

Abstract
Reported herein is the isolation and structure elucidation of three highly modified peptides, actinoramides A-C (1–3), which are produced by a marine bacterium closely related to the genus Streptomyces. The planar structures of the actinoramides, which are composed of the unusual amino acids 2-amino-4-ureidobutanoic acid and 4-amino-3-hydroxy-2-methyl-5-phenylpentanoic acid, were assigned by chemical transformations and by interpretation of spectroscopic data, while the absolute configuration of these new peptides were defined by application of the advanced Marfey’s and Mosher’s methods.
Keywords: Marine Streptomyces, peptide hydrolysis, unusual amino acids, Advanced Marfey’s method, Advanced Mosher’s method
1. Introduction
Natural products from terrestrial actinomycetes have been used for developing and discovering new medicines for the last 50 years.1 However, a decline in the discovery of new chemical structures from terrestrial actinomycetes has led to a reduced pipeline of new drugs for pharmaceutical research.2 We have been investigating marine-derived actinomycetes as a source of novel secondary metabolites. Studies of marine sediments collected off the coast of southern California have revealed considerable actinomycete diversity including many new groups that, to date, have only been reported from marine samples.3 Included in one of these groups is the seawater-requiring strain CNQ-027. This strain shares only 97.6% 16S rRNA gene sequence identity with a marine Streptomyces species, S. marinus, suggesting it may represent a new Streptomyces species. The genus Streptomyces is mainly a terrestrial genus and is abundantly distributed in soil with literally hundreds of species reported. In our studies of the marine environment, we have repeatedly isolated seawater-requiring Streptomyces strains indicating that a subdivision of this genus is likely adapted to life in marine systems.3 In culture, Streptomyces strain CNQ-027 produced a series of unprecedented peptides that we have named actinoramides A-C. Herein, we describe the isolation and structural elucidation of these highly modified peptides.
2. Results and Discussion
Actinoramide A (1) was isolated as a yellow oil, which analyzed for the molecular formula C31H47N7O9 based on a pseudomolecular ion peak at m/z 662.3510 [M+H]+ in the HRESIMS and on interpretation of 13C NMR data. The 13C NMR spectrum of 1 in combination with data from 135° DEPT spectra revealed four methyl, seven methylene, thirteen methine, and seven fully substituted carbons. The 1H and gradient (g)COSY NMR spectra from 1 displayed the typical features for a peptide with three amide proton signals [δH 8.10, 7.59, 7.43], and four α-amino protons [δH 5.50, 4.95, 4.38, 4.13]. The 1H NMR spectrum also revealed mono-substituted benzene protons [δH 7.23 (2H), 7.21, 7.14 (2H)], two oxygenated methine protons [δH 3.44, and 3.43], three methyl doublets [δH 1.05, 0.87 and 0.83], and an O-methyl group [δH 3.37].
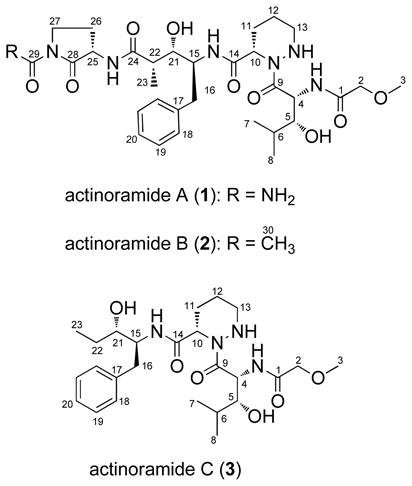
Analysis of gCOSY, gHSQC, and gHMBC NMR spectroscopic data allowed five distinct fragments to be discerned, including two rarely reported amino acids: 3-hydroxyleucine (Hle) and piperazic acid (Pip) and two unusual amino acids, 4-amino-3-hydroxy-2-methyl-5-phenylpentanoic acid (Ahmppa) and 2-amino-4-ureidobutanoic acid (Auba). The Hle and Pip residues were identified by interpretation of gCOSY and gHMBC NMR data. For Hle, gCOSY NMR crosspeaks permitted construction of the spin system from H-4 through H-8 [H-4 (δH 5.50) - H-5 (δH 3.43) - H-6 (δH 1.64) - H-7 (δH 0.87) and H-8 (δH 0.83)]. The gCOSY NMR correlations observed from H-5 to 5-OH (δH 4.79) indicated that the alcohol should be attached to C-5. The Hle residue was also confirmed by gHMBC NMR correlations from H-5 (δH 3.43) to C-9 (δC 173.0), C-4 (δC 50.4), C-6 (δC 29.8), C-7 (δC 20.8), and C-8 (δC 16.1). The connectivity from H-10 through H-13 in the Pip residue was established by analysis of the gCOSY crosspeaks [H-10 (δH 4.95) - H-11 (δH 2.09, 1.63) - H-12 (δH 1.34) - H-13 (δH 2.79)]. The NH (δH 4.50) proton coupled to H-13 (δH 2.79) displayed a gHMBC correlation to C-9 (δC 173.0). gHMBC correlations were also observed from the α proton H-10 (δH 4.95) to two carbonyl carbons C-14 (δC 171.0) and C-9 (δC 173.0). However, the α proton, H-10 (δH 4.95), was not coupled to the NH (δH 4.50), as revealed by interpretation of gCOSY spectral data. On this basis, this residue was assigned as a piperazic acid moiety. The Ahmppa residue was also constructed by analysis of gCOSY NMR correlations from methylene protons H-16 (δH 2.79) and an oxygenated proton H-21 (δH 3.44) to the α proton H-15 (δH 4.13), and from the oxygenated proton H-21 (δH 3.44) and the methyl doublet H-23 (δH 1.05) to the methine proton H-22 (δH 2.23), and by long-range gHMBC correlations from H-16 (δH 2.79) to carbons C-15 (δC 53.3), C-17 (δC 139.9), C-18 (δC 129.8), and C-21 (δC 72.4) and from the methyl doublet H-23 (δH 1.05) to carbons C-21 (δC 72.4), C-22 (δC 43.7), and C-24 (δC 174.8). The cyclic 2-amino-4-ureidobutanoic acid (Auba) residue was assigned by interpretation of gCOSY crosspeaks from the α proton H-25 (δH 4.38) and H-27 (δH 3.71, 3.43) toH-26 (δH 2.15, 1.93), along with gHMBC correlations from the α proton H-25 (δH 4.38) to a carbon C-28 (δC 175.2) and from proton H-27 (δH 3.71) to carbons C-25 (δC 52.8), C-26 (δC 23.9), C-28 (δC 175.2), and C-29 (δC153.4). Lastly, the 2-methoxyacetamide moiety was constructed by analysis of gHMBC correlations from the downfield methylene protons [H-2 (δH 3.81)] to C-1 (δC 169.0) and C-3 (δC 59.2).
