

Abstract
Interest in the synthesis of the C(23)-C(40) fragment 2 of tetrafibricin prompted us to develop a new method for the synthesis of 1,5-syn-(E)-diols. Toward this end, the kinetically controlled hydroboration of allenes 6, 33, ent-39, 42 and 45 with the Soderquist borane 25R were studied. Tetrabutylammonium allenyltrifluoroborate 45 gave superior results and was utilized in a double allylboration sequence with two different aldehydes to provide the targeted 1,5-syn-(E)-diols in generally high yields (72–98%), and with high enantioselectivity (>95% e.e.), diastereoselectivity (d.r. >20:1), and (E)/(Z) selectivity (>20:1). This new method was applied to the synthesis of the C(23)-C(40) fragment 2 of tetrafibricin.
Keywords: Studies on the synthesis of tetrafibricin; Double allylboration; Allene hydroboration, kinetically controlled; Tetrabutylammonium allenyltrifluoroborate; Stereoselective synthesis of syn-1,5-E-diols
1. Introduction
The 1,5-syn-(E)-diol substructure is found in a variety of natural products.1 One of the strategies used to access 1,5-syn-(E)-diols, developed by Cossy and co-workers in their work on the total synthesis of tetrafibricin (Figure 1), involves an iterative allyltitanation-cross metathesis-allyltitanation sequence.2,3 This strategy provides the 1,5-syn-(E)-diol motif with good levels of diastereoselectivity. In 2004, Leighton published a method for synthesis of 1,5-syn-(E)-diols via a unique intramolecular alkyne silylformylation-allylsilylation reaction sequence that proceeds in modest yield and with modest diastereoselectivity.4 A key sequence in Nicolaou’s total synthesis of marinomycin A involved a Horner-Wadsworth-Emmons olefination and a syn-reduction of the carbonyl group that provided the 1,5-syn-(E)-diol unit with good diastereoselectivity.5 In 2010, Friestad published a method for synthesis of the 1,5-(E)-diol unit of tetrafibricin involving the iterative Julia-Kociensky olefination of chiral aldehydes using a chiral α-silyloxy-γ-sulfononitrile.6 As an alternative, we anticipated that use of a bifunctionalized allylmetal reagent could provide convenient, one-pot access to the 1,5-syn-(E)-diol motif.7 Only a few chiral bifunctionalized allylmetal reagents have been reported that provide a high level of stereochemical control in such processes.8 Our group has developed several chiral 1,3-bifunctionalized allylborane reagents,8c–e and have successfully utilized these reagents in studies on the synthesis of polyhydroxylated natural products.9
Figure 1.

Tetrafibricin (1).
We report herein the development of a new 1,3-bifunctionalized allylborane reagent that enables the targeted 1,5-syn-(E)-diol motif to be synthesized with excellent stereochemical control. We demonstrate the utility of this new method in the synthesis of the C(23)-C(40) fragment of tetrafibricin (1).
2. Background
2.1 Studies on the Synthesis of Tetrafibricin
The present research was motivated by our interest in the synthesis of tetrafibricin, and especially the C(23)-C(40) fragment 2 (Scheme 1). Tetrafibricin (1) is a structurally unique natural product isolated in 1993 from Streptomyces neyagawaensis.10 It displays potent anti-aggregation properties against human platelets by blocking the glycoprotein (GP)IIb/IIIa receptor on the platelet surface, which is important for blood clotting.11 The stereochemistry was assigned by Kishi based on 1H NMR database technology, including NMR measurements in chiral solvents.1b The total synthesis of tetrafibricin has not yet been reported, which is necessary to confirm the relative stereochemistry assignment and to set the stage for biological structure-function studies. Our group reported the synthesis of the tetrafibricin C(1)-C(19) fragment,12 and contributions from several other laboratories have also appeared.2, 6, 13 We recently published a preliminary communication describing the synthesis of the C(23)-C(40) fragment 2,14 via the fragment coupling of 3 and 4 with a new bifunctional allylborane reagent. We provide herein the details of this effort, focusing on the studies leading to the development of the new double allylborating agent.
Scheme 1.

Retrosynthetic Analysis of the C(23)-C(40) Fragment 2 of Tetrafibricin
2.2 The Double Allylboration Reaction
In 2002 our group reported highly diastereo- and enantioselective syntheses of 1,5-anti-(E) (10) and 1,5-syn-(Z) (15) diols via a double allylboration sequence (Scheme 2).8c
Scheme 2.

Synthesis of 1,5-anti-(E) 10 and 1,5-syn-(Z) 15 Diols via the Double Allylboration Reaction
Hydroboration of allenes 68c and 118c using d-diisopino- campheylborane (dIpc2BH)15 provides the kinetic hydroboration products 7 and 12, which undergo rapid (Z) to (E) isomerization via a [1,3]-boratropic shifts16 to give the observed (thermodynamic) allylboranes 8 and 13.
The allylboration reaction of these reagents with aldehydes proceeds via chair-like transition states TS-1 or TS-3 to generate anti-β-alkoxyallylboronates 9 and 14, respectively. Treatment of anti-β-alkoxyallylboronate 9 with a second aldehyde proceeds via the chair-like transition state TS-2 with the methallylboronate α-substituent in an equatorial position. This leads to the formation of the 1,5-anti-diol 10 with an intervening (E)-olefin. However, due to severe steric interactions the α-substituent in methallylboronate 14 occupies an axial position in the second allylboration transition state TS-4, leading to the formation of the 1,5-syn diol 15 with an embedded (Z)-olefin. Accordingly, we turned our efforts to the synthesis of the 1,5-syn-(E)-diol configuration, which is not accessible via these first-generation double allylboration reactions, but which is required for the synthesis of the C(23)-C(40) fragment of tetrafibricin.
3. Results and Discussion
3.1 Initial Attempts to Extend the Double Allylboration Reaction to the Synthesis of 1,5-syn-(E)-Diols 17 via Kinetically Controlled Hydroboration of Allenylboronates with Ipc2BH
We anticipated that if the [1,3]-boratropic shifts16 of 7 and 12 could be prevented by performing the hydroboration of 6 or 11 at low temperatures, we could access to the syn-β-alkoxyallylboronates 16 and 18, respectively. If so, we expected (by analogy to transition states TS-2 and TS-4 in Scheme 2) that the subsequent allyboration of 16 or 18 would provide the 1,5-syn-(E)-diol 17 and 1,5-anti-(Z)-diol 19 with synthetically useful selectivity (Scheme 3).
Scheme 3.

1,5-syn-(E)-Diol 17 and 1,5-anti-(Z)-Diol 19
Accordingly, we attempted to generate (Z)-allylborane 21 via the kinetic hydroboration of allene 20 with lIpc2BH15 under a range of reaction conditions (including variations of the solvent, reaction temperature, and time), including the use of Wilkinson’s catalyst to perform the hydroboration at low temperature.17 However, when the reagent generated under these conditions was treated with hydrocinnamaldehyde at −78 °C, the targeted 1,5-syn-(E)-diol ent-17a was consistently the minor product and was not obtained with acceptable selectivity (Scheme 4 and Table 1).
Scheme 4.

Studies of the Kinetic Hydroboration Reaction of Allene 20 with lIpc2BH
Table 1.
Results of Hydroboration-Allylboration of Allene 20 with lIpc2BH a
| Entry | Catalyst | T (°C) | Time | Ratio ent-17a : ent-10ab : ent-19gc |
Yieldd |
|---|---|---|---|---|---|
| 1 | none | −20 | 30 min | 1 : 11 : 1 | 40% |
| 2 | none | −30 | 1 h | 1 : 6.2: 0.8 | 36% |
| 3 | (Ph3P)3RhCl (5%) |
−78 | 4 h | 1 : 0.9 : 1.2 | 17% |
Hydroboration reactions of 20 were performed in THF at the indicated temperatures and times. The resulting allylborane was then treated with hydrocinnamaldehyde as specified in Scheme 4 to give the indicated products.
Product ratio determined by 13C NMR analysis.
Product ratio determined by 1H NMR analysis.
Isolated yields.
When the hydroboration reaction of 20 was performed at −20 °C for 30 min followed by treatment with hydrocinnamaldehyde at −78 °C, a mixture of the 1,5-syn-(E)-diol ent-17a, the 1,5-anti-(E)-diol diastereomer ent-10a, and the 1,5-anti-(Z)-diol 19g were obtained with 1:11:1 selectivity, with favor of ent-10a predominating (entry 1, Table 1). This result can be explained by a rapid [1,3]-boratropic shift16 that converts 21 to 22. This isomerization also occurred rapidly at −30 °C (entry 2), as the hydroboration performed at this temperature resulted in a 1 : 6.2 : 0.8 mixture of products, again favoring the 1,5-anti-(E)-diol ent-10a. Allene hydroboration experiments performed using Wilkinson’s catalyst at −78 °C (entry 3) gave a somewhat improved ratio of ent-17a to ent-10a, however the ratio these two diols was only ca. 1: 1, and a comparable amount of ent-19g was also obtained. These experiments were compromised by the poor solubility of lIpc2BH especially at low temperatures, resulting in incomplete conversion and poor overall yields.
3.2. Synthesis of 1,5-anti-(Z)-Diols via the Kinetically Controlled Hydroboration of 11 using Soderquist’s Borane
The inability to generate the bis-boryl reagent 21 with acceptable selectivity for use as a precursor to 1,5-syn-(E)-diols ent-17a prompted us to reevaluate our strategy. It was apparent from the results in Table 1 that we needed to identify a chiral dialkylborane for the allene hydroboration reaction such that the kinetic allylic borane products would undergo the 1,3-boratropic shift very slowly, if at all. Soderquist had published a series of papers in which allylboration reactions were performed with crotylboranes containing the 10-TMS-9-borabycyclo[3.3.2]decane auxiliary (TBBD).18 These reagents demonstrated remarkable stability towards thermal isomerization:19 the (Z)-crotyl-TBBD 23 isomer is reported to isomerize to (E)-crotyl-TBBD 24 with a half-life of approximately one week at room temperature (Scheme 5).
Scheme 5.

B-Crotyl-10-TMS-9-borabyciclo[3.3.2]decanes (Z)-23 and (E)-24 undergo (Z) to (E) isomerization slowly at 25 °C in CDCl3.
This observation suggested that it might be possible to slow the isomerization of 7 to 8, or of 12 to 13 (Scheme 2) by replacing Ipc2BH in the hydroboration step with the Soderquist borane, 10-TMS-9-borabyciclo[3.3.2]decane hydride (TBBD-H; 25). If this hypothesis was confirmed, it would allow us to access the 1,5-syn-(E)-diol 17 starting from allene 6 and the 1,5-anti-(Z)-diol 19 starting from the allene 11 with the tetraphenylethylene glycol boronate ester.
We began by studying the hydroboration of allene 11 with the Soderquist borane 25, en route to the 1,5-anti-(Z)-diol ent-19. The unstable TBBD-H 25R8d, 20, 21 was generated in situ from the borohydride 26R (which we store in a glove box) by treatment with TMSCl. The resulting solution of borane 25R was then treated with allene 11 at −10 °C for 5 h. At that point, the solution of in situ generated reagent 27 was subjected to a standard double allylboration protocol, involving treatment of 27 with an aldehyde (R1CHO) at −78 °C typically for 4–12 h and then with a second aldehyde (R2CHO) at 0 °C to 20 °C for 12–24 h. Pleasingly, this protocol provided 1,5-anti-(Z)-diols ent-19 in 67–86% yields, with ca. 12–13:1 diastereoselectivity and with 94–96% enantiomeric excess (% e.e.) (Scheme 6).8d
Scheme 6.

Synthesis of 1,5-anti-(Z)-Diols ent-19a–f using TBBD-H and Allene 11
3.3. Studies of the Kinetically Controlled Hydroboration of 2-Allenyl-(1,3,2)-dioxaborinane 6 with the Soderquist Borane
In view of the success demonstrated in Scheme 6 for the highly stereoselective synthesis of 1,5-anti-(Z)-diols ent-19 via the kinetically controlled hydroboration of 11 using the Soderquist borane 25,8d we attempted to extend this chemistry to the kinetically controlled hydroboration of allene 6. Based on the examples summarized in Scheme 2 for reagent 8,8c we expected that double allylborations of 28 (Scheme 7) would provide the targeted 1,5-syn-(E)-diols ent-17a with synthetically useful selectivity, assuming that the hydroboration of 6 proceeded with kinetic control as demonstrated in Scheme 6 for 11. Accordingly, studies of the hydroboration of 6 were initiated with the experiments summarized in Scheme 7. For these initial studies, the stereoselectivity of the hydroboration was assessed by subjecting the derived allylboranes 28/29 to an allylation-oxidation sequence with benzaldehyde, which provided 30 and its anti diastereomer 31.
Scheme 7.

Hydroboration Studies with TBBD-H 25R and Allene 6
Best results were obtained when the hydroboration reaction of 6 was performed at −10 °C for 1 h with the Soderquist borane 25R (entry 1, Scheme 7). This resulted in the formation of syn diol 30 with 10:1 diastereoselectivity. However, increasing the reaction time from 1 h to 5 h led in an erosion of the diastereoselectivity, with the 1,2-syn diol 30 being favored by only 1.5:1 over its anti diastereomer (entry 2). This result indicates that the [1,3]-boratropic rearrangement16 of the kinetically formed (Z)-γ-boryl allylborane 28 occurs rapidly at −10 °C to give the thermodynamically favored (E)-γ-boryl allylborane 29. Thus, control of the reaction time and temperature for the hydroboration reaction of 6 is crucial.
By using the conditions for the hydroboration of 6 defined in Scheme 7, we examined the double allylboration sequence of 28 using benzaldehyde and hydrocinnamaldehyde (Scheme 8). This experiment provided the targeted 1,5-syn-(E)-diol ent-17b as the major product in 95% e.e., but as a ca. 4:1 mixture with the 1,5-anti-(Z)-isomer ent-19a.
Scheme 8.

Double Allylboration Sequence using TBBD-H 25R, Allene 6, Benzaldehyde and Hydrocinnamaldehyde.
This result (which is consistent with the results presented in Scheme 4) indicated that there was incomplete control of the transition states, with ca. 80% of the product (ent-17b) deriving from TS-5, and the remaining ca. 20% deriving from TS-6. While the reasons for the poor stereoselectivity of this transformation are not certain, it is conceivable that non-bonded interactions between the (1,3,2)-dioxaborinane unit and the group α to boron might destabilize TS-5 compared to TS-6, which are diastereomeric to TS-2 and TS-4 in Scheme 2 (discounting the differences in the structures of the Ipc2B— unit in TS-2/TS-4 and the TBBD unit in TS-5/TS-6).
We attempted to improve the reaction diastereoselectivity by changing the reaction solvent and using hydrocinnamaldehyde as the substrate (Scheme 9). However, these experiments resulted in significant erosion of the reaction diastereoselectivity, compared to the results summarized in Scheme 8 for reactions performed in CH2Cl2.
Scheme 9.

Attempted Optimization of the Double Allylboration Sequence using TBBD-H 25R, Allene 6, and Hydrocinnamaldehyde.
These results prompted us to abandon studies of allene 6 as a precursor to the targeted 1,5-syn-(E)-diols ent-17a, and to explore other options for their synthesis.
3.4. Diisopropyl Allenylboronate 33 as a Possible Precursor to 1,5-syn-(E)-Diols ent-17a
In view of our speculation that non-bonded interactions involving the (1,3,2)-dioxaborinane unit of 32 might contribute to the destabilization of TS-5 (Scheme 8), we decided to examine allenylboronate 33 with a conformationally flexible diisopropylboronate ester as the starting point for these experiments. The hydroboration reaction of 33 with the Soderquist borane 25R was optimized by using an allylation-oxidation sequence with benzaldehyde similar to the one described with the allene 6 (Scheme 10).
Scheme 10.

