

1. Introduction
The Nazarov cyclization1 is described as the 4 π electron conrotatory cyclization of a pentadienyl cation. The past 20 years have brought resurgent interest in this reaction most likely due to the realization that it can provide rapid and efficient access to multisubstituted cyclopentenones with excellent stereochemical control at two (or even three) contiguous ring carbon atoms.2
Typically, stoichiometric or super-stoichiometric Brønsted or Lewis acid is used to generate the conjugate acid 2 of pentadienone 1 reversibly (Scheme 1). 3 Electrocyclization subsequently leads to allylic cation 3 in which the trans relationship between substituents R2 and R4 is dictated by the conrotation. The process is normally terminated through proton loss from cation 3, leading typically either to cyclopentenone 4 or to 5, or to a mixture of the two. The trans stereochemistry between R1 and R2 in 4 or between R3 and R4 in 5 reflects the thermodynamically more stable product. The trans relationship between R2 and R4 in 3 can be preserved in the final product if loss of an exocyclic proton from R1 or R3 terminates the reaction.
Scheme 1.

Acid Catalyzed Conrotatory Cyclization.
Several obstacles must be overcome in order to render such a process general and synthetically useful. Super-stoichiometric strong acids must be avoided so as to suppress undesired competing processes, e.g. Wagner-Meerwein rearrangements. The absolute stereochemistry of the product is determined by the absolute sense of conrotation: the stereochemistry indicated for 3 is a consequence of clockwise conrotation of 2, viewed from the bottom of the structure as it is shown in Scheme 1. Therefore in an enantioselective Nazarov cyclization the absolute sense of conrotation, or the torqoselectivity, must be controlled. The course of the termination step must also be controlled so as to avoid the production of isomeric mixtures of cyclopentenones. Some bias must be present to direct proton loss from 3 so that it does not take place indiscriminately. This Report will examine these constraints in the context of the asymmetric Nazarov cyclization. Enantioselective Nazarov cyclizations that employ asymmetric chiral effectors either catalytically or stoichiometrically will be examined, as well as processes in which transfer of chirality to the cyclic product takes place.
2. Organocatalytic Reactions
Rueping and coworkers reported the first enantioselective organocatalytic Nazarov cyclization in 2007.4 Exposure of a toluene solution of dienone 6 for 2 hours at 0 °C to 10 mol% of (R)-BINOL-derived N-triflylphosphoramide 7 led to the formation of a 6:1 mixture of cis and trans cyclopentenones 8 (87% ee) and 9 (95% ee), respectively (eq 1). The stereochemistry of the phenyl-bearing β-carbon atom is determined by the absolute sense of conrotation, whereas stereochemistry at the α-carbon atom is determined during the final proton transfer step. The preponderance of cis product 8 from this reaction indicates that it takes place under kinetic control. The authors proposed a plausible catalytic cycle that is reproduced in Scheme 2. Proton transfer from the chiral Brønsted acid 7 to dienone 1 leads to an ion pair consisting of pentadienyl cation 10 and 11, the conjugate base of 7. Conrotatory cyclization of 10 takes place in the counterclockwise direction viewed from the bottom of structure 10 as shown to produce cyclic oxyallyl cation 12. Proton loss from 12 leads to enol 13 that undergoes successive proton transfer reactions that lead to product 14 with the regeneration of organocatalyst 7. The regiochemical preference for proton loss from oxyallyl cation 12 is dictated by the presence of the pyranyl ether oxygen atom.
 |
[1] |
Scheme 2.

Catalytic Cycle With Chiral Brønsted Acid
The presence of a pyranyl ether or other α-oxygen atom substituent is a common feature of many substrates for catalytic Nazarov reactions where it plays a dual role. It serves to localize the double bond in the cyclopentenone product, but it also lowers the activation energy for the cyclization by polarizing the dienone. The pyranyl ether substrates utilized by Rueping and coworkers had been used to good effect by Trauner and Liang in the catalytic asymmetric Nazarov some years earlier (vide infra, eq 6). The beneficial effect of an α-ether oxygen atom on the rate of Nazarov cyclizations had been demonstrated by Kocienski,5 Frontier6 and others.7 For example (eq 2) dienone 15 has been shown to undergo Nazarov cyclization to 16 upon treatment at ambient temperature with dilute hydrochloric acid. Frontier and coworkers and others have shown that polarization of the dienone in the opposite sense through the substitution of an α–electron withdrawing group also lowers the barrier for cyclization. Dienones that are polarized in a complementary sense at both the β-carbon atoms (i.e. C1 and C5 in 15) are predictably the most reactive of all.
 |
[2] |
As is commonly the case, Rueping and coworkers determined the optimal catalyst structure empirically. The BINOL-derived phosphates were examined with good results; however, both reactivity and enantioselectivity were highest with the N-triflylphosphoramide organocatalysts. A screen of reaction solvents indicated that chloroform was optimal. Since the induction of asymmetry depends upon the formation of a tight ion pair (e.g. 10/11 in Scheme 2) it should not be surprising that non-polar solvents were found to work best.
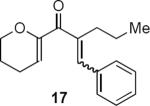
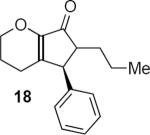
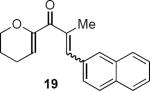
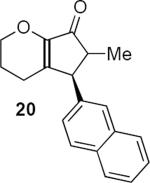
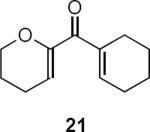
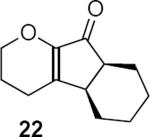
Table 1 summarizes a few of Rueping and coworkers' results. Rueping's work is noteworthy for several reasons. The low catalyst loading (2 mol %) compares favorably not only with most other organocatalytic reactions but also with many metal-catalyzed reactions. The kinetically formed cis diastereomers could be isomerized to the more stable trans diastereomers with no loss of optical purity by stirring in dichloromethane at room temperature for 24 hours in the presence of basic alumina. As mentioned above, the dihydropyran ring that is common to all of the examples reported by Rueping and coworkers activates the substrates for cyclization. Substrates 17 and 19 additionally benefit from the presence of the β-aryl group that contributes to the favorable complementary polarization of the dienone. Remarkably, the reaction is also successful in the case of 21 that lacks the β-phenyl group, and leads to a single cis isomer 22 in good yield and enantioselectivity.
 |
[3] |
Table 1.
Examples of the Organocatalyzed Asymmetric Nazarov Cyclizationa
| Entry | Substrate | Product | t(h) | Yield | cis:trans | ee(cis),ee(trans) |
|---|---|---|---|---|---|---|
| 1 |
|
|
1 | 85% | 3.2:1 | 93, 91 |
| 2 |
|
|
2 | 92% | 9.3:1 | 88, 98 |
| 3 |
|
|
4.5 | 68% | cis | 86, - |
2 mol% 7, CHCI3, 0 °C.