The sequence of amino acids in actinoramide A was determined by interpretation of key long-range gHMBC correlations. The linkage of Pip to Hle was achieved on the basis of gHMBC correlations from the α-amino proton of Hle [H-4 (δH 5.50)] and the α-amino proton of Pip [H-10(δH 4.95)] to the Hle amide carbonyl [C-9 (δC 173.0)]. The gHMBC correlation observed from the α-amino proton of Hle [H-4 (δH 5.50)] to the acetamide carbon [C-1 (δC 169.0)] established the connectivity of those two residues. The gHMBC correlations from both the α-amino proton of Ahmppa [H-15 (δH 4.13)] and the α-amino proton of Pip [H-10 (δH 4.95)] to the Pip amide carbonyl [C-14 (δC 171.0)] revealed Ahmppa to be adjacent to Pip. Lastly, gHMBC correlation of the α-amino proton of Auba [H-25 (δH 4.38)] to the Ahmppa amide carbonyl [C-24 (δC 174.8)] allowed the connection between Auba and Ahmppa to be established as shown below (Figure 1).
Figure 1.

Structure of actinoramide A (1) with COSY (bold lines) and key HMBC (solid arrows) correlations.
Actinoramide B (2) was isolated as a yellow oil, which analyzed for the molecular formula C32H48N6O9 based on HRESIMS data (a pseudomolecular ion peak at m/z 661.3550 [M+H]+). The 1H NMR spectrum of 2 was almost identical to that of 1 except that 2 displayed an additional methyl singlet. The observation of a gHMBC correlation from the methyl singlet to the C-29 carbon indicated that 2 had a methyl group at C-29 instead of an NH2 group. Thus, 2 was composed of a cyclic 4-acetamido-2-aminobutanoic acid (Aaba) unit. Analysis of 2D NMR spectroscopic data allowed the planar structure of 2 to be assigned as shown.
The molecular formula of actinoramide C (3) was assigned as C25H40N4O6 based on a pseudomolecular ion peak at m/z 493.2999 [M+H]+ observed in the HRESIMS data. The 1H NMR spectrum of 3 had similar features to that of 1, however differences included the absence of the α-amino proton and two methylene protons for Auba and the presence of a methyl triplet. Further interpretation of gCOSY, gHSQC, and gHMBC NMR spectroscopic data indicated that 3 contained a 2-amino-1-phenylpentan-3-ol moiety (App) in place of the Ahmppa adjacent to the Pip in 1. Interpretation of gCOSY NMR crosspeaks allowed the construction of two spin systems consisting of a five carbon unit (H-23-H-22-H-21-H15-H-16) and a benzene ring [H-18 (H-18′)-H-19 (H-19′)-H-20)]. The connection between C-16 and C-17 was established on the basis of gHMBC correlations from the methylene protons H-16 to carbons C-15, C-17, C-18, and C-21. These data permitted the assignment of the App residue.
The absolute configurations of actinoramides were assigned by a combination of spectral and chemical methods. We proposed that actinoramides A-C possessed the same absolute configurations based on their simultaneous isolation and similar NMR and CD spectroscopic data (Figure S1, Supporting Information).
The absolute configuration of C-4 in Hle was determined as R by acid hydrolysis followed by the application of the advanced Marfey’s method. The 1-fluoro-2,4-dinitrophenyl-5-L-leucinamide (L- FDLA) derivative of Hle was detected by LC-MS analysis at 46.3 min, while the L- and D-FDLA derivatives of Hle were detected by LC-MS analysis at 46.3 and 39.3 min, respectively. Due to the structural similarity of Hle and threonine (Thr), the elution behavior of FDLA derivatives of Hle was expected to be similar to Thr. The L-FDLA derivative of R-Thr eluted later than D-FDLA derivative of R-Thr.4 This result allowed the configuration at C-4 of Hle to be assigned as 4R. To determine the absolute configuration of C-5 for Hle, the advanced Mosher’s method was applied.5 Treatment of 1 with (R)- and (S)-MTPA-Cl [α-methoxy-α-(trifluoromethyl) phenylacetyl chloride] yielded the (S)- and (R)-MTPA ester derivatives, respectively. Calculation of 1H ΔδS-R values of bis-MTPA esters of 1 from proton NMR data established the absolute configuration of C-21 of Ahmppa as S. However, determination of the absolute configuration at C-5 of Hle proved to be more difficult. The 1H ΔδS-R values of the bis-MTPA esters (4a/b) were inconsistent (Figure 2a), thus we prepared mono-MTPA esters using (R)- and (S)- MTPA-Cl (5a/b and 6a/b). Unfortunately, 1H ΔδS-R values for C-5 of Hle in the mono-MTPA esters were also inconsistent (The 1H ΔδS-R values for C-21 of Ahmppa in the mono-MTPA esters further confirmed the assignment of S revealed by the analysis of the bis-MTPA esters, as shown in Figure 2b). Finally, calculation of 19F ΔδS-R values for the mono-MTPA esters allowed the absolute configuration at C-5 of Hle to be assigned as S (Figure 2c).6 Unfortunately, it became subsequently clear that many configurations have been incorrect when assigned on the basis of 19F NMR data alone.5,7 Therefore, we prepared the L-FDLA derivative of (2R, 3S)-Hle from lobocyclamide B, a cyanobacterial metabolite isolated from a complex cyanobacterial mat containing Lyngbya confervoides.8a The retention time of the L-FDLA derivative of (2R, 3S)-Hle of lobocyclamide B was 50.8 min. This result was not consistent for the assigned absolute configuration of C-5 of actinoramide A from the analysis of 19F NMR data.