Studies of the Kinetic Hydroboration of Allene 33 with TBBD-H 25R.
A 5.3:1 mixture of 1,2-syn-diol 30 and the 1,2-anti diastereomer 31 was obtained when the hydroboration of 33 with 25R was performed at −10 °C for 1 h (entry 1, Scheme 10). Increasing the hydroboration time from 1 h to 2 h led to a slightly decreased diastereoselectivity (4.3:1 favoring 30, entry 2). This result suggests that the [1,3]-boratropic rearrangement16 of the kinetically formed (Z)-γ-boryl allylborane 34 to the (E)-γ-boryl allylborane 35 is competitive with the hydroboration at −10 °C. The optimal diastereoselectivity was obtained by performing the hydroboration at −20 °C for 1 h (entry 3). By using these optimized hydroboration conditions, the double allylboration sequence using hydrocinnamaldehyde leading to 1,5-syn-(E)-diol ent-17a was examined (Scheme 11). Unfortunately, the product was obtained with only 2.7:1 diastereoselectivity (ratio of ent-17a:ent-19g). Once again, it became necessary to explore alternative strategies to improve the diastereoselectivity of the second step of the double allylboration sequence leading to ent-17a.
Scheme 11.

Double Allylboration Sequence using Allene 33, TBBD-H 25R, and Hydrocinnamaldehyde
3.5. Studies of the Kinetic Hydroboration and Allylboration Reactions of Pinanediol Allenylboronates 39 and ent-39
An (E)-selective allylboration of aldehydes leading to (E)-5-hydroxy-2-pentenes 37 was developed in our laboratory (Scheme 12),22 via the BF3•OEt2 promoted reaction of the α-alkyl allylboronate 36 with aldehydes.23 Of the two possible transition states, TS-7 and TS-8, we suggested that the Lewis acid selectively coordinates with the most accessible α-face lone pair of electrons on the oxygen atom distal to the angular methyl group to minimize steric interactions. The alternative TS-8, in which the Lewis acid coordinates with the β-face oxgen non-bonded lone pair, is disfavored due to a 1,3-syn-pentane interaction between the pseudoaxial methyl group and the BF3 unit.
Scheme 12.

(E)-Selective Lewis Acid Mediated Allylboration of the Methallylboronate 36
These results suggested than an analogous strategy might be applicable to the synthesis of targeted the 1,5-syn-(E)-diols 17. Thus, hydroboration of the pinanediol allenylboronates 39 or its enantiomer ent-39 with TBBD-H 25R, followed by double allylboration with the second allylboration step performed in a presence of a Lewis acid, was expected to proceed via a transition state similar to TS-7 to give the targeted 1,5-syn-(E)-diols ent-17a. Accordingly, we optimized the hydroboration reaction by using an allylation-oxidation sequence with benzaldehyde (Scheme 13).
Scheme 13.

Studies of the Hydroboration-Allylboration Reactions of Allenes 39 and ent-39.
The hydroboration reaction of allene 39 with borane 25R at 0 °C for 2 h resulted in the formation of 30 in less than 30% yield and with a diastereoselectivity of 3.2:1, but with excellent enantioselectivity (96% e.e.) (entry 1, Scheme 13). The same reaction was performed, switching the allene 39 for its enantiomer ent-39 to determine if there is a double diasteroselection24 component to the selectivity of the allylation reaction. This experiment led to the formation of the diol 30 with 48% yield, 3.2:1 diastereoselectivity and 97% e.e. (entry 2). These results showed that the chirality of the pinanediol moiety does not affect the stereoselectivity of the steps leading to diol 30. By decreasing the reaction time from 2 h to 40 min led to an improvement of the diastereoselectivity, 8.1:1 in favor of the 1,2-syn-diol 30 (entry 3). Performing the hydroboration reaction at −10 °C for 1 h resulted in an increase in the diastereoselectivity of the overall reaction process leading to 30 (12:1) (entry 4). However, increasing the reaction time from 1 h to 2 h resulted in a lower selectivity (8.1:1) (entry 5). Use of hydrocinnamaldehyde in the allylboration sequence gave diol 40 in 85% yield with a diastereoselectivity of 7.3:1 (entry 6). Unfortunately, when these optimized hydroboration conditions were applied in a double allylboration sequence using hydrocinnamaldehyde and BF3•Et2O during the second allylboration reaction, the 1,5-syn-(E)-diol ent-17a and 1,5-anti-(Z)-diol ent-19g were obtained in 83% yield but with a disappointing 2.7:1 ratio favoring the (Z)-diol ent-19g (Scheme 14). Attempts to increase the selectivity of the double allylboration reaction by performing the second allylboration in the presence of other Lewis or Brønsted acids (TiCl4, SnCl4, Et2AlCl, BCl3, TfOH, TFA, Sc(OTf)3) were unsuccessful.
Scheme 14.

Double Allylboration Sequence using Allene ent-39, TBBD-H 25R, and Hydrocinnamaldehyde
3.6. Studies of Double Allylmetallation Reactions Starting with Allenyltributylstannane 42
Although double allylboration reactions starting with allenylboronates 6, 20, 33, and 39 with the Soderquist borane 25R failed to provide 1,5-syn-(E)-diol ent-17 with acceptable diastereoselectivity, the hydroboration step proved to be efficient and kinetically controlled for formation of the required (Z)-allylic borane species. Accordingly, we decided to explore the use of 25R as a hydroborating reagent with other allenes that could be employed as precursors to the targeted 1,5-syn-(E)-diols. In particular, we were attracted to the use of allenylstannane 42 since the Lewis acid mediated reactions of allylstannanes with aldehydes are well established.25 With the aim of employing syn-β-alkoxyallylstannanes 44a and 44b as double allylmetallation intermediates, our attention turned to the hydroboration of the readily available allenylstannane 4226 with 25R which would provide the kinetic (Z)-bimetallic allylborane 43. This intermediate was expected to undergo an allylboration reaction with an aldehyde to provide 44a or 44b, which could react with a second aldehyde in the presence of Lewis acid. Two transition states TS-9 and TS-10 could be envisioned for the second allylation step. The synclinal transition state25d TS-9 would provide the desired 1,5-syn-(E)-diol ent-17a, ent-17c or ent-17b. Alternatively, the antiperiplanar transition state25 TS-10 would give the undesired 1,5-anti-(E)-diol ent-10a, ent-10c or ent-10b, depending on the aldehydes employed (Scheme 15).
Scheme 15.

Double Allylmetallation Sequence Involving Allenylstannane 42.
We first studied the hydroboration of 42 with the Soderquist borane 25R by using an allylboration sequence with hydrocinnamaldehyde. This provided the unstable syn-β-alkoxyallylstannane 44a with >15:1 selectivity when the hydroboration reaction was performed in dichloromethane at 0 °C for 1 h. In order to confirm the stereochemistry of 44a, we performed a subsequent allylstannation of 44a with benzaldehyde in presence of BF3•Et2O at −78 °C for 4 h, which resulted in the formation of a 4:1 mixture of the 1,5-syn-(E)-diol ent-17c and 1,5-anti-(E)-diol ent-10c in 81% yield; the enantiomeric purity of ent-17c was 94% e.e. (Mosher analysis). In an attempt to improve the diastereoselectivity, we examined the influence of the solvent on the reaction. When the hydroboration reaction of 42 was performed in Et2O, product ent-17c was obtained in only 2% yield, but with >15:1 diasteroselectivity. When the reaction was made in toluene, we obtained a 2:1 mixture of ent-17c and ent-10c in 40% yield (entry 3, Scheme 15). We also examined other Lewis Acids such as Ti(OiPr)4/S-Binol,27 Yb(OTf)3,28 ZnBr2, AgOTf/S-Binap,29 FeCl3 to improve the selectivity of the second allylstannation reaction, but without success. These experiments resulted either in no reaction, degradation or Peterson type elimination30 of the syn-β-alkoxyallylstannane 44a.
Attempts to improve the reaction diastereoselectivity by performing the second allylation at lower temperatures met with some success. When the second allylation reaction of 44a was performed at 0 °C, we obtained allylation products in 72% yield but with diminished selectivity (2:1 in favor of ent-17c, entry 1, Table 2). However, performing the second allylation reaction at −90 °C (entry 2) enabled us to obtain the 1,5-diol products in 81% yield, with slightly better selectivity (5:1). Further reductions of the reaction temperature, to −110 °C, required that we use a co-solvent such as Et2O or pentane that does not freeze at this temperature. Use of Et2O as the co-solvent led to improvement in the selectivity (11:1) but the yield was only 30% (entry 3). Use of pentane as co-solvent provided ent-17c in 66% yield and 10:1 selectivity (entry 4). Because of the difficulty maintaining the cold bath at such a low temperature for a long period, we ran the reaction in a mixture of dichloromethane and pentane (1:3) at −78 °C for 3 h and were pleased to find that the 1,5-syn-(E)-diol ent-17c was produced in 73% yield with 6:1 selectivity (entry 5). Investigations by Keck and Denmark suggest that a synclinal arrangement of the π-systems (C=O and C=C) in the transition state (comparable to TS-9) is favored as this allows stabilization of the transition state through frontier orbital interactions and minimization of charge transfer;31 thus, performing the reactions at low temperatures in non-polar solvent systems might accentuate these effects. Further reduction of the solvent polarity by increasing the ratio of pentane-dichloromethane to 4:1 at −78 °C provided 1,5-syn-(E)-diol ent-17c with 7:1 d.s. (entry 6).
Table 2.
Attempts to Optimize the Second Allylboration Reaction of 44a and 44b with Benzaldehyde or Hydrocinnamaldehyde and BF3•Et2O as per Scheme 15
| Entry | Solvent | T (°C), Time |
Product Ratioa | Yieldb |
|---|---|---|---|---|
| 1 | CH2Cl2 | 0, 4 h |
ent-17c:ent-10c (2:1) |
72% |
| 2 | CH2Cl2 | −90, 3 h |
ent-17c:ent-10c (5:1) |
81% |
| 3 | CH2Cl2/Et2O (1/2) |
−110, 3 h |
ent-17c:ent-10c (11:1) |
30% |
| 4 | CH2Cl2/Pent (1/3) |
−110, 2.5 h |
ent-17c:ent-10c (10:1) |
66% |
| 5 | CH2Cl2/Pent (1/3) |
−78, 3 h |
ent-17c:ent-10c (6:1) |
73% |
| 6 | CH2Cl2/Pent (1/4) |
−78, 4.25 h |
ent-17c:ent-10c (7:1) |
81% |
| 7 | CH2Cl2/Pent (1/4) |
−78, 4.25 h |
ent-17a:ent-10a (7:1) |
57% |
| 8 | CH2Cl2/Pent (1/4) |
−78, 4.25 h |
ent-17b:ent-10b (4:1) |
51% (93% e.e.)c |
Determined by 1H NMR analysis.
Isolated yields.
determined by Mosher ester analysis.
Similar diastereoselectivity but a lower yield (57%) was obtained when hydrocinnamaldehyde was used instead of benzaldehyde (compound ent-17a and ent-10a; entry 7). However, when benzaldehyde was used for first allylboration, followed by use of hydrocinnamaldehyde in the second allylstannation step, using the same conditions described as above, a 51% yield of a 4:1 mixture of 1,5-syn-(E)-diol ent-17b and 1,5-anti-(E)-diol ent-10b was obtained favoring of ent-17b (93% e.e.; entry 8). While these results were encouraging, we sought a significantly more stereoselective method for synthesis of the targeted 1,5-syn-(E)-diols.
3.7. Development of the Double Allylborating Agent 46 Starting from Tetrabutylammonium Allenyltrifluoroborate 45
We were intrigued by several publications from Batey’s laboratory, who reported allylation and crotylation of aldehydes using potassium allyl and crotyltrifluoroborates.32 These data stimulated the idea that it should be possible to generate a double allylborating reagent, specifically the (Z)-γ-boryl allyltrifluoroborate 46, via the kinetically controlled hydroboration of tetrabutylammonium allenyltrifluoroborate 45 with the Soderquist borane, 25S (Scheme 16). Based on the Batey precedent, it was expected that allylic trifluoroborate 47 could be converted to allylboron difluoride 48 upon treatment with an appropriate Lewis acid. Further, and most importantly, it was anticipated that 48 would undergo aldehyde allylboration reactions with much greater stereoselectivity than with 32 previously studied (Scheme 8), owing to the smaller size of the difluoroborane unit of 48 compared to the (1,3,2)-dioxaborinane unit in 32 (e.g., compare TS-11 for 48 with TS-5 for 32).
Scheme 16.