The Rueping group used the same approach to develop the first organocatalyzed asymmetric electrocyclization-protonation process,8 although Trauner and Liang had demonstrated this concept in a metal catalyzed process (vide infra, eq 6). Exposure of enones 23 that lack β substitution to 5 mol % of phosphoramide 24 at −10 °C leads to products 25 in yields varying between 49% and 93% and ee's between 67% and 78% (eq 3). The catalytic cycle of this reaction is presumably the same in its essential features as the one that has been summarized in Scheme 2. The key difference between the reactions of equations 1 and 3 is that in equation 1 the electrocyclization step takes place enantioselectively and is followed by a diastereoselective kinetic protonation, whereas in equation 3 enantioselective protonation of the reaction intermediate is the key step.
 |
[4] |
Implicit in the preceding discussion is the fact that the optimal catalyst structure is determined empirically through a process of trial and error. To the extent that the changes at two or more sites of the catalyst structure exercise their effects on the optical purity of the product independent of one another, progress toward identifying the optimal catalyst can be rapid. Given the successes of the Rueping group in optimizing the phosphoramide organocatalysts it is somewhat surprising that phosphorodithioic acids were found by Blanchet and coworkers to be poor catalysts for the Nazarov cyclization of 6 and not amenable to optimization (eq 4).9 For example, exposure of dienone 6 to 10 mol % of phosphorodithioic acid 26 in chloroform at 20 °C led to a 9:1 mixture of 8 and 9 in only 10% ee and 18% ee, respectively.
 |
[5] |
Tius and coworkers developed a mechanistically distinct organocatalytic Nazarov cyclization. Exposure of diketoester 27 in toluene at 23 °C for 10 days to 20 mol % of bifunctional organocatalyst 28 led in 60% yield to cyclopentenone 29 in an enantiomeric ratio of 95.5:4.5 (eq 5).10 The product was formed as a single diastereomer. A noteworthy feature of this reaction is the formation of products bearing two adjacent asymmetric carbon atoms one of which is an all-carbon quaternary stereocenter. The relative and absolute stereochemistry of both stereogenic carbon atoms is determined during the electrocyclization. The mechanistic hypothesis that accounts for the catalytic action that was proposed by the authors is indicated by structure 30 (Figure 1). A cooperative process whereby the thiourea engages the ketone carbonyl oxygen atom in a double hydrogen bonding interaction while the primary amine engages the enol form of the β-ketoester as a Brønsted base rationalizes the observation that thiourea organocatalysts lacking a basic amine do not convert substrate to cyclic product. This raises the question of how stereochemical information is transmitted from the organocatalyst to the substrate since several bond lengths separate the stereogenic carbon atoms of the catalyst from the developing carbon-carbon bond. If the mechanism implicit in structure 30 is valid, as it appears to be, the catalyst may be exercising its effect through the imposition of helicity from torsion of the C3-C4 bond.
Figure 1.

Cooperative mechanism.
This catalytic system was designed to induce complementary polarization at each of the terminal carbon atoms of the diketoester as indicated in 30. Exploiting the tendency of the substrate to form only E enol so as to minimize electron pair – electron pair repulsions from the enol hydroxyl and ester carbonyl oxygen atoms made it unnecessary to control the geometry of a tetrasubstituted alkene. The product yields varied between 58% and 95% whereas enantiomeric ratios varied between 90:10 and 98.5:1.5 (Figure 2). The long reaction times are very likely an indication of strong product inhibition of the catalyst. Since the starting material and the product are both α-hydroxyenones, the organocatalyst can likely engage the product through interactions that are analogous to those indicated for 30. A limitation of this reaction is the requirement for aryl substitution at C6 that is common for many substrates for the Nazarov reaction.
Figure 2.

Examples of cyclizations catalyzed by 28.
3. Metal Catalyzed Cyclizations
A larger body of work describes transition metal salt – catalyzed asymmetric Nazarov cyclizations. Trauner and Liang used Sc(OTf)3-PYBOX complex (1S,2R)-35 to catalyze the cyclization of dienones 23 to cyclopentenones ent-25 in good yields and optical purities (eq 6).11 As was the case in equation 3, the absolute
 |
[6] |
stereochemistry of the products was not determined during the electrocyclization step, but instead during the protonation of the scandium enolate. Sterically more demanding α-substituents R in 23 generally led to products of higher optical purity, as might be expected given the closer proximity of the bulky indane-PYBOX to the α-carbon atom. The influence of the PYBOX ligand is not felt
 |
[7] |
strongly at the β-carbon atoms of the dienone as revealed by the result that is summarized in equation 7. A comparison of the results of eqs 6 and 7 suggests that the presence of a β methyl group of either configuration in the cyclic enolate reverses the streochemical outcome of the protonation step. The Nazarov cyclization of 36 is only moderately diastereoselective, leading in 62% yield to syn product 37 (40% ee) and in 18% yield to anti isomer 38 (79% ee). Trauner has interpreted this result by invoking double diastereoselection during the protonation of the enolate following the electrocyclization. This limitation notwithstanding, Trauner's work represents the first example of catalytic asymmetric Nazarov reactions that proceed with high levels of enantioselectivity. It should also be noted that when the issue of diastereoselectivity was circumvented through the use of 39 (the five membered ring precludes the formation of anti product), then a reasonable yield and modest enantioselectivity was observed for product 41 (eq 8) under the influence of scandium(III)-PYBOX catayst 40.12
 |
[8] |
Transition metal BOX and PYBOX complexes are attractive structural motifs for catalysts, providing the substrate can offer an opportunity for bidentate chelation with the metal. Jun-An Ma and coworkers have described a tandem enantioselective Nazarov cyclization – electrophilic fluorination sequence that leads to fluoroindanones (eq 9).13 Development of a tandem process introduces another element of complexity to the process. In Ma and coworkers' reaction, fluorination of the enolate represents the termination step of the process. Exposure of phenone 42 to N-fluorobenzenesulfonimide 43 and 10 mol % Cu(II)-BOX complex 44 for 8 hours at 80 °C in 1,2-dichloroethane led to fluoroindanones 45. Although it is difficult to make generalizations on the basis of the three examples of the asymmetric version of the process that Ma and coworkers have reported, it appears that all reactions show excellent trans selectivity and that the optical purity of the product is highest in the case in which the β-aryl group in 42 is 2,4,6-trimethoxyphenyl. The synthesis of a fluorine-substituted quaternary carbon atom in high optical purity that has been described in this work is noteworthy.