Figure 2.

ΔδS-R of 1H for S- and R-bis-MTPA esters of actinoramide A (a). ΔδS-R of 1H for S- and R-mono-MTPA esters of Ahmppa (b), and ΔδS-R of 1H (plain) and 19F (bold italic) for S- and R-mono-MTPA esters of Hle (c).
Next, we prepared L-FDLA derivatives of authentic (2S, 3R)- and (2S, 3S)-Hle from dentigerumycin a metabolite produced by Pseudonocardia sp.8b The retention times of the L-FDLA derivatives of (2S, 3R)- and (2S, 3S)-Hle were 36.8 and 39.3 min, respectively. The elution order of L-FDLA derivatives of four stereoisomers of Hle were consistent with those reported by MacMillan et al., who demonstrated the elution behaviors of 1-fluoro-2,4-dinitrophenyl-5-L-alaninamide (L-FDAA) derivatives of Hle.8a Furthermore, while Hle from actinoramide A has 2R and 3R configurations, the D-FDLA derivative of (2S, 3S)- and L-FDLA derivative of (2R, 3R)-Hle were expected to have same retention time due to enantiomers. The D-FDLA derivative of (2S, 3S) was observed at 46.3 min which was the elution time of L-FDLA derivative of Hle from actinoramide A. These data suggested that the absolute configurations of Hle from actinoramide would be 4R, and 5R, respectively.
The configuration of Pip was determined as S by acid hydrolysis and application of Marfey’s method using standards of known configuration. The L-FDLA derivative of Pip in actinoramide A (1) was detected at 38.5 min by LC-MS analysis. L-FDLA derivatives of authentic R-Pip and S-Pip were eluted by LC-MS analysis at 35.3 and 38.5 min, respectively.
The absolute configuration of Auba was assigned as S by acid hydrolysis and comparison of the advanced Marfey’s products by LC-MS. The L-FDLA derivative of Auba with m/z = 456 [M+H]+ was detected by LC-MS analysis at 31.5 min, while the D- and L-FDLA derivatives of Auba were detected at 29.0 and 31.5 min, respectively. The structure of the L-FDLA derivative of Auba (7) was further characterized by NMR (Scheme 1). Although, there is no standard available for Auba, we identified two amino acids (ureido-alanine, and citrulline) with similar structures to Auba. Ureido-alanine has a side chain that is one carbon shorter than Auba, while citrulline (Cit) has a side chain that is one carbon larger than Auba. It is reasonable to expect the elution behavior of FLDA derivatives of these two amino acids to be similar to those of Auba. L-FDLA derivatives of commercial S-Cit eluted later than that of R-Cit. Furthermore, the amino L-FDLA derivatives of S-ureido-alanine and L-FDLA derivatives of S-Cit were detected by LC-MS analysis at 30.5 and 32.7 min, respectively, while the L-FDLA derivative of Auba was detected at 31.5 min. These results allowed the absolute configuration of Auba to be assigned as S.
Scheme 1.

Reaction sequence for determination of the absolute configuration of 2-amino-4-ureidobutanoic acid
The relative configurations of the remaining stereocenters in Ahmppa were determined by spectroscopic data analysis. 5-Benzyl-4-hydroxy-3-methylpyrrolidin-2-one (8) was isolated from the acid hydrolysis products of actinoramide A after multiple modifications of the hydrolysis conditions (Scheme 2). The relative configuration of 8 was assigned by interpretation of NOESY correlation data. The methyl protons H-3 (δH 0.99) showed strong correlations with H-4 (δH 3.84), and H-5 (δH 3.65) in the NOESY spectrum, which allowed the relative configuration to be designated as (2R*, 4R*, 5R*). On the basis of their relative configurations and the previously described application of advanced Mosher’s method, the absolute configuration of Ahmppa in actinoramide A was assigned as 15S, 21S, 22S.
Scheme 2.

Reaction for determination of the absolute configuration of 4-amino-3-hydroxy-2-methyl-5-phenylpentanoic acid
Actinoramide A is unique in its incorporation of the uncommon amino acids 2-amino-4-ureidobutanoic acid (Auba), and 4-amino-3-hydroxy-2-methyl-5-phenylpentanoic acid (Ahmppa), and rare known amino acids, piperazic acid (Pip) and 3β-hydroxyleucine (Hle). The Auba unit was previously reported in the synthetic literature, as a reagent for preparing peptidomimetics9 and the Ahmppa unit was also reported in the synthesis of bleomycin analogs.10 This is the first report of Auba and Ahmppa components in a natural product.
The crude culture extract of Streptomyces strain CNQ-027 showed NF-κB inhibitory activity, but unfortunately this activity was lost and actinoramides were found not to be active. The actinoramides were evaluated in limited bioassays. They displayed no significant activity against HCT-116 human colon carcinoma, methicillin-resistant Staphylococcus aureus, or the breast cancer-related enzyme aromatase.
3. Experimental
3.1. General experimental procedures
Optical rotations were measured using a Rudolph Research Autopol III polarimeter with a 10-cm cell. UV data were recorded using a Varian Cary UV-visible spectrophotometer with a path length of 1 cm. The IR spectrum was recorded on a Perkin-Elmer 1600 FT-IR spectrometer. Proton and 2D NMR spectra were recorded in DMSO-d6 or methanol-d4 containing Me4Si as internal standard on Varian Inova spectrometers at 500 or 600 MHz. C-13 NMR spectra were acquired on Varian Inova spectrometers at 75 or 150 MHz. High resolution mass TOF spectra were acquired at The Scripps Research Institute, La Jolla, CA. Low resolution LC-MS data were obtained using a Hewlett-Packard series 1100 LC/MS system with a reversed-phase C18 column (Phenomenex Luna, 4.6 mm × 100 mm, 5 μm) at a flow rate of 0.7 mL/min.
3.2. Collection and phylogenetic analysis of strain CNQ-027
The marine Streptomyces strain CNQ-027 was isolated from a marine sediment sample collected at a depth of 50 m off the coast of San Diego, CA. NCBI blastn analysis of the partial (1480 base pairs) 16S rDNA gene sequence of CNQ-027 (GenBank accession number EU214912) indicates that this strain shares only 97.6% sequence identity to Streptomyces marinus, thus is likely to be a new marine species.