Synthesis of 1,5-syn-(E)-Diols 17 from Allenyltrifluoroborate 45.
Studies of the hydroboration of 45 commenced with the experiments summarized in Table 3. Initially, the hydroboration of allene 45 (1.3 equiv) was performed with the Soderquist borane, 25S, at 0 °C for 1 h. The solution was cooled to −78 °C and then benzaldehyde (0.7 equiv) was added. The reaction was maintained at −78 °C for 4 h, then was worked up oxidatively. This provided 1,2-syn-diol ent-30 as a 5.7:1 mixture of diastereomers, along with the unexpected (E)-1,5-syn-diol 17d (Table 3, entry 1). The formation of 17d suggested that allylboron difluoride 48 was forming during the reaction.
Table 3.
Hydroboration of Allene 45 and Allylboration Reactions of the Derived Reagent 46
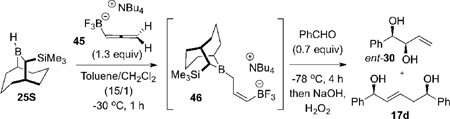 | ||||
|---|---|---|---|---|
| entry | solvent | hydroboration temp / time |
% yield of ent-30a (syn/anti)b |
ratio ent- 30:17dc |
| 1 | CH2Cl2 | 0 °C / 1 h | 39 (5.7:1) | 3.0:1 |
| 2 | CH2Cl2 | −10 °C / 1 h | 38 (16:1) | 2.9:1 |
| 3d | CH2Cl2 | −10 °C / 1 h | 39 (12:1) | 3.3:1 |
| 4 | CH2Cl2 | −10 °C / 3 h | 41 (5.1:1) | 3.2:1 |
| 5 | CH2Cl2 | −30 °C / 1 h | 52 (> 20:1) | 3.7:1 |
| 6 | CH2Cl2 | −30 °C / 3 h | 52 (> 20:1) | 4.3:1 |
| 7 | Et2O/CH2Cl2e | −30 °C / 1 h | 50 (> 20:1) | 7.1:1 |
| 8 | toluene/THFf | −30 °C / 1 h | 51 (> 20:1) | >30:1 |
| 9 | toluene/CH2Cl2g | −30 °C / 1 h | 87h (> 20:1) | >30:1 |
Isolated yields.
Determined by 1H NMR analysis.
Determined after isolation of ent-30 and 17d.
Dibal-H (2 equiv) added before the oxidation step.
Et2O/CH2Cl2 (2:1).
Toluene/THF (2:1).
Toluene/CH2Cl2 (15:1).
ent-30 obtained in 97% e.e., determined by Mosher ester analysis.
A significant improvement of the product diastereoselectivity (16:1) was realized by performing the hydroboration at −10 °C for 1 h (entry 2), suggesting that the rate of boratropic isomerization of 46 can be controlled by keeping the reaction temperature below −10 °C. However, ent-30 and 17d were still formed in ca. 3:1 ratio. Attempts to reduce any residual benzaldehyde, by addition of DIBAL prior to the oxidative workup did not eliminate 17d as a reaction product (entry 3), suggesting that 17d is formed during the allylboration reaction at −78 °C and not during workup. The best diastereoselectivity for ent-30 (> 20:1) was obtained from experiments in which the hydroboration reaction was run at −30 °C (entries 5, 6), which suggested that under these conditions the [1,3]-boratropic isomerization of 46 to the corresponding (E)-isomer is essentially suppressed. Examination of the reaction in other solvents led to the discovery that the competitive abstraction of fluoride ion from 47 to give 48 (the precursor to 17d) could also be suppressed (entries 7, 8, and 9). Best results were obtained (entry 9) when the hydroboration/allylboration sequence was performed in a 15:1 mixture of toluene and CH2Cl2 (CH2Cl2 is needed owing to the poor solubility properties of 45 in toluene). Under these conditions, the 1,2-syn-diol ent-30 was obtained in 87% yield, with 97% e.e., and >20:1 d.r. after oxidative workup. These data demonstrate that 46 is obtained with very high stereoselectivity via the kinetically controlled hydroboration of 45, and also that the first allylboration reaction provides the intermediate β-alkoxyallyltrifluroborate 47 with excellent stereochemical control.
Next, we applied these optimized reaction conditions to the double allylboration reaction leading to the targeted 1,5-syn-(E)-diols 17 (Table 4). This next phase of the reaction development was facilitated by addition of BF3•OEt2 to the intermediate β-alkoxyallyltrifluroborate 47.33 This smoothly generated the allylic boron difluoride intermediate 48, which reacted with the second aldehyde to give the targeted 1,5-syn diol 17. The amount of the aldehyde used in the first allylboration reaction proved to be important for avoiding the homo-coupling double allylboration products 17d (deriving from the first aldehyde) and 17a (deriving from the second aldehyde). Optimized reaction conditions (Table 4, entry 2) were found by using 1.3 equiv of the allene 45, 0.85 equiv of the first aldehyde, 1.5 equiv of BF3•Et2O and 1.2 equiv of second aldehyde. This combination provided the 1,5-syn-diol-(E) 17 in 73% yield, with excellent enantioselectivity (>95% e.e.), diastereoselectivity (d.r. >20:1), and (E)/(Z) ratio (>20:1).
Table 4.
Optimization of the Double Allylboration Reaction of 46 Generated by the Hydroboration of Allene 45a
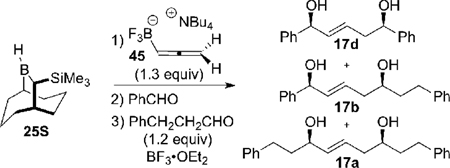 | |||||
|---|---|---|---|---|---|
| entry | PhCHO (equiv) |
BF3•OEt2 (equiv) |
% yield 17bb | Ratio (E)/(Z)c |
ratio 17d/17b/17ac |
| 1 | 0.75 | 1.5 | 60 | >20:1 | 0:89:11 |
| 2 | 0.85 | 1.5 | 73 | >20:1 | 0:100d:0 |
| 3 | 0.95 | 1.5 | 51 | >20:1 | 8:92:0 |
| 4e | 0.85 | 1.5 | 30 | 6:1 | 0:100:0 |
| 5f | 0.85 | - | 77 | 6:1 | 0:100:0 |
Reaction conditions: solvent: toluene/CH2Cl2; hydroboration: −30 °C, 1 h; first allylboration: −78 °C, 4 h; second allylboration: −78 °C, 4 h; workup: pH 7 buffer (KH2PO4/NaOH).
Isolated yields.
Determined by 1H NMR analysis.
17b obtained with > 20:1 d.r. and 97% ee, determined by Mosher ester analysis.
Second allylboration: 0 °C, 2 h.
Second allylboration: −78 to 20 °C, 12 h.
By using these optimized reaction conditions (Table 4, entry 2), a series of double allylboration reactions were performed to demonstrate the scope and utility of this new procedure (see Scheme 17). These results demonstrate that the long-sought 1,5-syn-(E)-diols 17 are obtained in 72–98% yields, >95% e.e., >20:1 d.r., and with >20:1 (E)/(Z) selectivity. Thus, use of the allenyltrifluoroborate 45 in combination with the Soderquist borane provides an efficient, highly stereoselective synthesis of the 1,5-syn-(E)-diols illustrated by the structures in Scheme 17.
Scheme 17.

1,5-Syn-(E)-Diols Synthesized By Using the Optimal Reaction Conditions.a
a All experiments were performed by using the conditions defined in entry 2 of Table 4.
3.8. Application of the New Double Allylboration Procedure to the Synthesis of the C(23)-C(40) Fragment of Tetrafibricin
The original objective of this methodology development project was the synthesis of a double allylboration reagent that would facilitate the stereoselective synthesis of the C(23)-C(40) fragment of tetrafibricin. As we presented in Scheme 1, we envisioned that this fragment could be assembled from aldehydes 3 and 4 by a double allylboration sequence. The synthesis of aldehyde 3 started from the known 4-azidobutanal34 49 which was subjected to an allylboration reaction with Brown’s (−)-di(isopinocampheyl)allylborane reagent.35 This provided secondary alcohol 50 as an inseparable mixture with isopinocampheol, 51. This mixture was treated with TBS-Cl and imidazole which provided the TBS ether 51 in 73% yield over the two steps. The enantiomeric purity of alcohol 50 was determined to be 91% e.e., by application of the Mosher ester analysis.36 This analysis also enabled us to confirm the absolute stereochemistry of 50. Reduction of the azide by treatment with Bu3P in wet diethyl ether, followed by protection of the primary amine as a tert-butyl carbamate (Boc) provided 53 in 70% yield.37 The cross olefin metathesis38 of 53 with acrolein and using the second generation Grubbs-Hoveyda catalyst39 gave aldehyde 3 in 94% yield ((E)/(Z) = 25:1). Aldehyde 3 corresponds to the C(33)-C(40) fragment of tetrafibricin.
The synthesis of aldehyde 4 began with cross-metathesis between readily available olefin 5440 and acrolein in the presence of Grubbs-Hoveyda second-generation catalyst.39 This reaction provided the unsaturated aldehyde 55 in 85% yield. Subjection of 55 to a Brown allylboration35 using (−)-di(isopinocampheyl)-allylborane afforded alcohol 56 in 83% yield and 96% e.e. as verified by Mosher ester analysis.36 Asymmetric epoxidation of alcohol 56 under the standard Sharpless epoxidation conditions41 delivered epoxy alcohol 57 in 69% yield as a single diastereomer. Reduction of the epoxide using Red-Al in the presence of MeOH42 provided anti-1,3-diol 58, the stereochemistry of which was verified by 13C NMR analysis of the derived acetonide according to Rychnovsky’s method.43 Protection of the diol in 58 with TBS-Cl and imidazole gave 59. Finally oxidative cleavage of the vinyl group of 59 by using Jin’s protocol (OsO4/NaIO4/2,6-lutidine)44 provided aldehyde 4, which corresponds to the C(23)-C(29) fragment of tetrafibricin (Scheme 19).
Scheme 19.

Synthesis of Aldehyde 4
Studies performed on the coupling of aldehyde fragments 3 and 4 to give the targeted tetrafibricin intermediate 2 are summarized in Scheme 20. Initial studies were performed using reagent 28 generated from allene 6 as described in Scheme 7. Use of 28 in this double allyboration sequence (using the conditions defined in Scheme 8) provided 60 in 67% yield, but as an inseparable 6:1 mixture of diastereomers (entry 1, Scheme 20). When reagent 43, generated from allene 42 as described in Scheme 15 was used, the double allylmetalation product 60 was obtained in 24% yield as an inseparable 4:1 mixture of diastereomers (entry 2, Scheme 20).
Scheme 20.

Synthesis of the C(23)-C(40) Fragment 2 of tetrafibricin.
Fortunately, excellent results were obtained when the double allylboration coupling of aldehydes 4 and 3 was performed using reagent 46 generated from allenyltrifluoroborate 45 (entry 3, Scheme 20). This reaction provided 60 in 83% yield, with exceptional diastereoselectivity (>20:1) and (E)/(Z) ratio (>20:1). The stereochemistry of the two hydroxyl groups in 45 were confirmed by using the Mosher method.36 This result demonstrates that the fragment assembly reaction using reagent 46 is compatible with protecting groups such as silyl ethers, PMB ethers, and acid labile Boc carbamates. Treatment of 60 with TBS-Cl and imidazole furnished the tetrafibricin C(23)-C(40) fragment 2 in 94% yield. Furthermore, replacement of 25R by 25S in the hydroboration reaction of allene 45 allowed the diastereomeric 1,5-diol 62 to be obtained in comparable yield and stereoselectivity (Scheme 21).
Scheme 21.