 |
[9] |
Shindo and Yaji have reported a Sc(OTf)3-PYBOX catalyzed asymmetric Nazarov reaction that appears to proceed through a unique mechanism. 14 Exposure of dienone 46 to 10 mol % of ent-40 and 0.7 equivalent of triethylamine in dichloromethane at room temperature led to a mixture of optically enriched cyclopentenone 47 and byproducts 48 and 49 (eq 10). Triethylamine was found
 |
[10] |
to be necessary so as to reduce the catalyst loading. The apparent migration of the alkoxy group from the β-carbon atom in 46 to the α-tertiary carbon atom in 47 is at first glance puzzling, but the appearance of byproducts 48 and 49 leave little doubt concerning the mechanism, which corresponds to an interrupted Nazarov cyclization. Scheme 3 summarizes the catalytic cycle proposed by Shindo and Yaji. Complexation of ent-40 with 46 leads to pentadienyl cation 50 that undergoes electrocyclization to oxyallylic cation 51. This intermediate partitions between elimination that proceeds via 52 leading ultimately to cross-conjugated cyclopentadienone 48 and nucleophilic trapping of 51 by alkoxide leading through 53 to major product 47. By means of crossover experiments intermolecular alkoxy migration was also shown to occur, providing an alternative pathway for the conversion of 51 to 47. Byproduct 49 is presumably the result of trapping of 51 by adventitious water. According to this mechanism, the absolute sense of the electrocyclization step is probably not under catalyst control whereas the termination step that leads from 51 to 53 is. In this sense the process resembles the reactions of equations 6 and 9. The yields for the aalkoxycyclopentenones varied from 34% to 71% whereas ee's varied between 0% and 91%. Controlling the stereochemical outcomes of both electrocyclization and trapping steps using a single catalyst would make it possible to form two stereogenic carbon atoms during the reaction, a process that would be very valuable in synthesis.
 |
[11] |
Scheme 3.

Catalytic Cycle of the Sc(OTf)3-PYBOX Catalyzed Process
Asymmetric Nazarov cyclizations catalyzed by Fe(II)- and Co(I)-PYBOX complexes have been described by Itoh, Kawatsura and coworkers.15 Exposure of triaryl pentadienone 54 to 50 mol% of the Fe(II) or Co(I) catalyst 55 led to the expected product 56 (eq 11). It appears that the C5 phenyl group in 54 is required and that electron poor aryl groups at C1 lead to low yields of cyclized product. Yields varied between 13% and 85% whereas ee's varied from 0% to 93%. It appears that this protocol may be limited to the subset of strongly polarized acyclic dienones. The high catalyst loadings may discourage its widespread adoption in synthesis.
A relatively small number of structural motifs of the metal ligand have been explored. The limitations of the BOX and PYBOX catalysts that are manifest in the earlier discussion led Tang and coworkers to develop a tris(oxazoline) ligand for Cu(II).16 They had hypothesized that incorporating a capping group into the structure of the BOX ligand would improve the enantiocontrol by extending the influence of the complex to the distal carbon atoms of the pentadienyl cation. A series of DFT calculations was not definitive, so the precise role of the capping group remains unknown. Nevertheless, the catalysts derived from the tris(oxazolines) proved to be highly effective both in terms of yield as well as enantioselectivity. All of the tris(oxazoline) ligands that were evaluated led to higher yields and far better optical purities of product than could be obtained from
 |
[12] |
35 or 55 (R = iPr, MXn = Cu(SbF6)2). For example, following treatment of divinyl β-ketoester 57 (eq 12) at 25 °C with the cationic Cu(II) complex derived from tris(oxazoline) 59, cyclopentenone 60 was formed in 90% yield and in 92% ee as essentially a single diastereomer (>99:1). A relatively small diminishment in yield and optical purity was observed when the diastereomeric ligand 58 was used in which the stereochemistry of the pendant indane-oxazoline is enantiomeric to that in 59. Catalyst loadings were modest (10 mol %) but the reaction times varied according to the electronic nature of the aryl substituent: cyclization of dienones bearing electron poor aryl groups were much slower than substrates bearing electron rich aryl rings. It is very significant that the aryl group in the substrate is not required and that the cyclization of 57 to 60 (R = cyclohexyl) in the presence of 59 took place in 88% yield (at 50% conversion) and 78% ee after 10 days at 40 °C in 1,2-dichloroethane. The complementary activation that is provided by the enol ether and the ester groups in 57 is sufficient to ensure that reaction takes place at a reasonable rate.
 |
[13] |
Since the Nazarov cyclization proceeds through the intermediacy of carbocations it should not be surprising that Wagner-Meerwein rearrangements can take place prior to the termination step. Indeed, such rearrangements are most often seen when the cyclization generates a quaternary carbon atom. Frontier and Huang have reported an interesting example of this chemistry (eq 13).17 Treatment of ketoester 61 with stoichiometric Cu(II) complex 62 led to spirocyclic product 65 in 69% yield in 40% ee. The initial cyclization to 63 is presumably followed by C-C bond migration to produce carbocation 64. [1,2]-Hydride migration converts 64 to 65.
 |
[14] |
Kobayashi and Kokubo have described a radically different catalyst system.18 Dodecylsulfate scandium (Sc(DS)3) was developed as a water-compatible Lewis acid as a part of a broader exploration of the use of water as a solvent for chemical reactions. This surfactant catalyst creates a hydrophobic environment in water in which reaction takes place. When dienone 23 was treated for 36 hours in water at room temperature with a catalyst prepared from 10 mol % Sc(DS)3 and homochiral bipyridine 66, Nazarov cyclization took place followed by trapping of the intermediate cation by water (eq 14). This resulted in cleavage of the dihydropyran ring and the formation of α-diketone 67 that was isolated in 75% yield in 32% ee.
 |
[15] |
Togni and Walz have reported a thorough exploration of an asymmetric Ni(II)-catalyzed Nazarov cyclization (eq 15).19 Togni had demonstrated through his earlier efforts the Lewis acidic properties of dicationic Ni(II)-Pigiphos (69) in diverse reactions, therefore the application of this catalyst system to the Nazarov cyclization represented a logical extension of his earlier work. A preliminary solvent screen indicated that dichloromethane was optimal and furthermore that the strongly coordinating acetonitrile completely suppressed the reaction. Exposure of 68 (R1 = CH3, R2 = CH2CH3, R3 = 2,4,6-trimethoxyphenyl) to 10 mol % Ni(II)-Pigiphos in dichloromethane led to 70 in 84% yield in 86% ee after 6–8 days reaction time. The catalyst was prepared in tetrahydrofuran but the solvent was evaporated and replaced with dichloromethane prior to the addition of the substrate. Likewise, exposure of 68 (R1 = Ph, R2 = CH2CH3, R3 = 2,4,6-trimethoxyphenyl) led to 71 in 85% yield and 87% ee. However, 68 (R1 = CH3, R2 = CH2CH3, R3 = 4-methoxyphenyl) led to product 72 in only 32% yield and in 71% ee after 9–15 days reaction time. Erosion of both yield and optical purity of the product was also observed for the cases in which R2 = Bn or R2 = 1-naphthyl (both R1 = CH3, R3 = 2,4,6-trimethoxyphenyl). In the case of R2 = 1-naphthyl no reaction took place after 1 month.