3.2. Cultivation and extraction
Marine Streptomyces sp. (strain CNQ-027) was cultured in forty 2.8 L Fernbach flasks each containing 1 L of a seawater-based medium (10 g starch, 4 g yeast extract, 2 g peptone, 1 g CaCO3, 40 mg Fe2(SO4)3•4H2O, 100 mg KBr) and shaken at 230 rpm at 27 °C. After seven days of cultivation, sterilized XAD-16 resin (20 g/L) was added to adsorb the organic substances, and the culture and resin were shaken at 215 rpm for 2 hours. The resin was filtered through cheesecloth and washed with deionized water, and eluted with acetone. The acetone was removed under reduced pressure, and the resulting aqueous layer was extracted with ethyl acetate (3 × 500 mL). The ethyl acetate-soluble fraction was dried in vacuo to yield 4.5 g of crude material from a 40 L culture.
3.3. Isolation of actinoramides A-C (1–3)
The crude extract (4.5 g) from strain CNQ-027 was fractionated by open column chromatography on silica gel (25 g) eluting with a step gradient of dichloromethane and methanol. The dichloromethane/methanol 10:1 fraction contained a mixture of actinoramide peptides, which were purified by reversed-phase HPLC (Phenomenex Ultracarb C30, 5 μm, 100 Å, 250 × 100 mm, 2.0 ml/min, UV = 210 nm), eluting with 35% CH3CN in H2O to afford actinoramide A (1, 374.0 mg), B (2, 5.0 mg) and C (3, 24.0 mg), as pale yellow oils.
3.4. Properties of actinoramides A-C (1–3)
3.4.1. Actinoramide A (1)
[α]D21 -18 (c 0.1, methanol); IR (KBr) ν max 3395, 2941, 1725, 1650, 1540, 1361 cm−1; UV (methanol) λmax (log ε) 211 (3.2), 252 (1.4) nm; 1H and 13C NMR data, See Table 1; ESI-TOF m/z 662 [M+H]+; HR ESI-TOF m/z 662.3510 (calcd. for C31H48N7O9, 662.3508).
Table 1.
NMR Data of 1 (DMSO-d6) a
| No. | δC, mult.b | δH (J in Hz) | COSY | HMBC | |
|---|---|---|---|---|---|
| 1 | 169.0, C | ||||
| 2 | 71.8, CH2 | 3.81, d (2.0) | 1, 3 | ||
| 3 | 59.2, CH3 | 3.37, s | 2 | ||
| Hle | 4-NH | 7.43, d (8.8) | 4 | 1, 9 | |
| 4 | 50.4, CH | 5.50, dd (8.8, 8.8) | 4-NH, 5 | 1, 5, 6, 9 | |
| 5 | 75.7, CH | 3.43* | 4, 5-OH, 6 | 4, 6, 7, 8, 9 | |
| 5-OH | 4.79, d (7.1) | 5 | |||
| 6 | 29.8, CH | 1.64, m | 5, 7, 8 | 4, 5, 7, 8 | |
| 7 | 20.8, CH3 | 0.87, d (7.1) | 6 | 5, 6, 8 | |
| 8 | 16.1, CH3 | 0.83, d (7.1) | 6 | 5, 6, 7 | |
| 9 | 173.0, C | ||||
| Pip | 13-NH | 4.50, br s | 13 | 9 | |
| 10 | 51.5, CH | 4.95, d (4.9) | 11 | 9, 11, 12, 14 | |
| 11 | 26.5, CH2 | 2.09, m, 1.63, m | 10, 12 | 10, 12, 13 | |
| 12 | 21.4, CH2 | 1.34, m | 11, 13 | 10, 11, 13 | |
| 13 | 46.7, CH2 | 2.79* | 12 | 11, 12 | |
| 14 | 171.0, C | ||||
| Ahmppa | 15-NH | 7.59, d (8.0) | |||
| 15 | 53.3, CH | 4.13, dd (8.0, 8.0) | 15-NH, 16, 21 | 14, 16, 17, 21, 22 | |
| 16 | 38.3, CH2 | 2.79* | 15 | 15, 17, 18, 21 | |
| 17 | 139.9, C | ||||
| 18, 18′ | 129.8, CH | 7.21, dd (7.4, 1.2) | 19 | 16, 17, 19, 20 | |
| 19, 19′ | 128.8, CH | 7.23, dd (7.4, 7.4) | 18, 20 | 17, 18, 20 | |
| 20 | 126.6, CH | 7.14, dd (7.4, 1.2) | 19 | 18, 19 | |
| 21 | 72.4, CH | 3.44* | 15, 21-OH, 22 | 15, 22, 23, 24 | |
| 21-OH | 5.08, d (7.1) | 21 | |||
| 22 | 43.7, CH | 2.23, q (6.6) | 21, 23 | 15, 21, 23, 24 | |
| 23 | 15.7, CH3 | 1.05, d (6.6) | 22 | 21, 22, 24 | |
| 24 | 174.8, C | ||||
| Auba | 25-NH | 8.10, q (10.3) | 25 | 24, 28 | |
| 25 | 52.8, CH | 4.38, dd (10.3, 8.1) | 25-NH, 26 | 24, 26, 28 | |
| 26 | 23.9, CH2 | 2.15, q (10.3), 1.93, q(10.3) | 25, 27 | 25, 27, 28 | |
| 27 | 42.1, CH2 | 3.71 dd (10.3, 10.3), 3.43* | 26 | 25, 26, 28, 29 | |
| 28 | 175.2, C | ||||
| 29 | 153.4, C | ||||
| 29- NH2 | 7.40, br s, 7.74, br s |
500 MHz for 1H NMR and 75 MHz for 13C NMR.
Numbers of attached protons were determined by analysis of DEPT spectra.
The coupling constant was not determined because of overlapping signals.