Synthesis of the diastereomer 62
In comparison with other methods2–7 described in the literature for synthesis of 1,5-diol units, our double allylboration procedure is highly convergent and has a high degree of stereochemical generality. In fact, the synthesis of several different diastereoisomers of a 1,5-diol system can now be achieved simply by changing the nature of the borane reagent (10-TMS-9-BBD-H or Ipc2BH) or the monosubstituted allene used to synthesize the double allylboration reagent (e.g., see Schemes 2, 6, 17, 20 and 21). Thus, we expect that our method will prove useful for the synthesis and confirmation of the relative and absolute stereochemistry of 1,5-diol units found in other natural products.
4. Conclusion
In summary, we have developed an efficient and highly diastereoselective double allyboration reaction sequence starting from the hydroboration of tetrabutylammonium allenyltrifluoroborate 45 and the Soderquist borane, TBBD-H 25. This method has enabled us to synthesize the C(23)-C(40) fragment 2 of tetrafibrincin and its C(29)-C(33) diastereoisomer 62 with very high stereoselectivity. Further progress toward the completion of the total synthesis of tetrafibricin 1 will be reported in due course.
5. Experimental Section
Tetrahydrofuran, dichloromethane, diethyl ether and toluene were purified by passing through a solvent column of activated alumina (A-1). Hexanes, acetonitrile and trimethylsilyl chloride were purified by distillation from calcium hydride. Anhydrous pentane and methanol were purchased from Aldrich Chemical Company. All commercially available aldehydes were purified by vacuum distillation before use. Unless indicated otherwise, other commercially available reagents were used as received without purification. Unless otherwise indicated, all reactions were conducted under an atmosphere of argon using flamed-dried or oven-dried (140 °C) glassware. Standard techniques for handling air-sensitive compounds were also employed for all operations. Celite 545® was dried at 140 °C for at least 12 h prior to use. Removal of solvents was accomplished on a rotary evaporator at reduced pressure. Reaction mixtures were maintained at low temperature by using a Thermo NESLAB CB-60 cold bath. Enantiomeric excess and absolute configurations of all new alcohol products were determined by using the Mosher36 method.
1H NMR spectra were recorded on a Bruker spectrometer at 400 MHz. 13C NMR spectra were recorded on a Bruker spectrometer at 100 MHz. The proton signal for non-deuterated solvent (δ 7.24 ppm for CHCl3) was used as an internal reference for 1H NMR spectra. For 13C NMR spectra, chemical shifts are reported relative to the δ 77.24 ppm resonance of CDCl3. Infrared (IR) spectra were recorded as thin films using CH2Cl2 as the solvent or neat on a Perkin-Elmer Spectrum 1000 FTIR. Optical rotations were measured on a Rudolph Autopol IV polarimeter using a quartz cell with 1 mL capacity and a 1 dm path length. Melting points were determined on a Mel-Temp II hot stage melting point apparatus and are uncorrected. High-resolution mass spectra were recorded on a spectrometer at the University of Florida Mass Spectrometry Laboratory.
Analytical thin layer chromatography (TLC) was performed on Kieselgel 60 F254 glass plates precoated with a 0.25 mm thickness of silica gel. TLC plates were visualized with UV light and/or by staining with cerium molybdate (5g Ce(SO4)2, 25g (NH4)Mo7O24.4H2O, 450 mL H2O, 50 mL H2SO4). Preparative thin layer chromatography (TLC) was performed on Kieselgel 60 F254 glass plates precoated with a 0.5 mm thickness of silica gel. Column chromatography was generally performed according to the method of Still45 using Kieselgel 60 (230–400 mesh) silica gel.
Procedure for the preparation of the allenylboronic esters (6, 20 and ent-39)
Freshly distilled trimethyl borate (21.7 mL, 196 mmol) was added to a round bottom flask containing 150 mL of ether placed in a −78 °C cold bath. A 0 °C solution of allenylmagnesium bromide (1 M in ether, 196 mmol) was added via syringe or canula at a rate as to keep the internal reaction temperature below −65 °C. A white precipitate formed during this addition. The reaction mixture was stirred 3 h at −78 °C, then was warmed to 0 °C over 1 h and stirred at 0 °C for an additional hour. Aqueous HCl (2M, 392 mmol) was added via an addition funnel. This mixture was stirred for 1 h and then the organic layer was separated using a separatory funnel. The aqueous layer was washed with ether (2 × 200 mL) and the combined organic phases were dried over Na2SO4 for 30 min. The solution was filtered under nitrogen and solvent removed in vacuo until ~250 mL of solvent remained (the rotary evaporator was flushed and backfilled using an argon balloon). The appropriate diol (137 mmol) was added to the flask and reaction stirred for 24 h at room temperature, at which time 10 g of MgSO4 was added and the mixture stirred for an additional 18 h at room temperature. The reaction was filtered under nitrogen using a coarse fritted funnel and solvent removed in vacuo using argon to backfill the rotary evaporator.
2-Allenyl-1,3,2-dioxaborinane (6)
The remaining clear yellow oil was distilled under vacuum (bp 20 mm Hg, 81–82°C) to yield 76% of 6 as a clear, colorless oil. The oil was transferred to small vials and stored under nitrogen in a dessicator in a −20 °C freezer. This material could be used neat over several months without decomposition. Spectroscopic data obtained for 6 matched that reported in the literature.8a
2-Allenyl-5,5-dimethyl-1,3,2-dioxaborinane (20)
The remaining clear yellow oil was distilled under vacuum (bp 46–48 °C/0.5 mbar) to yield 69% of 20 as a clear, colorless oil. The oil was transferred to small vials and stored under nitrogen within a dessicator in a −20 °C freezer and could be used neat over several months without decomposition: IR (neat) 2963, 2935, 2889, 1936, 1479, 1426, 1379, 1350, 1331, 1259, 1227, 1184, 1088, 1009, 870, 814, 681, 666 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.83 (t, J = 6.9 Hz, 1H), 4.61 (d, J = 7.0 Hz, 2H), 3.66 (s, 4H), 0.98 (s, 6H); 13C (100 MHz, CDCl3) δ 217.9, 72.5, 69.7, 31.8, 21.8; HRMS (ESI) calc. for C8H13BO2 (M+Na+) 175.0906, found 175.0908.
Pinanediol allenylboronate (ent-39)
Purified by flash column chromatography (96/4 : hexanes/EtOAc); [α]D26 = +10.4 (c 0.52, CHCl3); IR (neat) 2971, 2918, 1937, 1423, 1368, 1203, 1189 cm−1; 1H (400 MHz, CDCl3) δ 4.94 (t, J = 6.9 Hz, 1H), 4.65 (d, J = 6.9 Hz, 2H), 4.34 (dd, J = 8.7, 2.0 Hz, 1H), 2.39-2.30 (m, 1H), 2.29-2.17 (m, 1H), 2.64-2.54 (dd, J = 6.0, 5.0 Hz, 1H), 1.94-1.87 (m, 2H), 1.42 (s, 3H), 1.29 (s, 3H), 1.17 (d, J = 11.0 Hz, 1H), 0.85 (s, 3H); 13C (100 MHz, CDCl3) δ 219.2, 86.4, 78.4, 70.2, 51.5, 39.7, 38.4, 35.6, 28.8, 27.3, 26.7, 24.2; HRMS (ESI) calcd for C13H19BO2 [M+Na]+ 241.1376, found 241.1380.
N,N,N-tributylbutan-1-aminium trifluoro(propa-1,2-dien-1-yl)borate (45)
An established procedure46 for synthesis of allylic trifluoroborate tetrabutylammonium salts was modified for preparation of allene 8. A 1 M aqueous solution of tetrabutylammonium hydroxide (20.0 mL, 20.0 mmol, 1 equiv) was added to a suspension of the known potassium allenyl trifluoroborate47 (2.92 g, 20.0 mmol, 1 equiv) in CH2Cl2 (30 mL). The reaction was stirred at room for 30 min. The reaction mixture was then diluted with CH2Cl2 (20 mL), the organic layer was separated, dried over Mg2SO4, filtrated, and concentrated in vacuo to afford tetrabutylammonium allenyl trifluoroborate 45 (5.81g, 83%) as a hygroscopic colourless solid which was stored in the glove box. IR (neat) 2963, 2936, 2876, 1930, 1473, 1382, 1248, 1071, 996, 968, 882, 795, 739, 600 cm−1; 1H (400 MHz, CDCl3) δ 4.76 (bs, 1H), 4.09-4.06 (m, 2H), 3.18-3.14 (m, 8H), 1.61-1.53 (m, 8H), 1.38 (sextuplet, J = 7.3 Hz, 8H), 0.95 (t, J = 7.3 Hz, 12H); 13C (100 MHz, CDCl3) δ 210.6 (q, J = 4.3 Hz), 86.8 (bs), 65.6, 58.6, 24.0, 19.8, 13.8; 19F (376 MHz, CDCl3) δ −137.1 (d, J = 48 Hz); HRMS (ESI) calcd for C3H3BF3 [M-NBu4]− 107.0280, found 107.0286.
Lithium B-H2-(10R)-trimethylsilyl-9-borabicyclo[3.3.2] decane diethyl etherate (26)
Soderquist’s literature procedure was modified as follows: a solution of ethyl acetate (0.488 mL, 5.00 mmol, 0.5 equiv) in Et2O (4.5 mL) was added dropwise (0.5 mL/min) to a 1 M solution of LiAlH4 in Et2O (10.0 mL, 10.0 mmol, 1 equiv). The resulting suspension of lithium aluminium monoethoxyaluminium hydride was stirred at 20 °C for 1 h (white precipitate). The reaction mixture was cooled to 0 °C and a solution of (−)-B-methoxy-10R-trimethylsilyl-9-borabicyclo[3.3.2.]decane (2.38 g, 10.0 mmol, 1 equiv) 64R21 in Et2O (20 mL) was added dropwise (1 mL/min). The reaction was stirred at 0 °C for 1 h then was allowed to reach room temperature. The suspension was filtered (fritted funnel with Celite) with care taken to prevent exposure of the borohydride to the open atmosphere. The solid dialkoxyalane was washed with Et2O (3 × 10 mL). The combined washings and supernatant were concentrated in vacuo to afford mono-diethyl etherate 26 as a white solid which was dried under high vacuum for at least 12 h. The known borohydride 26 can be stored without decomposition in a glove box. 1H (400 MHz, CDCl3) δ 3.67 (q, J = 7.1 Hz, 4H), 2.37 (s, 1H), 1.66-1.37 (m, 13H), 1.29 (t, J = 7.1 Hz, 6H), 0.91 (s, 1H), 0.45 (bs, 1H), 0.00 (s, 1H), −0.07 s, 9H); 13C (100 MHz, CDCl3) δ 66.5, 37.3, 35.5, 34.8, 33.3, 33.0, 24.2, 23.4, 14.8, −0.1.
Procedure for titration of the borohydride 26
To a solution of borohydride 26 (0.073 g, 0.250 mmol, 1 equiv) in Et2O (1.0 mL) at 0 °C was added TMSCl (0.048 mL, 0.375 mmol, 1.5 equiv). The reaction mixture was stirred at 0 °C for 10 min before 4-methoxybenzaldehyde (0.121 mL, 1.0 mmol, 4 equiv) was added. The reaction mixture was stirred at 20 °C for 3 h then acetic acid (0.286 mL, 5.00 mmol, 20 equiv) was added and the mixture was stirred for an additional 4 h. The solvent was removed under reduced pressure (rotary evaporator) and the residue was analyzed by 1H NMR spectroscopy. The yield of in situ generated 25 was determined from the integrations of the methoxy groups of 4-methoxybenzyl alcohol (Int1) and 4-methoxybenzaldehyde (Int2). The titration reaction was run simultaneously in two different flasks. The average yields were generally in the range of 90–93%.
Procedure A. Synthesis of 1,5-diols ent-17a and ent-10a from the hydroboration reaction of allene 20 with lIpc2BH
lIpc2BH (287 mg, 1.0 mmol) was weighed in a glove box into a round bottom flask containing a stir bar (Note: The crystalline Ipc2BH should be crushed and pulverized with a glass rod prior to use in order to ensure efficient hydroboration). The flask was capped with a rubber septum and removed from the glove box and placed in bath at the appropriate reaction temperature (Table 1). THF (2 mL) was added to the flask followed by the allenyl boronic ester 20 (148 mg, 1.0 mmol) as a solution in THF (1 mL). For hydroboration reaction time and temperatures, see Table 1. After the hydroboration reaction was terminated, the reaction mixture was cooled to −78 °C. Distilled hydrocinnamaldehyde (71 µL, 0.54 mmol) was added dropwise via a microliter syringe. The flask was stirred at −78 °C for 4 h and then additional hydrocinnamaldehyde (184 µL, 1.40 mmol) was added dropwise via microliter syringe. The reaction was stirred an additional 2 h. The flask was then removed from the −78 °C bath and allowed to warm to room temperature and stirred overnight. The reaction mixture was cooled in an ice bath and 1 mL of 3M NaOH solution was added dropwise followed by 0.4 mL of a 50% H2O2 solution. This mixture was stirred for 4 h at room temperature at which time it was diluted with 40 mL of CH2Cl2 and 20 mL of a saturated aqueous solution of NaHCO3 and 5 mL of a saturated aqueous solution of NaCl. The organic layer was separated and the aqueous washed with CH2Cl2 (2 × 10 mL). The combined organic extracts were dried over Na2SO4, then filtered and the solvent removed in vacuo. The crude products were then purified by flash chromatography.
Procedure B. Synthesis of 1,2-diols 30 and 40 from allenes 6, 33, 39 and ent-39