Togni's work highlights several of the challenges that are commonly encountered. The lack of reactivity of the ligated Lewis acidic metal makes it necessary to use highly activated substrates such as 68, thereby limiting the reaction scope. Even with reactive substrates a complete reaction requires 1–2 weeks with 10 mol % catalyst. A 31P NMR study showed that the major Ni(II)-Pigiphos species that was observed during the reaction corresponded to the catalyst-product adduct. This provides an indication that the concentration of active catalyst remained very low and identifies the problem as product inhibition. These two problems, low catalyst reactivity and product inhibition, have contributed to the difficulty in defining a general and efficient catalytic asymmetric Nazarov cyclization.
4. Stoichiometric Reactions
There are a small number of Nazarov reactions that make use of stoichiometric or nearly stoichiometric chiral effectors again underscoring the problem of product inhibition of the catalytic cycle. Aggarwal and Belfield have described one such stoichiometric (or nearly stoichiometric) process (eq 16).20 Treatment of ketoester 73 (R = CH3) with one equivalent of the Cu(II) hexafluoroantimonate adduct of PYBOX ligand 74 at room temperature in dichloromethane led to product 75 in 73% yield and 76% ee. Yield and enantioselectivity both improved for the reaction leading to 76 (98% yield, 86% ee). As anticipated, the presence of the additional phenyl group resulted in activation of the acyclic substrate. This became clearer from the results of the reaction that used substoichiometric Cu(II) complex. When 73 (R = CH3) was treated with 0.5 equivalent of reagent the yield of 75 dropped to 42% whereas the optical purity of product increased slightly to 78% ee. By contrast when 73 (R = Ph) was treated with 0.5 equivalent of the Cu(II) complex the optical purity of 76 was unaltered (86% ee) and the yield was essentially the same (96%), indicating that the catalyst had turned over once. This reaction is subject to the same limitations that have been disclosed earlier in this review: in the absence of phenyls at both distal carbon atoms of 73, erosion of either the yield or the optical purity, or both, is observed. Aggarwal and Belfield observed similar trends with substrates bearing the N,N-diethylamide group in place of the carboethoxy group in 73. Their work is noteworthy as it represents the first asymmetric Nazarov cyclization that is promoted by a chiral non-racemic Lewis acid complex and that achieves high levels of enantioselectivity.
 |
[16] |
Tius and coworkers adopted a different approach making use of iminium ion catalysis that provides an attractive strategy for controlling the absolute stereochemical outcome of the Nazarov cyclization.21 Unfortunately, absent some compensating factor, the acyclic iminium ion that is derived from the pentadienone is energetically favored over the cyclic allylic cation. This problem of unfavorable energetics was overcome through the use of diamine mono-triflate 78 (eq 17). Exposure of α-diketone 77 to 1.05 equivalents of 78 in wet acetonitrile at room temperature led to slow consumption of the starting material with the formation of cyclic product 79 in 62% yield in >99:1 er after a reaction time of 7.5 days. The diamine could be isolated and reused without erosion of the optical purity of the product. This reaction presumably succeeds by engaging both carbonyl groups of 77 with reagent 78 to form enamine-iminium species 77b. Following cyclization, enamine-iminium species 77c is formed, rendering the energetics of the cyclization favorable. Cyclopentenone 79 is liberated from the complex with 78 during aqueous workup. Notably this Nazarov cyclization leads to products in very high optical purity and in moderate yield even in the absence of activating aryl substituents. Shortcomings of the process are that it is not completely general and it is very slow.
 |
[17] |
5. Asymmetry Transfer
There are a few examples that demonstrate transfer of asymmetry during the Nazarov cyclization. The first example was provided by Denmark, Wallace and Walker who showed that cyclization of optically active β'-silyldivinyl ketone 80 (eq 18) under the influence of ferric chloride led to conrotatory ring closure to provide tricyclic ketone 81 in 72% yield.22 The optically enriched starting material for the synthesis of 80 was prepared by chromatographic separation of diastereomeric carbamates derived from (S)-(1-phenylethyl)isocyanate. This reaction took place with no erosion of optical purity, indicating that the trimethylsilyl group in 80 directed the electrocyclization in an anti-SE' sense with essentially perfect control. The observed absolute sense of the conrotation is the one that allows continuous overlap of the C-Si bond with the developing allyl cation. The trimethylsilyl substituent is lost during the termination step of the cyclization leading to the exocyclic (with respect to the five membered ring) double bond in 81.
 |
[18] |
Tius and coworkers first demonstrated axial to tetrahedral chirality transfer from an allene during the Nazarov cyclization (eq 19).23 The enantiomers of morpholino enamide 82 were separated by semi-preparative HPLC. Exposure of 82 (93% ee) to vinyllithium species 83 in tetrahydrofuran at −78 °C provided a mixture of cyclopentenones 84 (R = n-Bu, 50% yield 78% ee) and 85 (R = n-Bu, 9% yield, 64% ee). The tetrahedral intermediate 83a was presumably converted to allenyl ketone 83b during workup with aqueous potassium dihydrogen phosphate. Cyclization of 83b to a mixture of diastereomers 84 and 85 took place spontaneously. Major product 84 was formed with 84% asymmetry transfer. As predicted, asymmetry transfer of >95% was observed when allenamide 86 was used as the starting material: the preferred sense of conrotation is the one that maintains the greatest distance between the substituent on the exocyclic methylene carbon atom (n-Bu or t-Bu) and the substituent on the sp3-hybridized ring carbon atom. This preference for the Z exocyclic double bond isomer is kinetic, since complete isomerization to the E isomer could be induced to take place. The lower optical purity of 85 (R = n-Bu) is a consequence of Z to E isomerization of 84 (R = n-Bu) that took place during column chromatography of the product mixture on silica gel.
 |
[19] |
An indication of mechanistic complexity that can be encountered in these reactions was manifested in work of Hoppe and coworkers (Scheme 4).24 Although there is at present no effective asymmetric synthesis of allenamides such as 82 or 86, Hoppe and coworkers had demonstrated a highly efficient asymmetric synthesis of enantiomerically enriched allenyl carbamates based on the (−)-sparteine-mediated isomerization of propargylic carbamates. Allenyl carbamate 87 (80% ee) was deprotonated with n-butyllithium/TMEDA in toluene. Deprotonation took place regiospecifically, directed by the carbamoyl group. The lithioallene is internally chelated and is configurationally stable. Addition of the lithioallene to morpholino enamide 88 at −78 °C led to presumptive intermediate 89. When the solution of 89 at −78 °C was transferred to ethanolic HCl solution at low temperature, Nazarov product 91 was isolated in 74% yield and 78% ee, representing 98% asymmetry transfer from 87. This is the product that one would have predicted based on the results shown in equation 19. If, however, the solution of 89 was allowed to warm to room temperature over the course of an hour before quenching with HCl, the outcome of the reaction was quite different. α-Morpholino cyclopentenones 92 and 93 were isolated in 50% yield (79% ee) and 24% yield (80% ee), respectively. Complete or nearly complete asymmetry transfer was observed in both products. Hoppe and coworkers suggested that upon warming the solution of 89, 1,2-migration of the carbamoyloxy group took place to produce a mixture of diastereomers of 90. Each diastereomer produced a single product, either 92 or 93. Whereas the stereochemical outcome of the cyclization that led to 91 was determined only by the axial chirality of the allene, in the case of 92 and 93 it was the stereochemistry of the tetrahedral carbon atom in 90 that controlled the reaction outcome. Hoppe and coworkers interpreted these results to suggest that the cyclization of 90 corresponds to a conrotatory process.
 |
[20] |
Scheme 4.

