/j.chembiol.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herm-Gotz A, Agop-Nersesian C, Munter S, Grimley JS, Wandless TJ, Frischknecht F, Meissner M. Nature methods. 2007;4:1003. doi: 10.1038/nmeth1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armstrong CM, Goldberg DE. Nature methods. 2007;4:1007. doi: 10.1038/nmeth1132. [DOI] [PubMed] [Google Scholar]
- 5.Banaszynski LA, Sellmyer MA, Contag CH, Wandless TJ, Thorne SH. Nat Med. 2008;14:1123. doi: 10.1038/nm.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muralidharan V, Oksman A, Iwamoto M, Wandless TJ, Goldberg DE. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4411. doi: 10.1073/pnas.1018449108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. Nature. 2002;418:387. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 8.Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Nature. 2003;425:686. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 9.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Nature. 2003;425:737. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 10.Chu BW, Banaszynski LA, Chen LC, Wandless TJ. Bioorg Med Chem Lett. 2008;18:5941. doi: 10.1016/j.bmcl.2008.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koltin Y, Faucette L, Bergsma DJ, Levy MA, Cafferkey R, Koser PL, Johnson RK, Livi GP. Mol Cell Biol. 1991;11:1718. doi: 10.1128/mcb.11.3.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brizuela L, Chrebet G, Bostian KA, Parent SA. Mol Cell Biol. 1991;11:4616. doi: 10.1128/mcb.11.9.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peter M, Gartner A, Horecka J, Ammerer G, Herskowitz I. Cell. 1993;73:747. doi: 10.1016/0092-8674(93)90254-n. [DOI] [PubMed] [Google Scholar]
- 14.Mendenhall MD, Richardson HE, Reed SI. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:4426. doi: 10.1073/pnas.85.12.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dohmen RJ, Wu P, Varshavsky A. Science. 1994;263:1273. doi: 10.1126/science.8122109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.