To a 0°C solution of borohydride 25R21 (81 mg, 0.250 mmol, 1 equiv, considering a 90% yield for the conversion of borohydride 26R to borane 25R based on the borane 25R titration8d) in CH2Cl2 (0.5 mL) was added TMSCl (32 µL, 0.250 mmol, 1 equiv). The reaction mixture was stirred for 10 min, then was cooled to −78°C and a solution of allene 6, 33, or 39 (0.325 mmol, 1.3 equiv) in CH2Cl2 (1.0 mL) was added dropwise. For hydroboration reaction times and temperatures, see Schemes 7, 10, and 13. The resulting reaction mixture was then cooled to −78 °C and a solution of benzaldehyde or hydrocinnamaldehyde (0.175 mmol, 0.7 equiv) in CH2Cl2 (0.3 mL) was added. The mixture was stirred for 4 h at −78 °C, then an aqueous 3 N NaOH solution (0.25 mL) and aqueous 50% H2O2 (0.1 mL) were added follow by THF (1 mL) and MeOH (0.5 mL). This mixture was then stirred at 20°C for 12 h. The biphasic mixture was poured into saturated aqueous NH4Cl, and the crude product was extracted with ethyl acetate (3×). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude products were then purified by flash chromatography.
Procedure C. Synthesis of 1,5-diols ent-17a, ent-17b, ent-19g from allenes 6, 33, and 39
To a 0 °C solution of borohydride 25R (81.0 mg, 0.250 mmol, 1 equiv, considering a 90% yield for the conversion of borohydride 26R to borane 25R based on the borane 25R titration8d) in CH2Cl2 (0.5 mL) was added TMSCl (32 µL, 0.250 mmol, 1 equiv). The reaction mixture was stirred for 10 min, then was cooled to −78 °C and a solution of allene 6, 33, or 39 (0.325 mmol, 1.3 equiv) in CH2Cl2 (1.0 mL) was added dropwise. For hydroboration reaction times and temperature, see Schemes 8, 11, 14 for optimal conditions. The resulting clear reaction mixture was then cooled to −78 °C and benzaldehyde or hydrocinnamaldehyde (0.175 mmol, 0.7 equiv) was added. The mixture was stirred at −78 °C for 4h, then a second aldehyde (0.375 mmol, 1.5 equiv) (followed by BF3•OEt2 (0.125 mmol, 0.5 equiv.) if using allene 39 was added and the reaction mixture was stirred for an additional 4 h. The reactions were quenched by using one of three general methods: (1) addition of ethanolamine (75 µL, 1.25 mmol, 5 equiv); (2) addition of aqueous 3 N NaOH solution (0.25 mL) and aqueous 50% H2O2 (0.1 mL); or (3) by addition of a pH 7 buffer solution (KH2PO4/NaOH) (1 mL) followed by THF (1.5 mL) and MeOH (0.5 mL). In the latter procedure, additional THF (1 mL) and MeOH (0.5 mL) were added, and the mixture was stirred at 20 °C for 12 h. The reaction mixture was then poured into saturated aqueous NH4Cl, and the crude product was extracted with ethyl acetate (3×). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude products were then purified by flash chromatography.
Procedure D. Synthesis of 1,5-diols ent-17a, ent-17b, ent-17c from allene 42
To a 0°C solution of borohydride 25R (81 mg, 0.250 mmol, 1 equiv, considering a 90% yield for the conversion of borohydride 26R to borane 25R based on the borane 25R titration8d) in CH2Cl2 (0.5 mL) was added TMSCl (32.0 µL, 0.250 mmol, 1 equiv). The reaction mixture was stirred for 10 min, then was cooled to 0 °C. At this point, a solution of allenyltributylstannane 42 (80% purity from Sigma-Aldrich, 121 µL, 0.325 mmol, 1.3 equiv) in CH2Cl2 (1.0 mL) was added and the reaction mixture was stirred for 1 h. The mixture was cooled to −78°C and hydrocinnamaldehyde or benzaldehyde (0.175 mmol, 0.7 equiv) was added. After 3 h, the reaction was diluted with pentane (see Table 2) and a second aldehyde (0.325 mmol, 1.3 equiv) was added followed by BF3•OEt2 (46 µL, 0.375 mmol, 1.5 equiv) and the reaction was stirred for an additional 4.25 h. Ethanolamine (75 µL, 1.25 mmol, 5 equiv) was added and the reaction mixture was allowed to reach 20 °C and stirred for 12 h. The mixture was poured into saturated aqueous NH4Cl solution (20 mL) and the crude product was extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with brine (20 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. The crude products were then purified by flash chromatography.
Procedure E. Synthesis of 1,5-diols 17 and ent-17 from allene 45
To a 0 °C solution of borohydride 25 (81.0 mg, 0.250 mmol, 1 equiv, considering a 90% yield for the conversion of borohydride 26 to borane 25 based on the borane 25 titration8d) in toluene (0.5 mL) was added TMSCl (32.0 µL, 0.250 mmol, 1 equiv). The reaction mixture was stirred for 10 min, then was cooled to −78 °C and a solution of allene 45 (0.114 g, 0.325 mmol, 1.3 equiv) in toluene (1 mL) / CH2Cl2 (0.1 mL) was added dropwise. The viscous mixture was stirred at −30 °C for 1 h. The resulting clear reaction mixture was then cooled to −78 °C and a solution of the first aldehyde (0.213 mmol, 0.85 equiv) in CH2Cl2 (0.25 mL) was added. After 4h, a solution of the second aldehyde (0.300 mmol, 1.2 equiv) in CH2Cl2 (0.25 mL) followed by BF3•OEt2 (47.0 µL, 0.375 mmol, 1.5 equiv) were added. The reaction mixture was stirred for an additional 4 h at −78 °C. A pH 7 buffer solution (KH2PO4/NaOH) (1 mL) followed by THF (1.5 mL) and MeOH (0.5 mL) were then added to the mixture, which was stirred at 20°C for 24 h. The biphasic mixture was poured into saturated aqueous NH4Cl solution, and the crude product was extracted with ethyl acetate (3×). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude products were then purified by flash chromatography.
(3R,4E,7S)-1,9-diphenylnon-4-ene-3,7-diol (17a)
[α]D25 = −6.4 (c 0.64, CHCl3); e.e. 96%; IR (neat) 3347, 3084, 3061, 3025, 2924, 2857, 1668, 1602, 1495, 1454, 1312, 1051, 1030, 971, 923, 746, 698 cm−1; 1H (400 MHz, CDCl3) δ 7.35-7.18 (m, 10H), 5.72-5.57 (m, 2H), 4.11 (q, J = 6.8 Hz, 1H), 3.72-3.62 (m, 1H), 2.85-2.65 (m, 4H), 2.34-2.13 (m, 2H), 1.84-1.67 (m, 4H); 13C (100 MHz, CDCl3) δ 142.0, 141.9, 136.5, 128.45, 128.41, 127.6, 125.88, 125.87, 72.1, 70.3, 40.5, 38.8, 38.6, 32.0, 31.8; HRMS (ESI) calcd for C21H26O2Na [M+Na]+ 333.1831, found 333.1825.
(3S,4E,7R)-1,9-diphenylnon-4-ene-3,7-diol (ent-17a)
[α]D25 +6.4 (c 0.64, CHCl3); 1H NMR, 13C NMR and IR and HRMS data were identical to 17a.
(3S, 4E, 7S)-1,9-diphenylnon-4-ene-3,7-diol (ent-10a)
[α]D24 = −12.8 (c 1.0, CHCl3); IR (thin film) 3350, 3026, 2928, 1603, 1495, 1454, 1051 cm−1; 1H (500 MHz, CDCl3) δ 7.35-7.18 (m, 10H), 5.70 (dt, J = 15.0, 7.0, 7.0 Hz, 1H), 5.63 (dd, J = 15.0, 6.0 Hz, 1 H), 4.11 (q, J = 6.8 Hz, 1H), 3.72-3.62 (m, 1H), 2.85-2.65 (m, 4H), 2.34-2.13 (m, 2H), 1.84-1.67 (m, 4H); 13C (125 MHz, CDCl3) δ 142.1, 142.0, 136.7, 128.45, 128.41, 127.5, 125.89, 125.87, 72.0, 70.3, 40.3, 38.8, 38.5, 32.1, 31.8; HRMS (CI, NH3) calcd for C21H30NO2 [M + NH4]+ 328.2277, found 328.2275.
(1S,2S)-1-Phenylbut-3-ene-1,2-diol (30)48
[α]D23 = +13.6 (c 1.17, CHCl3); IR (neat) 3391, 2919, 1710, 1453, 1197, 1125, 995, 928, 762, 700 cm−1; 1H (400 MHz, CDCl3) δ 7.34-7.25 (m, 5H), 5.68 (ddd, J = 17.4, 10.6, 5.6 Hz, 1H), 5.20 (dt, J = 17.2, 1.5 Hz, 1H), 5.10 (dt, J = 10.6, 1.5 Hz, 1H), 4.44 (d, J = 7.1 Hz, 1H), 4.19-4.15 (m, 1H), 2.98 (bs, 2H); 13C (100 MHz, CDCl3) δ 140.4, 136.5, 128.5, 128.3, 127.2, 117.2, 77.8, 77.1; HRMS (ESI) calcd for C10H12NaO2 [M+Na]+ 187.0735, found 187.0730.
(1R,2R)-1-phenylbut-3-ene-1,2-diol (ent-30)
[α]D25 = −4.2 (c 0.66, CHCl3); e.e. 97%; 1H NMR, 13C NMR and IR and HRMS data were identical to 30.
(1S,2E,5R)-1,5-diphenylpent-2-ene-1,5-diol (17d)
[α]D25 = +49.6 (c 0.23, CHCl3); e.e. 98%; IR (neat) 3360, 3085, 3061, 3029, 2921, 1667, 1602, 1493, 1452, 1318, 1197, 1028, 1007, 969, 913, 758, 699 cm−1; 1H (400 MHz, CDCl3) δ 7.27-7.17 (m, 10H), 5.69-5.66 (m, 2H), 5.08-5.06 (m, 1H), 4.63 (t, J = 6.6 Hz, 1H), 2.56 (bs, 2H), 2.42-2.39 (m, 2H); 13C (100 MHz, CDCl3) δ 144.1, 143.1, 136.3, 128.7, 128.1, 127.8, 127.7, 126.4, 126.0, 75.1, 73.8, 42.3; HRMS (ESI) calcd for C17H18O2Na [M+Na]+ 277.1204, found 227.1205.
(1S,2E,5S)-1,7-diphenylhept-2-ene-1,5-diol (17b)
[α]D25 = +2.7 (c 1.28, CHCl3); e.e. 97%; IR (neat) 3360, 3084, 3061, 3026, 2923, 2854, 1602, 1494, 1453, 1319, 1069, 1048, 1030, 969, 748, 698 cm−1; 1H (400 MHz, CDCl3) δ 7.33-7.28 (m, 4H), 7.25-7.21 (m, 3H), 7.15-7.12 (m, 3H), 5.74-5.70 (m, 2H), 5.13 (d, J = 4.8 Hz, 1H), 3.66-3.60 (m, 1H), 2.77-2.70 (m, 1H), 2.66-2.58 (m, 1H), 2.28-2.22 (m, 1H), 2.17-2.09 (m, 1H), 1.75-1.70 (m, 2H); 13C (100 MHz, CDCl3) δ 143.2, 142.2, 136.1, 128.8, 128.63, 128.61, 128.1, 127.9, 126.4, 126.0, 75.2, 70.5, 40.6, 38.8, 32.2; HRMS (ESI) calcd for C19H22NaO2 [M+Na]+ 305.1517, found 305.1515.
(1R,5R,2E)-1,7-diphenylhept-2-ene-1,5-diol (ent-17b)
[α]D23 = −14.1 (c 0.70, CHCl3); 1H NMR, 13C NMR and IR and HRMS data were identical to 17b.
(1R, 5S, 2E)-1,7-diphenylhept-2-ene-1,5-diol (ent-10b)
[α]D24 = −16.52 (c 0.96, CHCl3); IR (neat) 3351, 3026, 2929, 1493, 1453 cm−1; 1H (400 MHz, CDCl3) δ 7.38-7.10 (m, 10H), 5.78-5.68 (m, 2H), 5.12 (d, J = 4.8 Hz, 1H), 3.68-3.58 (m, 1H), 3.20 (d, J = 3.2 Hz, 1H); 2.78-2.69 (m, 1H), 2.66-2.56 (m, 1H), 2.48 (d, J = 4.4 Hz, 1H), 2.28-2.21 (m, 1H), 2.18-2.08 (m, 1H), 1.78-1.69 (m, 2H); 13C (100 MHz, CDCl3) δ 143.0, 141.9, 135.7, 128.4, 128.3, 128.3, 127.52, 127.47, 126.1, 125.7, 74.6, 70.2, 39.9, 38.3, 31.2; HRMS (CI, NH3) calcd for C19H26NO2 [M+NH4]+ 300.1964 found 300.1974.
(3S, 7S, 4Z)-1,9-Diphenylnon-4-ene-3,7-diol (ent-19g)
[α]D24 = −38.5 (c 1.04, CHCl3); IR (neat) 3351, 3025, 2930, 1602, 1495, 1454, 1030 cm−1; 1H (500 MHz, CDCl3) δ 7.38-7.28 (m, 4H), 7.22-7.19 (m, 6H), 5.71-5.66 (m, 1H), 5.64-5.58 (m, 1H), 4.42 (ddd (app q), J = 7.0 Hz, 1H), 3.77-3.71 (dddd (app quintet), J = 6.5 Hz, 1H), 2.83-2.66 (m, 4H), 2.45-2.38 (dddd, J = 14.0, 8.5, 4.5, 1.0 Hz, 1H), 2.37 (s, OH broad, 1H), 2.28-2.21 (m, 1H), 2.06 (s, OH broad, 1H), 1.99-1.92 (dddd, J = 13.5, 9.5, 7.0, 6.5 Hz, 1H), 1.86-1.77 (m, 3H); 13C (125 MHz, CDCl3) δ 142.1, 142.0, 136.1, 128.7, 128.63, 128.61, 128.60, 127.8, 126.1, 126.0, 70.4, 66.8, 38.9, 38.2, 35.1, 32.4, 31.9; HRMS (FAB, Na) calcd for C21H26O2 [M+Na]+ 333.1831, found 333.1817.
(3S,4S)-6-Phenylhex-1-ene-3,4-diol (40)48
[α]D23 = −18.5 (c 0.99, CHCl3); IR (neat) 3386, 2922, 1711, 1496, 1454, 1046, 992, 926, 749, 699 cm−1; 1H (400 MHz, CDCl3) δ 7.29-7.15 (m, 5H), 5.83 (ddd, J = 17.2, 10.8, 6.4 Hz, 1H), 5.33 (dt, J = 17.2, 1.2 Hz, 1H), 5.23 (dt, J = 10.8, 1.2 Hz, 1H), 3.94 (tt, J = 6.0, 1.2 Hz, 1H), 3.49 (ddd, J = 9.2, 6.0, 3.2 Hz, 1H), 2.88-2.81 (m, 1H), 2.73-2.65 (m, 1H), 1.90 (bs, 2H), 1.88-1.71 (m, 2H); 13C (100 MHz, CDCl3) δ 142.1, 137.7, 128.61, 128.57, 126.0, 117.8, 76.5, 73.8, 34.8, 32.0; HRMS (ESI) calcd for C12H16NaO2 [M+Na]+ 215.1048, found 215.1042.
(1R,3E,5R)-1,7-diphenylhept-3-ene-1,5-diol (17c)
[α]D25 = +31.2 (c 0.47, CHCl3); e.e. 97%; IR (neat) 3356, 3061, 3027, 2925, 2858, 1602, 1494, 1453, 1308, 1051, 1029, 970, 748, 699, 546 cm−1; 1H (400 MHz, CDCl3) δ 7.32-7.27 (m, 4H), 7.25-7.20 (m, 3H), 7.15-7.12 (m, 3H), 5.64-5.52 (m, 2H), 4.65 (td, J = 6.6, 3.1 Hz, 1H), 4.02 (q, J = 6.3 Hz, 1H), 2.66-2.52 (m, 2H), 2.43 (t, J = 6.3 Hz, 2H), 2.15 (bs, 2H), 1.86-1.68 (m, 2H); 13C (100 MHz, CDCl3) δ 144.1, 142.1, 136.8, 128.67, 128.65, 128.5, 127.92, 127.86, 126.01, 125.99, 73.9, 72.4, 42.3, 38.8, 31.9; HRMS (ESI) calcd for C19H22O2Na [M+Na]+ 305.1517, found 305.1524.
(1S,5S,3E)-1,7-diphenylhept-3-ene-1,5-diol (ent-17c)
[α]D24 = −21.1 (c 0.75 CHCl3); 1H NMR, 13C NMR and IR and HRMS data were identical to 17c.
(1R,3E,5R)-1-cyclohexyl-7-phenylhept-3-ene-1,5-diol (17e)
[α]D25 = +7.5 (c 0.59, CHCl3); e.e. 96%; IR (neat) 3352, 3085, 3062, 3026, 2924, 2852, 1495, 1450, 1311, 1057, 1030, 969, 892, 746, 698 cm−1; 1H (400 MHz, CDCl3) δ 7.28-7.23 (m, 2H), 7.18-7.13 (m, 3H), 5.65 (ddd, J = 15.4, 7.3, 6.1 Hz, 1H), 5.58 (dd, J = 15.4, 6.6 Hz, 1H), 4.08 (q, J = 6.3 Hz, 1H), 3.38-3.33 (m, 1H), 2.74-2.60 (m, 2H), 2.30-2.24 (m, 1H), 2.13-2.05 (m, 1H), 1.93 (bs, 1H), 1.91-1.72 (m, 5H), 1.66-1.63 (m, 2H), 1.37-1.28 (m 1H), 1.27-094 (m, 5H); 13C (100 MHz, CDCl3) δ 142.1, 136.3, 128.8, 128.65, 128.57, 126.0, 75.4, 72.4, 43.4, 38.9, 37.4, 32.0, 29.3, 28.3, 26.7, 26.5, 26.3; HRMS (ESI) calcd for C19H28O2Na [M+Na]+ 311.1987, found 311.1990.
(1E,3R,5E,7R)-1,9-diphenylnona-1,5-diene-3,7-diol (17f)