Mechanistic Complexity in the Carbamoyloxy Allene Nazarov Cyclization
Hoppe and coworkers have demonstrated that this process is general. For example, enantiomerically pure carbamoyloxyallene 94 was deprotonated at −78 °C and was added to enone 95 (eq 20).25 After 1 hour, the reaction mixture was warmed to room temperature for 1 hour and was subsequently quenched with saturated aqueous ammonium chloride. This led to cyclopentenone 96 in 33% yield as the major product. None of the E geometrical isomer of 96 was observed, although other reaction products were formed. It seems unlikely that the addition of the lithioallene derived from 94 to enone 95 took place with complete diastereoselectivity. The isolation of a single isomer in moderate yield suggests that only one of the tetrahedral intermediates was able to undergo cyclization. This reaction is noteworthy not only because of the structural complexity of allene 94, but also because it demonstrates simultaneous control of the absolute stereochemistry of the ring carbon atom and control of the geometry of the exocyclic double bond.
6. Diastereoselective Nazarov Cyclizations of Non-Racemic Precursors
A modest number of examples have been reported in which a Nazarov cyclization has been performed on a chiral, non-racemic substrate. Williams and coworkers26 have reported the total synthesis of (+)-fusicoauritone that makes use of a diastereospecific Nazarov cyclization in the key step. The cyclization of α-diketone 97 took place in the presence of boron trifluoride etherate in 1,2-dichloroethane at reflux to produce cyclic product 98 in 77% yield (eq 21).27
 |
[21] |
Williams and coworkers reported that their early efforts had led to mixtures of 98 and isomeric α-diketone 99. Exposure of 99 to boron trifluoride etherate led to 98 suggesting that 99 was the kinetic product whereas 98 was the result of thermodynamic equilibration of 99. For the purposes of the (+)-fusicoauritone synthesis, divinyl ketone 100 was prepared (eq 22) and was exposed to catalytic tosic acid in 1,2-dichloroethane to produce enone 101 in 92% yield as a mixture of C6 diastereomers. Epimerization at C6 took place readily through the formation of the extended enol. This reactivity was exploited in the fusicoauritone synthesis. Autoxidation of chloroform solutions of the enol led to the C6 hydroperoxide that was reduced to the alcohol in 95% overall yield for the two steps from 101.
 |
[22] |
The synthesis of marine sponge metabolite nakiterpiosin made use of a rarely utilized photochemical Nazarov reaction of a phenone. Gao, Wang and Chen prepared phenone 102 through a carbonylative Stille reaction (eq 23).28
 |
[23] |
Irradiation of a solution of 102 in acetonitrile at 350 nm led to a 1:1 mixture of C9 epimers of the desired Nazarov product. Brief exposure of this mixture to Hünig's base in methanol at 50 °C led to the trans ring fused product 103 in 60% yield for the two steps. In subsequent work, Chen and coworkers improved their original nakiterpiosin synthesis by deferring the photochemical step to the very end of the synthesis.29 Irradiation of phenone 104 at 350 nm (eq 24) under the same conditions as were used for 102, followed by epimerization and fluorodesilylation led to nakiterpiosin 105 in 55% yield for the three steps. The success of this stereospecific photochemical Nazarov cyclization in the context of the polyfunctional nakiterpiosin precursor 104 testifies to the mildness of the conditions.
 |
[24] |
Giannis and coworkers reported a conceptually related photochemical Nazarov cyclization in their recent synthesis of terpendole E analogues.30 Protected 3-indole ketone 106 (eq 25) was irradiated at 350 nm in acetonitrile to produce cyclopentenone 107 in 80% yield as a single diastereomer. The angular methyl group at C4 probably determines the stereochemistry of the product of equation 25, ring closure taking place so as to move the methyl group at C3 away, leading to the trans relationship of the two adjacent angular methyl groups in 107. The stereochemistry at C16 was determined during the proton transfer step that took place following cyclization.
 |
[25] |
The mechanism of the photochemical Nazarov cyclizations of equations 23, 24 and 25 deserves some comment. It is known that in the case of cyclic cross-conjugated dienones ring size influences the mechanism.31,32 In 4,4-disubstituted 2,5-cyclohexadienones the photochemical Nazarov cyclization takes place from an electronically excited state by means of a disrotation, as predicted by the orbital symmetry rules. However, medium-sized ring cross-conjugated dienones have been shown to follow a different pathway in which photochemical Z to E isomerization of one of the two double bonds of the dienone leads to a highly strained ground state intermediate that undergoes a thermal Nazarov electrocyclization. Both the disrotatory ring closure from the excited state of the dienone as well as the conrotatory process from the strained E isomer of the ground state would lead to the same stereoisomer of the product.33 1-Cyclohexenyl(phenyl)methanone has been shown to undergo photochemical Z to E isomerization followed by thermal ring closure.34 This suggests that irradiation of ketones 102, 104 and 106 also results in thermal, conrotatory ring closure from a highly distorted ground state intermediate. The sense of conrotation cannot be ascertained from the structures of the respective products 103, 105 or 107. Proton loss to regenerate the aromatic ring in each case obliterates the stereocenter that would have distinguished between the two modes of electrocyclization. The unique advantage of the photochemical Nazarov cyclization is that it makes it possible to selectively activate the enone function under neutral conditions within a large, polyfunctional molecule.
When one of the alkene groups of the dienone is part of a bridged, bicyclic system, conrotation is inevitably driven to minimize steric interactions. This was clearly seen in West and coworkers' results with bornenyllithium-derived dienone 108 (eq 26) that cyclized to produce exo cyclopentenone 109 as the nearly
 |
[26] |
exclusive reaction product.35 The trimethylsilyl group in 108 controls the location of the double bond in the enone product. The strong preference for exo diastereomer 109 is consistent with the observed preference for exo addition of electrophiles to norbornene. West and coworkers pointed out that although the interactions of the distal carbon atoms of the pentadienyl cation that is derived from 108 are not completely analogous to an electrophilic addition to norbornene, the process should be subject to the same constraints and should therefore be expected to lead to the same stereochemical outcome. This was certainly the case.