3.4.2. Actinoramide B (2)
[α]D21 -27 (c 0.1, methanol); IR (KBr) ν max 3394, 2940, 1724, 1651, 1542, 1360 cm−1; UV (methanol) λmax (log ε) 211 (3.2), 252 (1.4) nm; 1H and 13C NMR data, See Table 2; ESI-TOF m/z 661 [M+H]+; HR ESI-TOF m/z 661.3550 (calcd. for C32H49N6O9, 661.3555).
Table 2.
NMR Data of 2 and 3 (DMSO-d6)a
| No. | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| δC, mult.b | δH (J in Hz) | δC, mult.b | δH (J in Hz) | |||
| 1 | 168.3, C | 169.0, C | ||||
| 2 | 71.1, CH2 | 3.81, d (2.0) | 71.1, CH2 | 3.81, d (2.0) | ||
| 3 | 58.6, CH3 | 3.37, s | 58.5, CH3 | 3.32, s | ||
| Hle | 4-NH | 7.44, d (8.8) | Hle | 7.48, d (8.4) | ||
| 4 | 49.9, CH | 5.50, dd (8.8, 8.8) | 50.0, CH | 5.48, dd (8.4, 8.4) | ||
| 5 | 75.1, CH | 3.43* | 75.0, CH | 3.40* | ||
| 5-OH | 4.93, br s | 4.84, br s | ||||
| 6 | 29.2, CH | 1.64, m | 29.1, CH | 1.64, m | ||
| 7 | 20.0, CH3 | 0.87, d (7.1) | 20.1, CH3 | 0.88, d (6.8) | ||
| 8 | 15.5, CH3 | 0.83, d (7.1) | 15.6, CH3 | 0.83, d (6.8) | ||
| 9 | 172.2, C | 172.7, C | ||||
| Pip | 13- NH | 4.50, br s | Pip | 4.43, br s | ||
| 10 | 51.0, CH | 4.95, d (4.9) | 50.6, CH | 4.95, d (4.9) | ||
| 11 | 25.9, CH2 | 2.09, m, 1.63, m | 26.0, CH2 | 2.03, m, 1.63, m | ||
| 12 | 20.7, CH2 | 1.34, m | 20.7, CH2 | 1.34, m, 1.15, m | ||
| 13 | 46.6, CH2 | 2.79* | 46.0, CH2 | 2.79* | ||
| 14 | 170.2, C | 170.5, C | ||||
| Ahmppa | 15-NH | 7.66, d (8.0) | App | 7.69, d (8.0) | ||
| 15 | 52.7, CH | 4.13, dd (8.0, 8.0) | 53.0, CH | 4.03, dd (8.0, 8.0) | ||
| 16 | 37.9, CH2 | 2.79* | 36.4, CH2 | 2.68, dd(14.0,6.5), 2.85, dd (14.0, 6.5) | ||
| 17 | 139.9, C | 139.9, C | ||||
| 18, 18′ | 129.1, CH | 7.21, dd (7.4, 1.2) | 129.1, CH | 7.21, dd (7.4, 1.2) | ||
| 19, 19′ | 128.8, CH | 7.23, dd (7.4, 7.4) | 128.1, CH | 7.23, dd (7.4, 7.4) | ||
| 20 | 126.6, CH | 7.14, dd (7.4, 1.2) | 126.6, CH | 7.14, dd (7.4, 1.2) | ||
| 21 | 71.8, CH | 3.44* | 71.8, CH | 3.21* | ||
| 21-OH | 4.82, br s | |||||
| 22 | 43.0, CH | 2.23, q (6.6) | 26.4, CH2 | 1.28, m | ||
| 23 | 14.9, CH3 | 1.05, d (6.6) | 11.3, CH3 | 0.79, t (6.8) | ||
| 24 | 174.2, C | |||||
| Aaba | 25-NH | 8.20, q (10.3) | ||||
| 25 | 52.1, CH | 4.38, dd (10.3, 8.1) | ||||
| 26 | 23.2, CH2 | 2.15, q (10.3), 1.93, q (10.3) | ||||
| 27 | 41.3, CH2 | 3.71 dd (10.3, 10.3), 3.43* | ||||
| 28 | 173.3, C | |||||
| 29 | 170.3, C | |||||
| 30 | 25.5, CH3 | 2.37, s | ||||
500 MHz for 1H NMR and 75 MHz for 13C NMR.
Numbers of attached protons were determined by analysis of 2D spectroscopic data.
The coupling constant was not determined because of overlapping signals.
3.4.3. Actinoramide C (3)
[α]D21 -52 (c 0.2, methanol); IR (KBr) ν max 3393, 2941, 1725, 1653, 1540, 1361 cm−1; UV (methanol) λmax (log ε) 211 (3.1), 252 (1.4) nm; 1H and 13C NMR data, See Table 2; ESI-TOF m/z 493 [M+H]+; HR ESI-TOF m/z 493.2999 (calcd. for C25H41N4O6, 493.3020).
3.5. Bis-MTPA esters of actinoramide A
Actinoramide A (1, 10.0 mg) was dissolved in freshly distilled dry pyridine (1 ml) with several dry crystals of dimethylaminopyridine. The mixture was stirred for 15 min at room temperature. Treatment with R(-)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (MTPA-Cl) at 60 °C yielded the S-MTPA ester after 3 days. The reaction was quenched with 1 mL of methanol and extracted with EtOAc. After removal of solvent under vacuum, the residue was purified by reversed-phase HPLC (Phenomenex Luna C18 (2), 5 μm, 250 × 100 mm, 2.0 ml/min, 100 Å, UV =254 nm) using a gradient solvent system (isocratic 70% aqueous CH3CN for 20 min; 70% aqueous CH3CN to 100% aqueous CH3CN over 40 min; 100% CH3CN for 15 min). S-MTPA ester was obtained at 42 min. R-MTPA ester was prepared with S-MTPA-Cl in the same manner. ΔδS-R values for bis- S- and R-MTPA esters of actinoramide were recorded in ppm in methanol-d4.