[α]D25 = +12.6 (c 0.45, CHCl3); e.e. 95%; IR (neat) 3350, 3060, 3026, 2923, 2857, 1494, 1452, 1316, 1100, 1030, 967, 747, 694 cm−1; 1H (400 MHz, CDCl3) δ 7.34-7.32 (m, 2H), 7.29-7.18 (m, 5H), 7.16-7.12 (m, 3H), 6.55 (d, J = 15.7 Hz, 1H), 6.19 (dd, J = 15.9, 6.6 Hz, 1H), 5.67 (dt, J = 15.4, 6.6 Hz, 1H), 5.60 (dd, J = 15.7, 6.1 Hz, 1H), 4.32-4.27 (m, 1H), 4.07 (q, J = 6.6 Hz, 1H), 2.70-2.58 (m, 2H), 2.41-2.28 (m, 2H), 2.10 (bs, 2H), 1.90-1.73 (m, 2H); 13C (100 MHz, CDCl3) δ 142.1, 136.9, 136.7, 131.8, 130.7, 128.8, 128.63, 128.55, 127.9, 127.5, 126.7, 126.0, 72.4, 72.2, 40.6, 38.8, 31.9; HRMS (ESI) calcd for C21H24O2Na [M+Na]+ 331.1674, found 331.1679.
(1E,3S,5E,7S)-1,9-diphenylnona-1,5-diene-3,7-diol (ent-17f)
[α]D25 = −14.0 (c 0.59, CHCl3); e.e. 95%; 1H NMR, 13C NMR and IR and HRMS data were identical to 17f.
(1R,2E,5R)-6-(benzyloxy)-1-cyclohexylhex-2-ene-1,5-diol (17h)
[α]D25 = −14.0 (c 0.59, CHCl3); e.e. 96%; IR (neat) 3399, 2923, 2852, 1451, 1097, 1027, 1001, 971, 735, 697 cm−1; 1H (400 MHz, CDCl3) δ 7.33-7.23 (m, 5H), 5.56 (dt, J = 15.4, 6.6 Hz, 1H), 5.49 (dd, J = 15.4, 6.3 Hz, 1H), 4.50 (s, 2H), 3.84-3.78 (m, 1H), 3.72 (t, J = 6.6 Hz, 1H), 3.43 (dd, J = 9.3, 3.3 Hz, 1H), 3.31 (dd, J = 9.3, 7.3 Hz, 1H), 2.41 (bs, 1H), 2.20-2.14 (m, 2H), 1.82-1.78 (m, 1H), 1.72-1.58 (m, 4H), 1.36-1.27 (m, 1H), 1.24-1.05 (m, 3H), 0.96-0.83 (m, 2H); 13C (100 MHz, CDCl3) δ 138.1, 135.2, 128.6, 128.1, 128.0, 127.9, 77.6, 74.2, 73.6, 70.1, 43.7, 36.6, 29.0, 28.9, 26.7, 26.32, 26.25; HRMS (ESI) calcd for C19H28O3Na [M+Na]+ 327.1936, found 327.1942.
(2S,3E,6S)-1-{[tert-butyl(dimethyl)silyl]oxy}-8-phenyloct-3-ene-2,6-diol (17i)
[α]D25 = −14.0 (c 0.59, CHCl3); e.e. 96%; IR (neat) 3383, 3027, 2951, 2928, 2857, 1471, 1462, 1455, 1361, 1254, 1111, 1052, 1006, 971, 837, 778, 747, 699 cm−1; 1H (400 MHz, CDCl3) δ 7.26-7.22 (m, 2H), 7.17-7.12 (m, 3H), 5.77-5.69 (m, 1H), 5.50 (ddd, J = 15.4, 6.1, 1.3 Hz, 1H), 4.13-4.09 (m, 1H), 3.65-3.60 (m, 1H), 3.59 (dd, J = 10.1, 3.8 Hz, 1H), 3.41 (dd, J = 10.1, 7.8 Hz, 1H), 2.77 (dt, J = 13.9, 7.6 Hz, 1H), 2.64 (dt, J = 13.9, 8.1 Hz, 1H), 2.28-2.22 (m, 1H), 2.14 (dt, J = 14.1, 7.8 Hz, 1H), 1.77-1.72 (m, 2H), 0.87 (s, 9H), 0.04 (s, 6H); 13C (100 MHz, CDCl3) δ 142.3, 132.3, 129.3, 128.64, 128.57, 126.0, 72.8, 70.2, 67.3, 40.9, 38.7, 32.2, 26.1, 18.5, −5.13, −5.16; HRMS (ESI) calcd for C20H34O3NaSi [M+Na]+ 373.2175, found 373.2184.
(1R, 5S, 3E)-1,7-diphenylhept-3-ene-1,5-diol (ent-10c)
[α]D24 = +28.4 (c 1.08, CHCl3); IR (neat) 3360, 3027, 2925, 1949, 1878, 1807, 1669, 1603, 1495, 1454 cm−1; 1H (500 MHz, CDCl3) δ 7.38-7.17 (m, 10H), 5.72-5.66 (m, 1H), 5.62 (dd, J = 15.5, 6.5 Hz, 1H), 4.67 (td, J = 6.6, 3.1 Hz, 1H), 4.04-3.96 (m, 1H), 2.74-2.62 (m, 2H), 2.54-2.52 (m, 2H), 2.01 (d, OH, J = 3.5 Hz, 1H), 1.92-1.76 (m, 2H), 1.54 (d, OH, J = 4.0 Hz, 1H); 13C (100 MHz, CDCl3) δ 144.0, 142.1, 136.6, 128.6, 128.5, 127.8, 127.5, 126.01, 125.97, 73.8, 72.1, 42.1, 38.7, 31.9; HRMS (CI, NH3) calcd for C19H26NO2 [M+NH4]+ 300.1964 found 300.1951.
Synthesis of aldehyde 4
(E)-5-(4-Methoxy-benzyloxy)-pent-2-en-1-al (55)
To a solution of alkene 5440 (3.12 g, 16.2 mmol) in CH2Cl2 (70 mL) under argon, were successively added acrolein (freshly distilled, 3.25 mL, 48.7 mmol) and Hoveyda-Grubbs 2nd generation catalyst39 (weighed in a glove box, 254 mg, 0.40 mmol) as a solution in CH2Cl2 (3 mL). The reaction mixture was stirred overnight at room temperature, then directly concentrated in vacuo. The residue was purified by flash chromatography (8/2 : hexanes/EtOAc) to give 55 (3.04 g, 85%) as a brown oil; IR (neat) 2999, 2934, 2907, 2855, 1614, 1586, 1514, 1464, 1442, 1361, 1302, 1247, 1209, 1173, 1097, 1036, 972, 821 cm−1; 1H (400 MHz, CDCl3) δ 9.51 (d, J = 7.8 Hz, 1H), 7.26 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 6.87 (dt, J = 16.1, 6.0 Hz, 1H), 6.17 (ddt, J = 15.7, 7.8, 1.4 Hz, 1H), 4.46 (s, 2H), 3.81 (s, 3H), 3.61 (t, J = 6.2 Hz, 2H), 2.62 (tdd, J = 6.4, 6.4, 1.5 Hz, 2H); 13C (100 MHz, CDCl3) δ 194.1, 159.5, 155.4, 134.3, 130.2, 129.5, 114.1, 73.0, 67.8, 55.5, 33.3; HRMS (ESI) calcd for C13H16O3 [M+Na]+ 243.0997, found 243.0995.
(S, E)-8-(4-Methoxy-benzyloxy)-octa-1,5-dien-4-ol (56)
(−)-Ipc2BOMe (5.68 g, 17.9 mmol) was weighed in a glove box. Et2O (72 mL) was added and the suspension was cooled to 0°C. Allylmagnesium bromide (1 M in Et2O, 16.6 mL, 16.6 mmol) was added dropwise. The mixture was stirred for 1 h, then was cooled to −78 °C and a solution of aldehyde 55 (3.04 g, 13.8 mmol) in Et2O (17.5 mL) was added via cannula. The mixture was stirred for 3 h, then MeOH (1.8 mL), followed by NaOH (3 N, 17.4 mL, 52.3 mmol) and H2O2 (30% in H2O, 17.3 mL, 169 mmol) were added. The solution was warmed to 23°C and stirred overnight. The mixture was cooled at 0°C and neutralize with HCl (3N). The aqueous phase was extracted with Et2O. The combined organic phases were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. Purification of the crude by flash chromatography (gradient elution 85/15 to 70/30 : hexanes/ethyl acetate) provided 56 (2.99 g, 83%) as a colorless oil: e.e. 96%; [α]D22 = −10.5 (c = 0.89, CHCl3); IR (neat) 3419 (br.), 2933, 2907, 2858, 1613, 1514, 1464, 1448, 1361, 1302, 1248, 1174, 1094, 1035, 972, 916, 821 cm−1; 1H (400 MHz, CDCl3) δ 7.28-7.24 (m, 2H), 6.90-6.86 (m, 2H), 5.80 (ddt, J = 17.3, 9.9, 7.1 Hz, 1H), 5.70 (dt, J = 15.5, 6.6 Hz, 1H), 5.57 (dd, J = 15.5, 6.5 Hz), 5.16-5.14 (m, 1H), 5.11 (br. s, 1H), 4.44 (s, 2H), 4.17-4.10 (m, 1H), 3.81 (s, 3H), 3.48 (t, J = 6.8 Hz, 2H), 2.35 (dt, J = 6.8, 6.7 Hz, 2H), 2.31-2.23 (m, 2H), 1.70 (m, OH); 13C (100 MHz, CDCl3) δ 159.4, 134.50, 134.1, 130.7, 129.5, 128.5, 118.3, 113.9, 72.7, 71.8, 69.6, 55.5, 42.1, 32.8; HRMS (ESI) calcd for C16H22O3 [M+Na]+ 285.1467, found 285.1468.
(S)-1-((2S,3S)-3-(2-(4-Methoxy-benzyloxy)-ethyl)oxiran-2-yl)-but-3-en-1-ol (57)
To a solution L-(+)-DET (freshly distilled, 1.19 mL, 6.86 mmol, dried over MS 4Å for 1.5 h) in CH2Cl2 (11.2 mL) was added Ti(OiPr)4 (freshly distilled, 1.75 mL, 5.72 mmol) at −40 °C, resulting in a slightly yellow solution. This mixture was stirred for 40 min, then a solution of alcohol 56 (3.0 g, 11.4 mmol, dried over MS 4Å for 2 h) in CH2Cl2 (9 mL) was added via cannula. The mixture was stirred for 45 min at −20 °C, then a cold solution of TBHP (5.5 M in decane, 2.08 mL, 11.4 mmol, dried over MS 4Å for 2 h) in CH2Cl2 (9 mL) was added via cannula. The solution was stirred at −20 °C for 7 h, then stored in a −20 °C freezer for 20 h. The mixture was allowed to warm to 0 °C, then NaOH (1 M, sat with NaCl, 17.6 mL) and H2O (11 mL) were added. This mixture was stirred for 2 h at room temperature. It was then filtered through a plug of Celite, which was rinsed with CH2Cl2. The combined filtrates were washed with brine. The aqueous phase was extracted with CH2Cl2, the combined organic extracts dried over MgSO4, filtered and concentrated under reduced pressure. Purification of the crude product by flash chromatography (gradient elution 85/15 to 70/30 : hexanes/ethyl acetate) afforded 57 (2.20 g, 69%) as a clear oil: 99% d.e.; [α]D22 = −5.5 (c = 1.02, CHCl3); IR (neat) 3437 (br.), 2933, 2912, 2862, 1745, 1642, 1613, 1586, 1514, 1465, 1442, 1363, 1302, 1249, 1175, 1096, 1035, 917, 821 cm−1; 1H (400 MHz, CDCl3) δ 7.27-7.25 (m, 2H), 6.86-6.90 (m, 2H), 5.86 (ddt, J = 17.1, 10.1, 7.1 Hz, 1H), 5.17-5.14 (m, 1H), 5.13-5.11 (m, 1H), 4.50-4.43 (m, 2H), 3.81 (br. s, 4H), 3.58 (t, J = 6.3 Hz, 2H), 3.14 (td, J = 9.1, 2.2 Hz, 1H), 2.84 (dd, J = 3.0, 3.0 Hz, 1H), 2.42-2.25 (m, 2H), 2.08 (bs, 1H), 1.91-1.76 (m, 2H); 13C (100 MHz, CDCl3) δ 159.4, 133.9, 130.4, 129.5, 118.3, 114.0, 73.0, 68.4, 66.8, 60.5, 55.5, 53.4, 38.3, 32.3; HRMS (ESI) calcd for C16H22O4 [M+Na]+ 301.1416, found 301.1415.
(3S,5S)-1-(4-Methoxy-benzyloxy)-oct-7-ene-3,5-diol (58)
To a solution of epoxide 57 (2.2 g, 7.90 mmol) in THF (39.5 mL) was added MeOH (0.16 mL) and Red-Al® (3.3 M in toluene, 6.0 mL, 19.8 mmol) at 0°C (gas evolution!) over a period of 10 min. After being warmed to 23 °C over 2 h, the solution was stirred for 16 h. NaOH (3 N, 23.7 mL) was added at 0°C followed by water. The aqueous phase was extracted with Et2O and the combined organic phases washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. Purification of the crude by flash chromatography (1/2 : hexanes/ethyl acetate) afforded 58 (2.06 g, 93%) as colorless oil: [α]D24 = +16.8 (c = 1.06, CHCl3); IR (neat) 3402, 2937, 1613, 1514, 1303, 1249, 1175, 1090, 1035, 821 cm−1; 1H (400 MHz, CDCl3) δ 7.25-7.23 (m, 2H), 6.90-6.86 (m, 2H), 5.83 (ddt, J = 17.1, 10.2, 7.1 Hz, 1H), 5.15-5.11 (m, 1H), 5.11-5.09 (m, 1H), 4.46 (s, 2H), 4.19-4.13 (m, 1H), 4.02-3.95 (m, 1H), 3.81 (s, 3H), 3.73 (q, J = 4.8 Hz, 1H), 3.67 (td, J = 9.2, 3.8 Hz, 1H), 3.51 (d, J = 2.6 Hz, OH), 2.84 (d, J = 3.9 Hz, OH), 2.26 (ddt, J = 6.7, 6.7, 1.2 Hz, 2H), 1.94-1.86 (m, 1H), 1.71-1.61 (m, 3H); 13C (100 MHz, CDCl3) δ 159.5, 135.1, 130.0, 129.6, 117.9, 114.1, 73.3, 69.8, 69.4, 68.3, 55.5, 42.3, 42.3, 36.4; HRMS (ESI) calcd for C16H24O4 [M+Na]+ 303.1572, found 303.1566.
(3S,5S)-3,5-Bis-(tert-butyldimethylsilanyloxy)-1-(4-methoxy-benzyloxy)-oct-7-ene (59)
To a solution of diol 58 (2.06 g, 7.35 mmol) in DMF (14.7 mL) was added imidazole (2.02 g, 29.4 mmol) and TBSCl (4.31 g, 27.2 mmol). The mixture was stirred overnight and then poured into water (100 mL) and diluted with Et2O. The aqueous phase was extracted with Et2O and the combined organic phases were washed several times with water, then with brine. The combined extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. Purification of the crude product by flash chromatography (95/5 : hexanes/ethyl acetate) afforded the 59 (3.2 g, 86%) as pale yellow oil: [α]D24 = +3.4 (c = 1.23, CHCl3); IR (neat) = 2945, 2929, 2895, 2857, 1614, 1514, 1472, 1463, 1361, 1250, 1099, 1041, 1004, 836, 807, 884 cm−1; 1H (400 MHz, CDCl3) δ 7.27-7.24 (m, 2H), 6.89-6.85 (m, 2H), 5.81 (mc, 1H), 5.06-5.02 (m, 2H), AB signal (δA = 4.40, δB = 4.42, JAB = 11.5 Hz), 3.87 (mc, 1H), 3.80-3.76 (m, 4H), 3.50 (t, J = 6.8 Hz, 2H), 2.28-2.14 (m, 2H), 1.83-1.53 (m, 4H), 0.87 (s, 9H), 0.86 (s, 9H), 0.05 (s, 9H), 0.03 (s, 3H); 13C (100 MHz, CDCl3) δ 159.3, 135.2, 130.9, 129.4, 117.1, 113.9, 72.9, 69.8, 67.7, 67.0, 55.5, 45.6, 42.4, 37.9, 26.1, 18.32, 18.28, −3.86, −3.91, −4.0, −4.1; HRMS (ESI) calcd for C28H52O4Si2 [M+Na]+ 531.3302, found 531.3305.
(3S,5S)-3,5-Bis-(tert-butyldimethylsilanyloxy)-7-(4-methoxy-benzyloxy)-heptane-1-al (4)
To a solution of alkene 59 (1.75 g, 3.44 mmol) in dioxane (26 mL) and water (8.6 mL) was added 2,6-lutidine (freshly distilled, 0.865 mL, 7.43 mmol), OsO4 (2.5 wt% in 2-methyl-2-propanol, 0.862 mL, 0.07 mmol) and NaIO4 (2.95 g, 13.7 mmol). This mixture was stirred for 5 h, then the brown suspension was diluted with water and CH2Cl2. The aqueous phase was extracted with CH2Cl2, the combined organic phases washed with brine, dried over MgSO4, and concentrated under reduced pressure. Purification of the crude product by flash chromatography (95/5 : hexanes/ethyl acetate) afforded 4 (1.57 g, 89%) as yellow oil: [α]D24 = −4.9 (c = 1.23, CHCl3); IR (neat) 2954, 2930, 2857, 1727, 1514, 1472, 1464, 1250, 1102, 1040, 1005, 837, 809, 776 cm−1; 1H (400 MHz, CDCl3) δ 9.78 (t, J = 2.5 Hz, 1H), 7.26-7.23 (m, 2H), 6.89-6.85 (m, 2H), AB signal (δA = 4.39, δB = 4.42, JAB = 11.5 Hz), 4.24 (dddd, app. tt, J = 6.0 Hz, 1H), 3.87 (dddd, app. tt, J = 6.1 Hz, 1H), 3.81 (s, 3H), 3.49 (t, J = 6.6 Hz, 2H), 2.57 (ddd, J = 15.4, 4.5, 2.1 Hz, 1H), 2.49 (ddd, J = 15.4, 6.5, 3.0 Hz, 1H), 1.84-1.64 (m, 4H), 0.87 (s, 9H), 0.86 (s, 9H), 0.07 (s, 3H), 0.06 (s, 3H), 0.054 (s, 3H), 0.047 (s, 3H); 13C (100 MHz, CDCl3) δ 202.3, 159.3, 130.7, 129.5, 113.9, 72.9, 67.5, 66.6, 66.5, 55.5, 51.5, 46.3, 38.0, 26.1, 25.9, 18.2, 18.1, −4.0, −4.1, −4.2; HRMS (ESI) calcd for C27H50O5Si2 [M+Na]+ 533.3095, found 533.3099.
Synthesis of aldehyde 3
(R)-7-Azido-4-[(tert-butyldimethylsilanyloxy]-1-hepten (52)