Under the appropriate reaction conditions it is sometimes possible to couple the Nazarov cyclization with subsequent reactions that exploit the reactivity of the intermediate cation. West and Wu have demonstrated the Nazarov/Wagner-Meerwein cascade process that is summarized in Scheme 5.36 Propargyl silyl ether 111 was derived in a straightforward sequence of steps from (−)-myrtenal. Base-catalyzed isomerization of 111 to allenyl ether 112 was followed by exposure to trifluoroacetic acid in dichloromethane at room temperature. This led to the production of enone 117 in 45% overall yield from 111. The cis relative stereochemistry of the phenyl group at C3 and the hydrogen atom at C3a indicates that sterically controlled protonation of the allenyl ether to produce pentadienyl cation 113 as the initially formed product was followed by Z to E isomerization to give cation 114. Conrotatory ring closure of 114 took place to produce cation 115 that rearranged to 116 in order to relieve the strain of the cyclobutane ring. Trapping of 116 with trifluoroacetate and silyl enol ether hydrolysis led to 117. The sense of conrotation of 114 was apparently controlled by the gem-dimethyl substituted bridge and took place so as to move the phenyl group away from the methyl. The counterintuitive consequence of this is that the cyclization then occurs from the same side of the pentadienyl cation as the gem-dimethyl substituted bridge.
 |
[27] |
Scheme 5.

Nazarov/Wagner-Meerwein Cascade Process
There are significant ambiguities that render the incorporation of such diastereoselective reactions into synthetic planning a challenge. Prandi, Occhiato and coworkers have nonetheless marked some significant accomplishments in the area. The protonation of racemic triene 118 led to a pentadienyl cation that underwent Nazarov cyclization to a mixture of diastereomeric enones 119 and 120 in a 16:1 ratio (eq 27).37 Spiroenone 121 is the product of an alternative Nazarov cyclization and accounted for 10% of the total product mixture. Major product 119 is the result of conrotation toward the pyran ring and does not correspond to the isomer that one would have predicted from the example of Scheme 5. Prandi and Occhiato do not offer an explanation for this observation beyond a suggestion that the sense of conrotation in this example may result from the interplay of steric and stereoelectronic factors. This is apparently also the case for the Nazarov cyclization of homochiral dienone 122 (eq 28); however, in this example it is the syn diastereomer 123 that is the major reaction product.
 |
[28] |
Although the reasons for the observed diastereoselectivity for these reactions may be opaque, Prandi, Occhiato and coworkers have used remote stereocontrol in a Nazarov cyclization that was directed toward a total synthesis of roseophilin (eq 29).38 Treatment of dienone 125 in neat trifluoroacetic acid for 20 hours led to cyclopentenone 126 in 43% yield as a 4:1 diasteromeric mixture. The structure of the minor component of the mixture was not confirmed, but the authors proposed that it might be derived from an interrupted Nazarov process in which the pendant alkene group has intercepted the intermediate allylic cation. Cyclizations of the type shown in equations 28 and 29 lead to enantiopure products in moderate yield but with excellent control of relative stereochemistry.
 |
[29] |
7. Chiral Auxiliaries in the Nazarov Cyclization
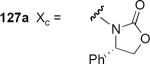
Pridgen and coworkers reported the first asymmetric Nazarov cyclization controlled by a chiral auxiliary group in 1999.39 Their work was motivated by the need for a practical and scalable asymmetric synthesis of indane endothelin receptor antagonists. The synthesis of starting materials 127a–c followed a conventional strategy that made use of a Knoevenagel condensation with piperonal. The phenyl- and isopropyl oxazolidinones 127a and 127b both underwent cyclization in dichloromethane or toluene at 0 °C in the presence of tin tetrachloride to produce 128a or 128b, respectively, as the major reaction product in good yield. The reasonable assumption that the stereochemical course of the cyclization was controlled through bidentate chelation of the Lewis acid with the carbonyl oxygen atoms of the oxazolidinone and the amide is probably not correct in this case. Cyclization of 127c that lacks the oxazolidinone ring takes place with even higher selectivity for 128c, and cyclization of 127a to 128a is catalyzed by methanesulfonic acid, arguing against a bidentate interaction of the substrate with the acid. These two results strongly suggest that bidentate metal-carbonyl complexation is not a prerequisite for high diastereoselectivity in this particular reaction. The authors suggest on the basis of computational work an unconventional model in which the two carbonyl groups in 127 adopt a transoid conformation in the reactive intermediate. The lack of an aromatic group in the chiral auxiliary group of 127b and the lack of a carbonyl group in the chiral auxiliary group of 127c preclude π-stacking interactions as the source of asymmetric induction.
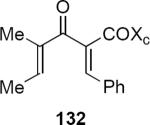
Flynn, Kerr and Metje have described a very useful extension of this work that introduces a one-pot palladium catalyzed tandem hydrostannylation-cross coupling process to prepare the oxazolidinone starting materials (Scheme 6).40 Palladium catalyzed syn hydrostannation of 130 was followed by coupling in situ with tigloyl chloride 131 to produce dienone 132 in 88% yield. Cyclization of 132 in the presence of methanesulfonic acid at low temperature led to the isolation of cis diastereomer 133 in 73% yield. This is evidently the kinetic product, since exposure of 132 to methanesulfonic acid in dichloromethane at 18 °C led to trans diastereomer 134. Trans product 134 was also isolated in 76% yield following treatment of 132 with Cu(II) triflate in dichloromethane at 18 °C. These are convenient protocols for forming the two isomeric series of products.
Scheme 6.

Hydrostannylation/Cross Coupling/Nazarov Cascade Process
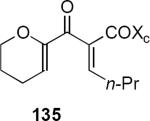
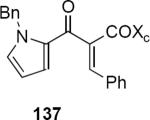
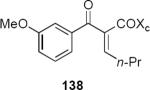
In subsequent and more extensive work Flynn and coworkers explored the full scope of these reactions and were able to extend it to the synthesis of cyclopentenones bearing only aliphatic substituents.41 A comprehensive screen of oxazolidinone chiral auxiliary groups revealed none that were more effective than the phenyloxazolidinone, therefore the (S)-phenyloxazolidinone group was used throughout. A partial listing of their results is summarized in Table 3. Product yields were generally high; however, torquoselectivity was modest. This was offset by the ease with which the diastereomeric products could be separated by chromatography. The sense of asymmetric induction for substrates bearing a β-phenyl (136, 132, 137) or a β-n-propyl group (135, 138) was reversed. In at least one case that is not discussed in this Report, reversal of torquoselectivity was observed in changing a Brønsted to a Lewis acid catalyst. A mechanistic hypothesis based on the relative sizes of the chiral auxiliary and the substituent groups has been proposed by the authors to rationalize the results. Nevertheless, these results highlight some of the challenges of incorporating these reactions into synthetic planning at this early stage of development.
Table 3.