3.5.1. Bis-S-MTPA ester (4a)
1H NMR (500 MHz, methanol-d4): δ 7.59 (d, 2H, J = 8.0 Hz), 7.49-7.38 (m, 8H), 7.25 (dd, 2H, J = 8.0, 8.0 Hz), 7.18 (m, 3H), 5.90 (d, 1H, J = 7.4 Hz), 5.52 (dd, 1H, J = 7.4, 3.2 Hz), 5.45 (dd, 1H, J = 7.4, 3.2 Hz), 4.78 (dd, 1H, J = 11.4, 8.4 Hz), 4.61 (m, 2H), 3.99 (d, 1H, J = 4.8 Hz), 3.91 (m, 2H), 3.60 (m, 1H), 3.53 (s, 3H), 3.51 (s, 3H), 3.40 (s, 3H), 2.93 (dd, 1H, J = 12.0, 4.3 Hz), 2.82 (m, 2H), 2.72 (d, 1H, J = 12.1 Hz), 2.56 (t, 1H, J = 12.1 Hz), 2.37 (m, 1H), 2.07 (m, 1H), 1.99 (t, 1H, J = 11.0 Hz), 1.88 (d, 1H, J = 13.0 Hz), 1.19 (d, 1H, J = 13.0 Hz), 1.09 (d, 3H, J = 8.0 Hz), 1.05 (d, 3H, J = 6.8 Hz), 1.03 (d, 3H, J = 6.8 Hz), 0.95 (m, 2H); ESI-MS: m/z 1095 [M+H]+, ESI-MS m/z 1117 [M+Na] +.
3.5.2. Bis-R-MTPA ester (4b)
1H NMR (500 MHz, methanol-d4): δ 7.63 (m, 2H), 7.55 (m, 2H), 7.46 (m, 3H), 7.42-7.41 (m, 3H), 7.22 (dd, 2H, J = 8.0, 8.0 Hz), 7.15 (dd, 1H, J = 8.0, 8.0 Hz), 7.09 (d, 2H, J = 8.0 Hz), 5.83 (d, 1H, J = 7.4 Hz), 5.51 (dd, 1H, J = 7.4, 3.2 Hz), 5.36 (dd, 1H, J = 7.4, 3.2 Hz), 4.77 (dd, 1H, J = 11.4, 8.4 Hz), 4.53 (m, 1H), 3.91 (s, 1H), 3.89 (m, 2H), 3.61 (m, 2H), 3.60 (s, 3H), 3.51 (s, 3H), 3.39 (s, 3H), 2.86 (m, 2H), 2.72 (m, 2H), 2.37 (m, 2H), 2.06-2.01 (m, 3H), 1.55 (m, 1H), 1.24 (d, 1H, J = 13.0 Hz), 1.22 (d, 3H, J = 7.0 Hz), 1.19 (m, 1H), 0.95 (d, 3H, J = 6.8 Hz), 0.86 (d, 3H, J = 6.8 Hz); ESI-MS: m/z 1095 [M+H]+, ESI-MS m/z 1117 [M+Na] +.
3.6. Mono-MTPA esters of actinoramide A
Actinoramide A (1, 3.0 mg) was dissolved in freshly distilled dry pyridine (1 ml) with several dry crystals of dimethylaminopyridine. The mixture was stirred for 15 min at room temperature. Treatment with R(-)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (MTPA-Cl) at room temperature yielded the S-MTPA ester after 11 hrs. The reaction was quenched with 1 mL of methanol and extracted with EtOAc. After removal of solvent under vacuum, the residue was purified by reversed-phase HPLC (Phenomenex Luna C18 (2), 5 μm, 100 Å, 250 × 100 mm, 2.0 ml/min, UV = 254 nm) using an isocratic solvent system with 60% aqueous CH3CN. The S-MTPA esters were obtained at 42 and 48 min, respectively. R-MTPA esters were prepared with S-MTPA-Cl in the same manner. The ΔδS-R values for S- and R-MTPA esters of actinoramide A were recorded in ppm in methanol-d4.
3.6.1. Ahmppa-S-MTPA ester of actinoramide A (5a)
1H NMR (600 MHz, methanol-d4): δ 7.51 (d, 1H, J = 8.0 Hz), 7.46 (d, 2H, J = 8.0 Hz), 7.38-7.36 (m, 2H), 7.17 (dd, 2H, J = 8.0, 8.0 Hz), 7.13 (d, 2H, J = 8.0 Hz), 7.05 (dd, 1H, J = 8.0, 8.0 Hz), 5.45 (dd, 1H, J = 8.0, 4.0 Hz), 5.24 (d, 1H, J = 8.0 Hz), 3.83 (m, 1H), 3.79 (m, 1H), 3.51 (dd, 1H, J = 8.0, 4.0 Hz), 3.47 (dd, 1H, J = 10.0, 4.0 Hz), 3.37 (s, 3H), 3.36 (m, 1H), 3.29 (s, 3H), 3.28 (m, 1H), 3.19 (m, 1H), 2.90 (m, 1H), 2.71-2.69 (m, 2H), 2.55 (m, 1H), 2.47 (m, 1H), 2.25 (m, 1H), 2.06 (d, 1H, J = 14.0 Hz), 1.91 (t, 1H, J = 14.0 Hz), 1.63 (m, 1H), 1.46-1.41 (m, 2H), 1.18-1.16 (m, 2H), 0.96 (d, 3H, J = 8.0 Hz), 0.90 (d, 3H, J = 6.8 Hz), 0.84 (d, 3H, J = 6.8 Hz); ESI-MS: m/z 878 [M+H]+, ESI-MS m/z 900 [M+Na] +.
3.6.2. Ahmppa-R-MTPA ester of actinoramide A (5b)
1H NMR (600 MHz, methanol-d4): δ 7.51 (d, 1H, J = 8.0 Hz), 7.47 (d, 2H, J = 8.0 Hz), 7.38 (m, 1H), 7.17 (dd, 2H, J = 8.0, 8.0 Hz), 7.11 (d, 2H, J = 8.0 Hz), 7.05 (dd, 1H, J = 8.0, 8.0 Hz), 7.05 (d, 1H, J = 8.0), 5.42 (dd, 1H, J = 8.0, 8.0 Hz), 5.35 (d, 1H, J = 8.0 Hz), 4.55 (dd, 1H, J = 8.0, 8.0 Hz), 4.49 (m, 1H), 3.76 (m, 1H), 3.46 (m, 3H), 3.44 (s, 3H), 3.31 (m, 1H), 3.30 (s, 3H), 2.76 (dd, 2H, J = 14.0, 2.0 Hz), 2.75 (dd, 1H, J = 8.0, 8.0 Hz), 2.58 (m, 2H), 2.48 (d, 1H, J = 8.0 Hz), 2.23 (m, 1H), 1.96 (d, 1H, J = 14.0 Hz), 1.90 (t, 1H, J = 14.0 Hz), 1.59 (m, 1H), 1.39 (m, 1H), 1.13 (m, 2H), 1.10 (d, 3H, J = 8.0 Hz), 0.85 (d, 3H, J = 6.8 Hz), 0.80 (d, 3H, J = 6.8 Hz); ESI-MS: m/z 878 [M+H]+, ESI-MS m/z 900 [M+Na]+.