(−)-Ipc2BOMe (6.64 g, 21.0 mmol) was weighed in a glove box. Et2O (84 mL) was added and the suspension was cooled to 0°C. Allylmagnesium bromide (1 M in Et2O, 19.3 mL, 19.3 mmol) was added dropwise. The mixture was stirred for 1 h, then was cooled to –78 °C and a solution of known aldehyde 4934 (3.04 g, 13.8 mmol) in Et2O (22.0 mL) was added via cannula. The mixture was stirred for 3 h at −78 °C, then MeOH (2.1 mL), followed by NaOH (3 N, 22.0 mL, 66.3 mmol) and H2O2 (30% in H2O, 22.0 mL, 215 mmol) were added. The solution was warmed to 23°C and stirred overnight. The mixture was cooled at 0°C and neutralize with HCl (3N). The aqueous phase extracted with Et2O. The combined organic phases were washed with brine, dried over MgSO4, filtered and carefully concentrated under reduced pressure (removal of solvent at 250 torr/0°C owing to the volatility of the product). The azidoalcohol 50 was obtained as a ca. 1:2 mixture with isopinocampheol 51 as a viscous oil. To this mixture of 50 and isopinocampheol 51 in DMF (10 mL) were added TBSCl (11.3 g, 71.4 mmol) and imidazole (5.27 g, 77.4 mmol). After being stirred overnight, the mixture was poured into water (100 mL) and Et2O. The aqueous phase was extracted with Et2O and the combined organic phases were washed several times with water, then with brine. The combined extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. Purification of the crude product by flash chromatography (95/5 : hexanes/ethyl acetate) afforded 52 (3.4 g, 73% over two steps) as a clear oil: e.e. 91%; [α]D26 = +13.1 (c = 1.09, CHCl3); IR (neat) 2955, 2931, 2858, 2093, 1472, 1464, 1361, 1256, 1092, 1041, 1005, 939, 914, 837, 809, 775 cm−1; 1H (400 MHz, CDCl3) δ 5.84-5.74 (m, 1H), 5.08-5.06 (m, 1H), 5.03 (br. s, 1H), 3.73 (dddd, app. tt, J = 5.7 Hz, 1H), 3.27 (t, J = 6.9 Hz, 2H), 2.22 (dd, app. t, J = 6.0 Hz, 2H), 1.74-1.43 (m, 4H), 0.89 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H); 13C (100 MHz, CDCl3) δ 134.8, 117.1, 71.3, 51.7, 41.9, 33.6, 25.9, 24.7, 18.1, −4.4, −4.6; HRMS (ESI) calcd for C13H27N3OSi [M+Na]+ 292.1821, found 292.1830.
Tert-butyl ((4R)-4-{[tert-butyl(dimethyl)silyl]oxy}hept-6-en-1-yl)carbamate (53)
To a room temperature solution of alkene 52 (1.0 g, 3.49 mmol) in wet Et2O (9 mL) was added tributylphosphine (1.07 mL, 4.19 mmol). The mixture was stirred for 1.5 h at ambient temperture and then cooled to −50°C. Di-tert-butyl dicarbonate (923 mg, 4.19 mmol) as a solution in Et2O (2.6 mL) was slowly added and the resulting mixture was stirred for 2 h. The reaction was quenched with a solution of saturated NaHCO3 (11.6 mL), and allowed to warm to room temperature and stirred overnight. The aqueous phase was separated and extracted with Et2O. The combined organic phases were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. Purification of the crude product by flash chromatography (gradient elution 98/2 to 80/20 : hexanes/ethyl acetate) afforded 53 (845 mg, 70%) as clear oil: [α]D27 = +10.6 (c = 0.5, CHCl3); IR (neat) 3349, 2929, 2857, 1693 (broad), 1513, 1365, 1251, 1172, 1117, 1002, 912, 835, 773 cm−1; 1H (400 MHz, CDCl3) δ 5.84-5.71 (m, 1H), 5.06-5.02 (m, 1H), 5.01 (bs, 1H), 4.54 (bs, 1H), 3.71 (app quint, J = 5.7 Hz, 1H), 3.17-3.02 (m, 2H), 2.23-2.18 (m, 2H), 1.43 (s, 9H), 1.60-1.39 (m, 4H), 0.88 (s, 9H), 0.044 (s, 3H), 0.037 (s, 3H); 13C (100 MHz, CDCl3) δ 156.2, 135.3, 117.1, 79.2, 71.7, 42.1, 40.9, 34.0, 28.6, 27.6, 26.1, 18.3, −4.2, −4.3; HRMS (ESI) calcd for C18H37NO3Si [M+Na]+ 366.2440, found 366.2449.
Tert-butyl ((4R,6E)-4-{[tert-butyl(dimethyl)silyl]oxy}-8-oxooct-6-en-1-yl)carbamate (3)
To a solution of alkene 53 (273 mg, 0.79 mmol) in CH2Cl2 (2.4 mL) under argon were successively added acrolein (freshly distilled, 0.159 mL, 2.38 mmol) and Hoveyda-Grubbs 2nd generation catalyst39 (weighed in a glove box, 12 mg, 0.020 mmol) as a solution in CH2Cl2 (1 mL). The reaction was stirred overnight at room temperature, then concentrated in vacuo and directly purified by flash chromatography (8/2 : hexanes/EtOAc). This afforded aldehyde 3 (276 mg, 94%) as a clear oil which solidified when stored in the freezer: [α]D27 = +10.0 (c = 0.69, CHCl3); IR (neat) 3360, 2930, 2858, 1693 (broad), 1520, 1365, 1252, 1172, 836, 775 cm−1; 1H (400 MHz, CDCl3) δ 9.51 (d, J = 8.0 Hz, 1H), 6.86 (dt, J = 15.6, 7.4 Hz, 1H), 6.13 (ddt, J = 15.6, 7.9, 1.3 Hz, 1H), 4.52 (bs, 1H), 3.87 (app quint, J = 5.6 Hz, 1H), 3.19-3.03 (m, 2H), 2.56-2.41 (m, 2H), 1.56-4.46 (m, 4H), 1.44 (s, 9H), 0.88 (s, 9H), 0.06 (s, 3H), 0.035 (s, 3H); 13C (100 MHz, CDCl3) δ 194.0, 156.2, 155.1, 135.1, 79.4, 70.8, 40.6, 34.5, 28.6, 26.0, 25.9, 18.3, −4.25, −4.27; HRMS (ESI) calcd for C19H37NO4Si [M+Na]+ 394.2390, found 394.2391.
Tert-butyl {(4R,6E,8S,10E,12S,14S,16S)-4,14,16-tris{[tert-butyl(dimethyl)silyl]oxy}-8,12-dihydroxy-18-[(4-methoxybenzyl)-oxy]octadeca-6,10-dien-1-yl}carbamate (60)
To a 0 °C solution of borohydride 26R (81.0 mg, 0.250 mmol, 1 equiv, considering a 90% yield for the conversion of borohydride 26R to borane 25R based on titration3) in toluene (0.5 mL) was added TMSCl (32.0 µL, 0.250 mmol, 1 equiv). The reaction mixture was stirred for 10 min, then was cooled to −78 °C and a solution of allene 45 (0.114 g, 0.325 mmol, 1.3 equiv) in toluene (1 mL) / CH2Cl2 (0.1 mL) was added dropwise. The viscous mixture was stirred at −30 °C for 1 h. The resulting clear reaction mixture was then cooled to −78 °C and a solution of 4 (0.213 mmol, 0.85 equiv) in CH2Cl2 (0.25 mL) was added. Four hours later, a solution of 3 (0.300 mmol, 1.2 equiv) in CH2Cl2 (0.25 mL) followed by BF3•OEt2 (47.0 µL, 0.375 mmol, 1.5 equiv) were added and the reaction mixture was stirred for an additional 4 h. A pH 7 buffer solution (KH2PO4/NaOH) (1 mL) followed by THF (1.5 mL) and MeOH (0.5 mL) were then added and the mixture was stirred at 20 °C for 24 h. The biphasic mixture was poured into a saturated aqueous NH4Cl solution, and the crude product was extracted with ethyl acetate (3×). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (SiO2, hexanes / ethyl acetate, gradient elution: 7:3 to 6:4 to 1:1) to afford 60 (163 mg, 83%) as a colorless oil:[α]D25 = −4.7 (c 0.64, CHCl3); IR (neat) 3375, 2953, 2929, 2896, 2856, 1694, 1514, 1472, 1463, 1389, 1365, 1250, 1172, 1098, 1041, 1005, 971, 836, 809, 774 cm−1; 1H (400 MHz, CDCl3) δ 7.24 (d, J = 8.6 Hz, 2H), 6.87 (d, J = 8.6 Hz, 2H), 5.70-5.47 (m, 4H), 4.63 (bs, 1H), 4.41 (m, overlap, 1H), 4.40 (dd, J = 12.0, 11.6 Hz, 2H), 4.10 (app q, J = 6.2 Hz, 1H), 4.06-3.99 (m, 1H), 3.82 (m, overlap, 1H), 3.79 (s, 3H), 3.74-3.66 (m, 1H), 3.50 (t, J = 6.8 Hz, 2H), 3.15-3.02 (m, 2H), 2.34-2.13 (m, 4H), 1.89-1.63 (m, 6H), 1.43 (s, 9H), 1.62-1.35 (m, 7H), 0.88 (s, 18H), 0.86 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H), 0.06 (s, 3H), 0.041 (s, 3H), 0.035 (s, 6H); 13C (100 MHz, CDCl3) δ 159.3, 156.2, 136.8, 134.9, 130.8, 129.5, 128.1, 126.2, 114.0, 79.23, 72.9, 72.1, 71.8, 69.4, 69.3, 67.7, 66.8, 55.5, 45.0, 42.8, 40.8, 40.6, 40.4, 37.5, 33.9, 28.7, 26.3, 26.1, 26.0, 18.3, 18.2, 18.1, −3.9, −4.1, −4.1, −4.3, −4.4; HRMS (ESI) calcd for C49H93NO9NaSi3 [M+Na]+ 946.6056, found 946.6063.
The absolute stereochemistry of the two hydroxyl groups in 60 were assigned by using the Mosher ester method.
Tert-butyl {(4R,6E,8S,10E,12S,14R,16S)-4,8,12,14,16-pentakis{[tert-butyl(dimethyl)silyl]oxy}-18-[(4-methoxybenzyl)oxy]octadeca-6,10-dien-1-yl}carbamate (2)
To a solution of alcohol 60 (20 mg, 0.022 mmol) in CH2Cl2 (0.25 mL) was added imidazole (12 mg, 0.18 mmol) and TBS-Cl (20 mg, 0.13 mmol). The mixture was stirred overnight, then concentrated in vacuo. The crude product was directly purified by flash chromatography (gradient elution 99/1 to 90/10 : hexanes/ethyl acetate) to give 2 (24 mg, 94%) as clear oil: [α]D27 = −3.5 (c = 0.45, CHCl3); IR (neat) 2953, 2928, 2887, 2856, 1718, 1513, 1471, 1462, 1248, 1171, 1064, 1040, 1004, 971, 832, 807, 771 cm−1; 1H (400 MHz, CDCl3) δ 7.24 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 5.59-5.37 (m, 4H), 4.53 (bs, 1H), 4.41 (dd, J = 11.6, 11.6 Hz, 2H), 4.21-4.14 (m, 1H), 4.07 (app q, J = 6.1 Hz, 1H), 3.91-3.78 (m, 2H), 3.80 (s, 3H), 3.73-3.64 (m, 1H), 3.50 (t, J = 6.8 Hz, 2H), 3.16-3.01 (m, 2H), 2.27-2.12 (m, 4H), 1.89-1.79 (m, 1H), 1.43 (s, 9H), 1.72-1.36 (m, 9H), 0.88 (s, 18H), 0.872 (s, 9H), 0.869 (s, 9H), 0.86 (s, 9H), 0.06 (s, 3H), 0.05-0.03 (m, 21H), 0.02 (s, 3H), 0.01 (s, 3H); 13C (100 MHz, CDCl3) δ 159.2, 156.1, 136.4, 135.8, 131.1, 129.4, 126.8, 126.4, 113.9, 79.2, 73.6, 72.7, 71.9, 71.3, 67.5, 67.4, 66.9, 55.5, 47.4, 46.9, 41.7, 40.9, 40.3, 37.5, 33.7, 28.7, 26.23, 26.19, 26.14, 26.11, 18.5, 18.4, 18.30, 18.25, −3.2, −3.3, −3.5, −3.9, −4.06, −4.13, −4.2, −4.38, −4.45; HRMS (ESI) calcd for C61H121NO9Si5 [M+Na]+ 1174.7785, found 1174.7780.
Tert-butyl {(4R,6E,8R,10E,12R,14S,16S)-4,14,16-tris{[tert-butyl(dimethyl)silyl]oxy}-8,12-dihydroxy-18-[(4-methoxy-benzyl)oxy]octadeca-6,10-dien-1-yl}carbamate (61)
To a 0 °C solution of borohydride 25S (81.0 mg, 0.250 mmol, 1 equiv, considering a 90% yield for the conversion of borohydride 26S to borane 25S based on titration8d) in toluene (0.5 mL) was added TMSCl (32 µL, 0.250 mmol, 1 equiv). The reaction mixture was stirred for 10 min, then was cooled to −78 °C and a solution of allene 45 (0.114 g, 0.325 mmol, 1.3 equiv) in toluene (1 mL) / CH2Cl2 (0.1 mL) was added dropwise. The viscous mixture was stirred at −30 °C for 1 h. The resulting clear reaction mixture was then cooled to −78 °C and a solution of 4 (0.213 mmol, 0.85 equiv) in CH2Cl2 (0.25 mL) was added. After 4h, a solution of 3 (0.300 mmol, 1.2 equiv) in CH2Cl2 (0.25 mL) followed by BF3•OEt2 (47 µL, 0.375 mmol, 1.5 equiv) were added and the reaction mixture was stirred for an additional 4 h. A pH 7 buffer solution (KH2PO4/NaOH) (1 mL) followed by THF (1.5 mL) and MeOH (0.5 mL) were then added. The resulting mixture was stirred at 20 °C for 24 h. The biphasic mixture was poured into saturated aqueous NH4Cl solution, and the crude product was extracted with ethyl acetate (3×). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (SiO2, hexanes / ethyl acetate, gradient elution: 7:3 to 6:4 to 1:1) to afford 61 (153 mg, 78%) as a colourless oil: [α]D25 = −7.8 (c 0.50, CHCl3); IR (neat) 3374, 2953, 2929, 2895, 2856, 1695, 1514, 1472, 1462, 1365, 1250, 1172, 1098, 1040, 1005, 971, 835, 808, 774 cm−1; 1H (400 MHz, CDCl3) δ 7.24 (d, J = 8.6 Hz, 2H), 6.86 (d, J = 8.6 Hz, 2H), 5.70-5.47 (m, 4H), 4.67 (bs, 1H), 4.40 (dd, J = 12.0, 11.6 Hz, 2H), 4.26-4.17 (m, 1H), 4.08 (app q, J = 6.3 Hz, 1H), 3.92-3.85 (m, 1H), 3.85-3.78 (m, 1H), 3.79 (s, 3H), 3.74-3.66 (m, 1H), 3.48 (t, J = 6.8 Hz, 2H), 3.15-3.02 (m, 2H), 2.31-2.14 (m, 4H), 2.10-1.90 (bs, 1H), 1.83-1.57 (m, 6H), 1.57-1.36 (m, 6H), 1.43 (s, 9H), 0.88 (s, 9H), 0.87 (s, 9H), 0.85 (s, 9H), 0.084 (s, 3H), 0.079 (s, 3H), 0.044 (s, 3H), 0.035 (s, 3H), 0.030 (s, 6H); 13C (100 MHz, CDCl3) δ 159.3, 156.2, 136.3, 134.9, 130.7, 129.5, 127.9, 126.6, 113.9, 79.2, 72.9, 71.8, 71.7, 71.3, 70.7, 67.5, 66.8, 55.4, 47.0, 44.6, 40.8, 40.6, 40.4, 37.6, 33.9, 28.6, 26.08, 26.06, 26.04, 25.9, 18.3, 18.2, 18.1, −3.6, −4.0, −4.2, −4.3, −4.4; HRMS (ESI) calcd for C49H93NO9NaSi3 [M+Na]+ 946.6056, found 946.6060.
The absolute stereochemistry of the two hydroxyl groups in 61 were assigned by using the Mosher ester method.
Tert-butyl {(4R,6E,8R,10E,12R,14R,16S)-4,8,12,14,16-pentakis{[tert-butyl(dimethyl)silyl]oxy}-18-[(4-methoxy-benzyl)oxy]octadeca-6,10-dien-1-yl}carbamate (62)
To a solution of alcohol 61 (20 mg, 0.022 mmol) in CH2Cl2 (0.25 mL) was added imidazole (12 mg, 0.176 mmol) and TBSCl (20 mg, 0.126 mmol). The mixture was stirred overnight and then concentrated in vacuo. The crude product was directly purified by flash chromatography (gradient elution 99/1 to 90/10 : hexanes/ethyl acetate) to give 62 (23 mg, 90%) as clear oil: [α]D27 = −5.3 (c = 1.47, CHCl3); IR (neat) 2953, 2928, 2886, 2856, 1709, 1513, 1471, 1462, 1363, 1248, 1171, 1078, 1039, 1004, 832, 807, 772, 733 cm−1; 1H (400 MHz, CDCl3) δ 7.24 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 5.59-5.37 (m, 4H), 4.54 (bs, 1H), 4.41 (dd, J = 11.6, 11.6 Hz, 2H), 4.21 (app q, J = 6.7 Hz, 1H), 4.07 (app q, J = 5.9 Hz, 1H), 3.89-3.81 (m, 1H), 3.80 (s, 3H), 3.78-3.71 (m, 1H), 3.71-3.64 (m, 1H), 3.48 (t, J = 6.8 Hz, 2H), 3.16-3.01 (m, 2H), 2.27-2.12 (m, 4H), 1.85-1.73 (m, 1H), 1.72-1.36 (m, 9H), 1.43 (s, 9H), 0.89-0.88 (m, 27H), 0.87 (s, 9H), 0.86 (s, 9H), 0.045 (s, 9H), 0.04 (s, 9H), 0.03 (s, 6H), 0.02 (s, 6H); 13C (100 MHz, CDCl3) δ 159.3, 156.1, 135.6, 135.5, 131.0, 129.4, 127.0, 126.4, 113.9, 79.2, 73.4, 72.8, 71.9, 71.1, 67.7, 67.6, 66.9, 55.5, 47.1, 46.8, 41.8, 41.0, 40.5, 37.8, 33.9, 28.7, 26.21, 26.16, 26.1, 25.9, 18.5, 18.4, 18.31, 18.27, −3.5, 3.7, −3.8, −3.9, −4.06, −4.14, −4.2, −4.3, −4.4, −4.5; HRMS (ESI) calcd for C61H121NO9Si5 [M+Na]+ 1174.7785, found 1174.7793.
Scheme 18.