Lewis and Brønsted Acid Catalyzed Nazarov Cychzations 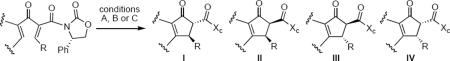
| Entry | Substrate | Conditionsa | Ib | IIb | IIIb | IVb |
|---|---|---|---|---|---|---|
| 1 |
|
A | 27 (32) | 0 | 65 (68) | 0 |
|
| ||||||
| 2 |

|
B | 48 (70) | (9) | 8 (13) | (8) |
| C | (7) | 64 (68) | (2) | (23) | ||
|
| ||||||
| 3 |
|
A | (31) | (41) | (17) | (11) |
| B | 65 (60) | (8) | 10 (16) | (2) | ||
| C | 0 | 70 (63) | 0 | (23) | ||
|
| ||||||
| 4 |
|
B | 51 (56) | 16 (18) | 18 (22) | (4) |
|
| ||||||
| 5 |
|
A | 26 (28) | (0) | 43 (46) | 0 |
A: MeSO3H (5 – 10 equiv), −78 °C to rt, CH2CI2; aq NaHCO3, 1 – 24 h; B: Cu(OTf)2 (1.1 equiv), −78 °C to rt, CH2CI2; aq NaHCO3, 1–48 h; C: MeSO3H (10 equiv), −78 °C to 0 °C, 0.5 – 2 h; aq NaHCO3.
isolated yield (1H NMR yield).
Harrington and Tius have developed a different type of chiral auxiliary controlled asymmetric Nazarov cyclization (Scheme 7).42 Addition of lithioallene 140 to morpholino enamide 139 at −78 °C in THF and in the presence of 4 equivalents of lithium chloride, followed by quenching into a solution of hydrochloric acid in hexafluoroisopropanol (HFIP) at 0 °C led to cyclopentenone 141 in 67% yield and 67% ee. In contrast to the oxazolidinone based chiral auxiliaries, the permethylated α-D-glucose derived chiral auxiliary in 140 is lost during the cyclization, so it is traceless. As a consequence, the timing of the cleavage of the chiral auxiliary along the reaction coordinate is important: cleavage must take place after the formation of the σ-bond that forms the cyclopentane ring has been initiated, and not before. Sugar derived chiral auxiliaries often suffer from a limitation due to the fact that in general only the D-sugars are available, thereby limiting access to only one enantiomeric series of product. This is not the case for the present example. Lithioallene 142 that is derived from the β-anomer of permethylated D-glucose leads to ent-141 in 71% yield and 82% ee. In this way both enantiomeric series of products are available from cheap D-glucose.
Scheme 7.

Glucose Derived Chiral Auxiliaries for the Allene Ether Nazarov
The permethylated D-glucose based reagents of Scheme 7 represent the first generation approach to the problem. Unexpectedly, lithioallene 144 (eq 30) that was derived from 2-deoxy-D-glucose proved to be far superior to both 140 and to 142, but also to the lithioallene that had been prepared from permethylated 2-deoxy-D-glucose.43 The combination of 144 with morpholino enamide 143 led to cyclopentenone 145 in 66% yield and 90% ee. The counterintuitive conclusion was that substitution at C2 of the chiral auxiliary had little effect on the stereochemical outcome of the cyclization whereas the nature of the ether protecting groups did have an effect. This observation led to the development of greatly improved second-generation chiral auxiliaries.
 |
[30] |
A noteworthy feature of all lithioallenes is their high reactivity. The asymmetric Nazarov cyclizations of the allenyl ethers take place readily at low temperature and they are not limited to substrates bearing aryl groups. The product yields reported for 144 varied between 53% and 85% whereas ee's varied between 86% and 93%.
 |
[31] |
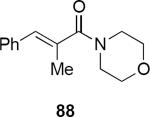
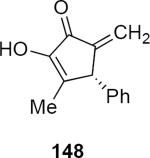
A systematic exploration of the factors that control the stereochemical outcome of the reactions of lithioallene 144 revealed that the silyloxy group at C3 exercised only a minimal effect on the product stereochemistry whereas the C4 silyloxy group had a significant effect (eq 31).44 For example, the reaction of 144 with morpholino enamide 88 led to cyclic product 148 in 84% yield and 93:7 er. Lithioallene 146 that was derived from 2,3-dideoxy-D-glucose led to 148 in 89% yield and 94:6 er, essentially the identical result that was obtained from 144 within experimental error. On the other hand 147 that was derived from 2,4-dideoxy-D-glucose led to 148 in 46% yield and with reduced er of 86.5:13.5. These results can be explained by postulating inversion of the pyran ring during the cyclization. Ring inversion has the effect of converting the C4 equatorial silyloxy group to an axial one that then shields one face of the acyclic pentadienyl cation, thereby strongly biasing the sense of conrotation.
 |
[32] |
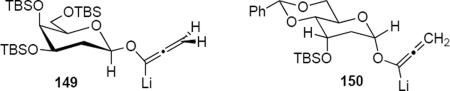
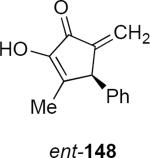
The pyran ring inversion that apparently takes place during the Nazarov cyclizations in the case of α-glycosidic allenyl ethers does not take place in the β-anomeric series. This is revealed by the result of equation 32. Lithioallene 149 that was derived from β-2-deoxy-D-galactose when combined with enamide 88 led to cyclopentenone ent-148 in 93% yield and with 96:4 er. This result can be contrasted with the outcome of the reaction of 88 with the lithioallene that was derived from persilylated β-2-deoxy-D-glucose. The yield of ent-148 was 68% yield but the er was only 89:11, suggesting that the C4 axial group in 149 plays the critical role in controlling the absolute stereochemistry of products. Therefore both in the α- and the β-anomer series the prerequisite for high enantioselectivity is the presence of an axial group that blocks one face of the pentadienyl cation.
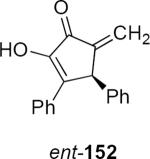
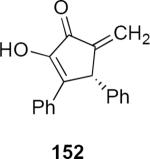
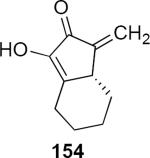
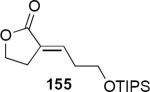
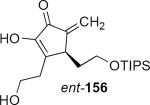
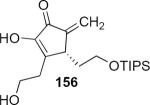
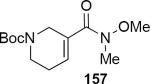
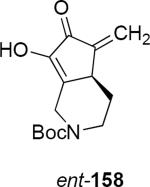
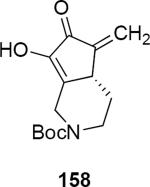
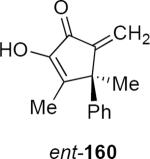
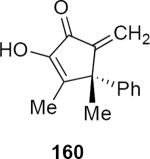
Lithioallene 150 was designed on the basis of the results presented above. Since conformational flexibility of the pyran ring is not a requirement for an effective chiral auxiliary, 150 was predicted to be an effective reagent for the asymmetric allene ether Nazarov cyclization because the silyloxy group at C3 is held in the axial position (Table 4). The results of Table 4 bear this out. It is noteworthy that an all carbon quaternary center can be incorporated into cyclopentenones 160 and ent-160 with 89:11 and 5:95 er, respectively. Low yields of 158 and ent-158 were observed because of the instability of these products that undergo decomposition through a β-elimination process involving the enol ether with the carbamate acting as a leaving group.
 |
[33] |
Table 4.