3.6.3. Hle-S-MTPA ester of actinoramide A (6a)
1H NMR (600 MHz, methanol-d4): δ 7.34 (d, 2H, J = 8.0 Hz), 7.31-7.28 (m, 3H), 7.15 (d, 2H, J = 8.0 Hz), 7.12 (d, 1H, J = 8.0 Hz), 7.10 (d, 1H, J = 8.0 Hz), 7.03 (dd, 1H, J = 8.0, 8.0 Hz), 5.41 (dd, 1H, J = 12.0, 2.0 Hz), 4.54 (dd, 1H, J = 11.4, 10.4 Hz), 4.49 (m, 1H), 4.19 (m, 1H), 3.79 (s, 3H), 3.77 (m, 2H), 3.48 (d, 1H, J = 10 Hz), 3.45 (d, 1H, J = 12 Hz), 3.41 (m, 2H), 3.40 (s, 3H), 3.17 (m, 1H), 2.84 (t, 1H, J = 14.0 Hz), 2.77 (dd, 1H, J = 14.0, 2.0 Hz), 2.44 (t, 1H, J = 14.0 Hz), 2.27 (m, 2H), 2.18 (m, 1H), 1.96 (m, 1H), 1.91 (m, 2H), 1.77 (d, 1H, J = 14.0 Hz), 1.00 (d, 3H, J = 8.0 Hz), 0.93 (d, 3H, J = 6.8 Hz), 0.89 (d, 3H, J = 6.8 Hz), 0.88 (m, 1H); ESI-MS m/z 878 [M+H]+, ESI-MS m/z 900 [M+Na]+.
3.6.4. Hle-R-MTPA ester of actinoramide A (6b)
1H NMR (600 MHz, methanol-d4): δ 7.46 (d, 1H, J = 8.0 Hz), 7.33 (d, 2H, J = 8.0 Hz), 7.24 (d, 2H, J = 8.0 Hz), 7.11 (d, 2H, J = 8.0 Hz), 7.05 (dd, 2H, J = 8.0, 8.0 Hz), 6.97 (dd, 1H, J = 8.0, 8.0 Hz), 5.63 (d, 1H, J = 8.0 Hz), 4.97 (dd, 1H, J = 8.0, 2.0 Hz), 4.50 (dd, 1H, J = 14.0, 8.0 Hz), 4.16 (m, 1H), 3.77 (m, 1H), 3.74 (s, 3H), 3.44 (d, 1H, J = 2.0 Hz), 3.42 (d, 1H, J = 2.0 Hz), 3.40 (dd, 1H, J = 8.0, 2.0 Hz), 3.33 (m, 1H), 3.29 (s, 3H), 3.10 (m, 1H), 2.79 (m, 1H), 2.73 (dd, 1H, J = 14.0, 2.0 Hz), 2.51 (d, 1H, J = 14.0 Hz), 2.26 (t, 1H, J = 14.0 Hz), 2.20 (m, 1H), 2.15 (m, 1H), 1.96 (m, 1H), 1.87 (m, 2H), 1.37 (m, 1H), 1.21 (d, 1H, J = 14.0 Hz), 1.01 (d, 3H, J = 6.9 Hz), 0.94 (d, 1H, J = 14.0 Hz), 0.76 (d, 3H, J = 6.8 Hz), 0.68 (d, 3H, J = 6.8 Hz); ESI-MS: m/z 878 [M+H]+, m/z 900 [M+Na] +.
3.7. Analytical-scale advanced Marfey’s reaction
Actinoramide A (1, 0.2 mg) was hydrolyzed in 0.4 mL of 6 N HCl at 110 °C for 1 h; the HCl was removed under vacuum; and the dry material was resuspended in 0.4 mL of water and dried three times to remove residual acid. The hydrolysate was divided into two portions and dissolved in 1 N NaHCO3 (100 μL). To the first portion was added 50 μL of 1% L-FDLA (1-fluoro-2,4-dinitrophenyl-5-L-leucinamide) in acetone; to the second portion was added 25 μl of 1%D- and L-FDLA mixtures in acetone. The reaction mixtures were incubated at 60 °C for 30 min, and quenched with 100 μL of 2 N HCl. A small portion (100 μL) of CH3CN was added to the solution to dissolve the reaction mixture. The resulting products were analyzed by reversed-phase LC-MS with a gradient solvent system (95 % aqueous CH3CN to 50% aqueous CH3CN (0.1% TFA) over 50 min; 50% aqueous CH3CN to 100% aqueous CH3CN (0.1% TFA) over 20 min; 100% CH3CN (0.1% TFA) for 15 min).
3.8. Preparatory-scale advanced Marfey’s reaction and isolation of 7 and 8
Actinoramide A (1, 20.0 mg) was hydrolyzed in 10 mL of 6 N HCl at 110 °C for 1h, and the HCl was removed under vacuum. The dry material was purified by reversed-phase HPLC (Phenomenex Ultracarb C30, 5 μm, 100 Å, 250 × 100 mm, 2.0 ml/min, detection at 254 nm) using a gradient solvent system from 5% to 40% CH3CN (0.05% TFA) over 50 min. Compound 8(5-benzyl-4-hydroxy-3-methylpyrrolidin-2-one) was obtained at 25.1 min. The residues from HPLC were dried under vacuum, dissolved in 1 N NaHCO3 (2 mL), and 2 mL of 1% L-FDLA solution was added. The reaction mixture was incubated at 60 °C for 30 min and quenched by neutralization with 2 mL of 2N HCl. The products were purified by reversed-phase HPLC (Phenomenex Ultracarb C30, 5 μm, 100 Å, 250 × 100 mm, 2.0 ml/min, detection at 338 nm) using a gradient solvent system from 5% to 40% CH3CN (0.05% TFA) over 60 min. Auba-L-FDLA (7) was obtained at 43.0 min.