Synthesis of Aldehyde 3
Acknowledgments
We thank the National Institutes of Health (GM038436) for support of this research, and the German Academic Exchange Service (DAAD) for a postdoctoral fellowship to A.S.
References and notes
- 1.Examples of natural products containing (E)-1,5-syn-diol units: (a) Amphidinol 3: Murata M, Matsuoka S, Matsumori N, Paul GK, Tachibana K. J. Am. Chem. Soc. 1999;121:870. (b) Tetrafibricin: Kobayashi Y, Czechtizky W, Kishi Y. Org. Lett. 2003;5:93. doi: 10.1021/ol0272895. (c) Marinomycins A–C: Kwon HC, Kauffman CA, Jensen PR, Fenical W. J. Am. Chem. Soc. 2006;128:1622. doi: 10.1021/ja0558948. (d) Aigialomycin F: Isaka M, Yangchum A, Intamas S, Kocharin K, Gareth Jones EB, Kongsaeree P, Prabpai S. Tetrahedron. 2009;65:4396. (e) Marinisporolides: Kwon HC, Kauffman CA, Jensen PR, Fenical W. J. Org. Chem. 2009;74:675. doi: 10.1021/jo801944d.
- 2.BouzBouz S, Cossy J. Org. Lett. 2004;6:3469. doi: 10.1021/ol048862i. [DOI] [PubMed] [Google Scholar]
- 3.(a) BouzBouz S, Cossy J. Org. Lett. 2001;3:1451. doi: 10.1021/ol0157095. [DOI] [PubMed] [Google Scholar]; (b) BouzBouz S, Simmons R, Cossy J. Org. Lett. 2004;6:3465. doi: 10.1021/ol049079t. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt DR, O’Malley SJ, Leighton JL. J. Am. Chem. Soc. 2003;125:1190. doi: 10.1021/ja0283201. [DOI] [PubMed] [Google Scholar]
- 5.Nicolaou KC, Nold AL, Milburn RR, Schindler CS, Cole KP, Yamaguchi J. J. Am. Chem. Soc. 2007;129:1760. doi: 10.1021/ja068053p. [DOI] [PubMed] [Google Scholar]
- 6.Friestad GK, Sreenilayam G. Org. Lett. 2010;12:5016. doi: 10.1021/ol1021417. [DOI] [PubMed] [Google Scholar]
- 7.Wender PA, Verma VA, Paxton TJ, Pillow TH. Acc. Chem. Res. 2008;41:40. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- 8.(a) Brown HC, Narla G. J. Org. Chem. 1995;60:4686. [Google Scholar]; (b) Barrett AGM, Braddock CD, de Koning PD, White AJP, Williams DJ. J. Org. Chem. 2000;65:375. doi: 10.1021/jo991205x. [DOI] [PubMed] [Google Scholar]; (c) Flamme EM, Roush WR. J. Am. Chem. Soc. 2002;124:13644. doi: 10.1021/ja028055j. [DOI] [PubMed] [Google Scholar]; (d) Kister J, DeBaillie AC, Lira R, Roush WR. J. Am. Chem. Soc. 2009;131:14174. doi: 10.1021/ja905494c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Cheng M, Handa M, Roush W. J. Am. Chem. Soc. 2009;131:14602. doi: 10.1021/ja904599h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Morgan JB, Morken JP. Org. Lett. 2003;5:2573. doi: 10.1021/ol034936z. [DOI] [PubMed] [Google Scholar]; (g) Pelz NF, Woodward AR, Burks HE, Sieber JD, Morken JP. J. Am. Chem. Soc. 2004;126:16328. doi: 10.1021/ja044167u. [DOI] [PubMed] [Google Scholar]; (h) Woodward AR, Burks HE, Chan LM, Morken JP. Org. Lett. 2005;7:5505. doi: 10.1021/ol052312i. [DOI] [PubMed] [Google Scholar]; (i) Sieber JD, Morken JP. J. Am. Chem. Soc. 2006;128:74. doi: 10.1021/ja057020r. [DOI] [PubMed] [Google Scholar]; (j) Gonzalez AZ, Roman JG, Alicea E, Canales E, Soderquist JA. J. Am. Chem. Soc. 2009;131:1269. doi: 10.1021/ja808360z. [DOI] [PubMed] [Google Scholar]
- 9.(a) Flamme EM, Roush WR. Org. Lett. 2005;7:1411. doi: 10.1021/ol050250q. [DOI] [PubMed] [Google Scholar]; (b) Owen RM, Roush WR. Org. Lett. 2005;7:3941. doi: 10.1021/ol0514303. [DOI] [PubMed] [Google Scholar]; (c) Hicks JD, Flamme EM, Roush WR. Org. Lett. 2005;7:5509. doi: 10.1021/ol052322j. [DOI] [PubMed] [Google Scholar]; (d) Lira R, Roush WR. Org. Lett. 2007;9:4315. doi: 10.1021/ol7018746. [DOI] [PubMed] [Google Scholar]
- 10.Kamiyama T, Umino T, Fujisaki N, Fujimori K, Satoh T, Yamashita Y, Ohshima S, Watanabe J, Yokose K. J. Antibiot. 1993;46:1039. doi: 10.7164/antibiotics.46.1039. [DOI] [PubMed] [Google Scholar]
- 11.(a) Satoh T, Yamashita Y, Kamiyama T, Watanabe J, Steiner B, Hadvary P, Arisawa M. Thromb. Res. 1993;72:389. doi: 10.1016/0049-3848(93)90239-k. [DOI] [PubMed] [Google Scholar]; (b) Satoh T, Yamashita Y, Kamiyama T, Arisawa M. Thromb. Res. 1993;72:401. doi: 10.1016/0049-3848(93)90240-o. [DOI] [PubMed] [Google Scholar]; (c) Satoh T, Kouns WC, Yamashita Y, Maiyama T, Steiner B. Biochem. J. 1994;301:785. doi: 10.1042/bj3010785. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Satoh T, Kouns WC, Yamashita Y, Kamiyama T, Steiner B. Biochem. Biophys. Res. Commun. 1994;204:325. doi: 10.1006/bbrc.1994.2463. [DOI] [PubMed] [Google Scholar]
- 12.Lira R, Roush WR. Org. Lett. 2007;9:533. doi: 10.1021/ol0629869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gudipati V, Bajpai R, Curran DP. Collect. Czech. Chem. Commun. 2009;74:771. doi: 10.1135/cccc2008195. Zhang K, Gudipati V, Curran DP. Synlett. 2010:667. doi: 10.1055/s-0029-1219376. Gudipati V, Curran DP. Tetrahedron Lett. 2011;52:2254. doi: 10.1016/j.tetlet.2011.01.086. (d) After our mansuscript was submitted for publication, Krisch’s synthesis of the C(21)-C(40) frragment of tetrafibricin was published: Kumpulainen ETT, Kang B, Krische MJ. Org. Lett. 2011;13:2484. doi: 10.1021/ol200735r.
- 14.Kister J, Nuhant P, Lira R, Sorg S, Roush WR. Org. Lett. 2011;13:1868. doi: 10.1021/ol2003836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown HC, Singaram B. J. Org. Chem. 1984;49:945. [Google Scholar]
- 16.(a) Mikhailov BM. Organomet. Chem. Rev., Sect. A. 1972;8:1. [Google Scholar]; (b) Kramer GW, Brown HC. J. Organomet. Chem. 1977;132:9. [Google Scholar]
- 17.(a) Beletskaya I, Pelter A. Tetrahedron. 1997;53:4957. [Google Scholar]; (b) Burgess K, Ohlmeyer MJ. Chem. Rev. 1991;91:1179. [Google Scholar]
- 18.Burgos CH, Canales E, Matos K, Soderquist JA. J. Am. Chem. Soc. 2005;127:8044. doi: 10.1021/ja043612i. [DOI] [PubMed] [Google Scholar]
- 19.(a) Munoz-Hernandez L, Soderquist JA. Org. Lett. 2009;11:2571. doi: 10.1021/ol900865y. [DOI] [PubMed] [Google Scholar]; (b) Ess DH, Kister J, Chen M, Roush WR. Org. Lett. 2009;11:5538. doi: 10.1021/ol902364d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Soderquist JA, Matos K, Burgos CH, Lai C, Vaquer J, Medina JR, Huang SD. ACS Symp. Ser. 2001;783:176. [Google Scholar]; (b) Gonzalez AZ, Roma´n JG, Gonzalez E, Martinez J, Medina JR, Matos K, Soderquist JA. J. Am. Chem. Soc. 2008;130:9218. doi: 10.1021/ja803119p. and references cited therein. [DOI] [PubMed] [Google Scholar]
-
21.Nonracemic borane 25R [10-TMS-9-BBD-H] is easily prepared from pseudoephedrine complex 63R by using Soderquist’s procedure.20 Both enantiomers of 63R are commercially available but also are easily prepared in two steps from racemic B-OMe-9-BBN 6420b We found, however, that generation of 25R from the mono-diethyl etherate borohydride 26R is best performed in the presence of allene, owing to the instability of 78d
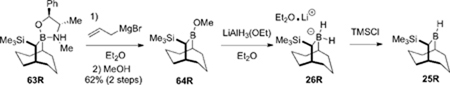
- 22.Chen M, Roush WR. Org. Lett. 2010;12:2706. doi: 10.1021/ol1007444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Carosi L, Lachance H, Hall DG. Tetrahedron Lett. 2005;46:8981. [Google Scholar]; (b) Carosi L, Hall DG. Angew. Chem., Int. Ed. 2007;46:5913. doi: 10.1002/anie.200700975. [DOI] [PubMed] [Google Scholar]; (c) Peng F, Hall DG. J. Am. Chem. Soc. 2007;129:3070. doi: 10.1021/ja068985t. [DOI] [PubMed] [Google Scholar]; (d) Peng F, Hall DG. Tetrahedron Lett. 2007;48:3305. [Google Scholar]; (e) Carosi L, Hall DG. Can. J. Chem. 2009;87:650. [Google Scholar]
- 24.Masamune S, Choy W, Petersen JS, Sita LR. Angew. Chem. Int. Ed. Engl. 1985;24:1. [Google Scholar]
- 25.(a) Keck GE, Boden EP. Tetrahedron Lett. 1984;25:265. [Google Scholar]; (b) Keck GE, Boden EP. Tetrahedron Lett. 1984;25:1879. [Google Scholar]; (c) Keck GE, Abbott DE. Tetrahedron Lett. 1984;25:1883. [Google Scholar]; (d) Keck GE, Savin KA, Cressman ENK, Abbott DE. J. Org. Chem. 1994;59:7889. [Google Scholar]; (e) Marshall JA. Chem. Rev. 1996;96:31. doi: 10.1021/cr950037f. and articles cited therein. [DOI] [PubMed] [Google Scholar]
- 26.Prevost C, Gaudemar M, Honigberg JCR. Acad. Sci. Vol. 230. Paris: 1950. p. 1186. [Google Scholar]
- 27.Keck GE, Tarbet KH, Geraci LS. J. Am. Chem. Soc. 1993;115:8467. [Google Scholar]
- 28.(a) Aspinall HC, Browning AF, Greeves N, Ravenscroft P. Tetrahedron Lett. 1994;35:4639. [Google Scholar]; (b) Aspinall HC, Greeves N, McIver EG. Tetrahedron Lett. 1998;39:9283. [Google Scholar]
- 29.Yanagisawa A, Nakashima H, Ishiba A, Yamamoto H. J. Am. Chem. Soc. 1996;118:4723. [Google Scholar]
- 30.Peterson DJ. J. Org. Chem. 1968;33:780. [Google Scholar]
- 31.(a) Denmark SE, Weber EJ, Wilson TM, Willson TM. Tetrahedron. 1989;45:1053. [Google Scholar]; (b) Keck GE, Dougherty SM, Savin KA. J. Am. Chem. Soc. 1995;117:6210. [Google Scholar]
- 32.Batey RA, Thadani AN, Smil DV. Tetrahedron Lett. 1999;40:4289. [Google Scholar]
- 33.BF3.OEt2 proved to be the most efficient Lewis acid in a previous study: Batey RA, Thadani AN, Smil DV, Lough AJ. Synthesis. 2000:990.
- 34.Hudlicky T, Frazier JO, Seoane G, Tiedje M, Seoane A, Kwart LD, Beal C. J. Am. Chem. Soc. 1986;108:3755. [Google Scholar]
- 35.(a) Jadhav PK, Bhat KS, Perumal PT, Brown HC. J. Org. Chem. 1986;51:432. [Google Scholar]; (b) Brown HC, Bhat KS, Randad RS. J. Org. Chem. 1989;54:1570. [Google Scholar]; (c) Racherla US, Brown HC. J. Org. Chem. 1991;56:401. [Google Scholar]
- 36.(a) Dale JA, Mosher HS. J. Am. Chem. Soc. 1991;95:512. [Google Scholar]; (b) Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J. Am. Chem. Soc. 1991;113:4092. [Google Scholar]
- 37.Afonso CAM. Tetrahedron Lett. 1995;36:8857. [Google Scholar]
- 38.Blackwell HE, O’Leary DJ, Chatterjee AK, Washenfelder RA, Bussmann DA, Grubbs RH. J. Am. Chem. Soc. 2000;122:58. [Google Scholar]
- 39.Kingsbury JS, Harrity JPA, Bonitatebus PJ, Hoveyda AH. J. Am. Chem. Soc. 1999;121:791. [Google Scholar]
- 40.Six Y. Eur. J. Org. Chem. 2003:1157. [Google Scholar]
- 41.Gao Y, Hanson RM, Klunder JM, Ko SY, Masamune H, Sharpless KB. J. Am. Chem. Soc. 1987;109:5765. [Google Scholar]
- 42.Page PCB, Rayner CM, Sutherland IO. J. Chem. Soc. Perkin Trans. 1990;1:2403. [Google Scholar]
- 43.Rychnovsky SD, Rogers B, Yang G. J. Org. Chem. 1993;58:3511. [Google Scholar]
- 44.Yu W, Mei Y, Kang Y, Hua Z, Jin Z. Org. Lett. 2004;6:3217. doi: 10.1021/ol0400342. [DOI] [PubMed] [Google Scholar]
- 45.Still WC, Kahn M, Mitra A. J. Org. Chem. 1978;43:2923. [Google Scholar]
- 46.(a) Batey RA, Quach TD. Tetrahedron Lett. 2001;42:9099. [Google Scholar]; (b) Thadani AN, Batey RA. Org. Lett. 2002;4:3827. doi: 10.1021/ol026619i. [DOI] [PubMed] [Google Scholar]
- 47.Bagutski V, Ros A, Aggarwal VK. Tetrahedron. 2009;65:9956. [Google Scholar]
- 48.Lombardo M, Morganti S, Trombini C. J. Org. Chem. 2003;68:997. doi: 10.1021/jo0262457. [DOI] [PubMed] [Google Scholar]