3.8.1. Auba-L-FDLA (7)
[α]D25 +31 (c 0.05, methanol); 1H NMR (600 MHz, DMSO-d6): δ 8.96 (s, 1H), 8.75 (d, NH, J = 6.8 Hz), 8.51 (d, NH, J = 6.8 Hz), 7.86 (s, NH), 7.40 (s, NH), 6.09 (s, NH), 5.76 (s, 1H), 5.53 (s, NH2), 4.48 (m, 1H), 4.27 (m, 1H), 3.05 (m, 2H), 1.97 (m, 2H), 1.71 (m, 3H), 0.98 (d, 3H, J = 6.9 Hz), 0.88 (d, 3H, J = 6.9 Hz); ESI-TOF-MS: m/z 456 [M+H]+; HRESI-TOF-MS m/z 456.1832 (calcd. for C17H25N7O8, 456.1837).
3.8.2. 5-benzyl-4-hydroxy-3-methylpyrrolidin-2-one (8)
[α]D25 -61 (c 0.1, methanol); 1H NMR (600 MHz, DMSO-d6): δ 7.56 (s, 2-NH), 7.27 (dd, H-9 and H-9′, J = 8.0, 8.0 Hz), 7.27 (d, H-8 and H-8′, J = 8.0 Hz), 7.19 (d, H-10, J = 8.0, 8.0 Hz), 5.39 (d, 4-OH, J = 5.1 Hz), 3.84 (dd, H-4, J = 11.7, 5.1 Hz), 3.65 (dd, H-5, J = 14.0, 6.3 Hz), 2.95 (dd, H-6a, J = 14.0, 6.3 Hz), 2.57 (dd, H-6b, J = 14.0, 6.3 Hz), 2.09 (qt, H-2, J = 6.9 Hz), 0.99 (d, H-3, J = 6.9 Hz) 13C NMR (150 MHz, DMSO-d6) δ 176.0 (C-1), 138.7 (C-7), 129.4 (C-9 and C-9′), 128.3 (C-8 and C-8′), 125.5 (C-10), 74.8 (C-4), 57.6 (C-5), 40.6 (C-2), 35.2 (C-6), 12.9 (C-3); ESI-TOF-MS: m/z 206 [M+H]+; HR ESI-TOF-MS m/z 206.1177 (calcd. for C12H15NO2, 206.1175).
Supplementary Material
Acknowledgments
Financial support was provided by the National Cancer Institute, NIH (under grant CA044848). We thank Dr. C. C. Hughes for his generous advice in the determination of stereochemistry of the actinoramides. We also thank Dr. D.-C. Oh (Harvard Medical School) for providing amino acids and Prof. T. Molinski (Department of Chemistry and Biochemistry, UCSD) for providing lobocyclamide B. We especially thank Dr. K. N. Maloney for providing the information for the strain CNQ-027, and Dr. J. Pezzuto (University of Hawaii, Hilo) for providing NF-κB and aromatase bioassay data.
Footnotes
The 1H, 13C, and 2D NMR spectra of actinoramide A, and 1H, and 13C for actinoramides B and C, and 1H NMR spectrum of bis-MTPA esters (4a, 4b), and mono-MTPA esters (5a, 5b, 6a, 6b), and 7, and, 1H, 13C, and 2D NMR spectra of 8 are available.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 2.(a) Koehn FE, Carter GT. Nat Rev Drug Discovery. 2005;4:206–220. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]; (b) Bérdy J. J Antibiot. 2005;58:1–26. doi: 10.1038/ja.2005.1. [DOI] [PubMed] [Google Scholar]; (c) Bull AT, Ward AC, Goodfellow M. Microbiol Mol Bol Rev. 2000;64:573–604. doi: 10.1128/mmbr.64.3.573-606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prieto-Davó A, Fenical W, Jensen PR. Aquat Microb Ecol. 2008;52:1–11. [Google Scholar]
- 4.(a) Fujii K, Ikai Y, Oka H, Suzuki M, Harada K. Anal Chem. 1997;69:3346–3352. [Google Scholar]; (b) Fujii K, Ikai Y, Oka H, Suzuki M, Harada K. Anal Chem. 1997;69:5146–5151. [Google Scholar]; (c) Marfey P. Carlsberg Res Commun. 1984;49:591–596. [Google Scholar]
- 5.(a) Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]; (b) Seco JM, Quiñoá E, Riguera R. Chem Rev. 2004;104:17–117. doi: 10.1021/cr2003344. [DOI] [PubMed] [Google Scholar]
- 6.Rieser MJ, Hui YH, Rupprecht JK, Kozlowski JF, Wood KV, McLaughlin JL, Hanson PR, Zhuang Z, Hoye TR. J Am Chem Soc. 1992;114:10203–10213. [Google Scholar]
- 7.Kusumi T, Ooi T, Ohkubo Y, Yabuuchi T. Bull Chem Soc Jpn. 2006;79:965–980. [Google Scholar]
- 8.(a) MacMillan JB, Ernst-Russell MA, de Ropp JS, Molinski TF. J Org Chem. 2002;67:8210–8215. doi: 10.1021/jo0261909. [DOI] [PubMed] [Google Scholar]; (b) Oh DC, Poulsen M, Currie CR, Clardy J. Nat Chem Biol. 2009;5:391–393. doi: 10.1038/nchembio.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flouret G, Chaloin O, Borovickova L, Slaninová J. J Pept Sci. 2006;12:347–353. doi: 10.1002/psc.733. [DOI] [PubMed] [Google Scholar]
- 10.Leitheiser CJ, Smith KL, Rishel MJ, Hashimoto S, Konishi K, Thomas CJ, Li C, McCormick MM, Hecht SM. J Am Chem Soc. 2003;125:8218–8227. doi: 10.1021/ja021388w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.