Sugar Derived Chiral Auxiliaries for the Allene Ether Nazarov
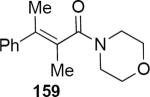
| Entry | Enamide | Product from 149 | yield/er | Product from 150 | yield/er |
|---|---|---|---|---|---|
| 1 |
|
|
92% 96.5:3.5 |
|
69% 97:3 |
| 2 |
|
|
57% 96:4 |
|
59% 95:5 |
| 3 |
|
|
93% 93:7 |
|
90% 97:3 |
| 4 |
|
|
83% 91:9 |
|
57% 93:7 |
| 5 |
|
|
59% 95:5 |
|
29% 93:7 |
| 6 |
|
|
78% 95:5 |
|
54% 89:11 |
The predictable stereochemical outcome of the allene ether Nazarov cyclizations makes their incorporation into synthetic planning straightforward. Camphor-derived lithioallene 161 was developed during the total synthesis of natural roseophilin (eq 33).45 The reaction of 161 with enamide 139 led to 141 in 78% yield and 86% ee, a significant improvement over the result with 140 (Scheme 7). Because of the array of useful functionality in 141 it was possible to convert it in only eight steps to (22R,23R)-roseophilin.
 |
[34] |
The camphor-derived lithioallene 161 has been shown to be generally useful for the asymmetric Nazarov cyclization, although the optical purity of products is somewhat lower than observed from the reactions of 149 or 150. The preparation of axially chiral substituted lithioallenes (e.g. 162, eq 34) as single diastereomers is straightforward and their reactivity with enamides follows that of 161.46 For example, addition of 162 to enamide 88 led to cyclopentenone 163 in 67% yield and 79% ee. Although 163 was isolated as a single E geometrical isomer at the exocyclic double bond, this was the result of acid catalyzed isomerization of the kinetically formed Z isomer. Substitution on the allene raises a matched-mismatched issue since the axial chirality of the allene also influences the torquoselectivity (see eq 18). In the case of methyl substitution at the distal allene carbon atom one would predict only a modest effect on the optical purity of the product. A much larger effect would be expected in the case of the allene that is substituted by a tert-butyl group (164, eq 35). This is indeed what was observed. Lithioallene 164 reacted with enamide 88 and led to cyclopentenone 165 in 80% yield and 96% ee. The axial stereochemistry of the allene in 164 and in 162 represents the matched case. In the mismatched case the axial chirality of the tert-butyl allene overwhelms the effect of the chiral auxiliary leading to the enantiomeric series of cyclopentenones. The products derived from 164 were much less prone to undergo Z to E isomerization during the reaction, making it possible to isolate the Z isomers as the major reaction products.
 |
[35] |
A somewhat different role for the camphor-derived chiral auxiliary group is shown in the diastereoselective interrupted Nazarov reaction that is summarized in Scheme 8.47 Addition of propargyllithium 166 to enamide 88 leads to enone 167 in 86% yield. Simply stirring 167 with 10 equivalents of phenethylamine 168 in the presence of dry Florisil in the absence of solvent leads to α-aminocyclopentenone 169 in 67% yield as a single diastereomer. This is a cascade process in which the first step is the amine-catalyzed isomerization of 167 to the allenyl ketone that then undergoes rapid cyclization. The cyclic cation is intercepted by the amine from the less hindered face of the molecule, leading ultimately to the observed product. There are several unusual features of this reaction. The first concerns the initial step that leads to the allene. The allene is stereogenic and since the distal TIPS substituent is very large, on the basis of the preceding discussion (e.g. eq 35) one would have to predict that it exerts a very large if not dominant effect on the torquoselectivity. This further suggests that the isomerization step is highly selective for the matched allene diastereomer and that the role of the chiral auxiliary is to influence the course of this isomerization step. The second unusual feature of this reaction concerns the nucleophilic trapping of the cation by the amine. The lifetime of the highly reactive intermediate cation must be long enough to allow effective trapping by the amine nucleophile in the absence of any solvent.
Scheme 8.

Asymmetric Interrupted Nazarov
The asymmetric interrupted Nazarov cyclization is reasonably general, as the examples of Figure 3 indicate. There appear to be no obvious limitations in terms of the enone starting material and both aliphatic and aromatic substituents are tolerated. It is noteworthy that secondary amines can also be used as nucleophiles as indicated by the examples of 171, 172 and 174. The acid labile acetal function in 170 was tolerated as well.
Figure 3.

Examples of the Asymmetric Interrupted Nazarov Reaction.
One example of the intramolecular version of this interrupted Nazarov reaction has been reported (Scheme 9). Reaction of an excess of 166 with enamide 175 leads to enone 176. Exposure of 176 to 10 equivalents of triethylamine on dry Florisil in the absence of solvent led to the anticipated product of intramolecular trapping 177 in 50% overall yield for the two steps, propargyllithium addition to the enamide and cyclization. Because the trapping step that leads to 177 takes place intramolecularly, the relative stereochemistry of the amino and phenyl groups in 177 is opposite that in 169.
Scheme 9.

Intramolecular Asymmetric Interrupted Nazarov
8. Summary and Outlook
It is curious that the first efforts to develop asymmetric versions of the Nazarov cyclization, a reaction that was first described in 1941, were reported only within the last ten years or so. Although a number of imaginative conditions for catalytic asymmetric cyclizations, as well as chiral auxiliary controlled processes have been described, each is subject to some limitations. At this time a truly general asymmetric Nazarov reaction, either catalytic or chiral auxiliary-controlled has not been described. One of the challenges is the requirement for an activated dienone. For example, in Rueping's seminal work polarization of the dienone by the enol ether is a requirement for the reaction. All the catalytic asymmetric or chiral auxiliary controlled methods that have been described to date succeed only for dienones that are substituted by either an α-ether or an α-carboxy group, or both. In addition, several of the methods are further limited by requiring β-substitution by one or more aryl groups. Another problem that bedevils the catalytic asymmetric Nazarov reactions is product inhibition.
The chiral auxiliary controlled allene ether cyclizations do not appear to be subject to any limitations in terms of the dienone, probably because of the activation inherent in the strain energy of the favorably polarized allene. A major shortcoming inherent in these reactions is that four of the five ring carbon atoms in the product are sp2 hybridized, affording the opportunity to control the absolute stereochemistry of only one ring carbon atom. Although chiral auxiliaries can provide highly practical solutions to problems in asymmetric synthesis, catalytic asymmetric processes have the advantage of atom economy. It seems likely that efforts in the future will focus on developing a truly general catalytic asymmetric Nazarov reaction.
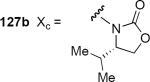
Table 2.
Chiral Auxiliary Controlled Aromatic Nazarov
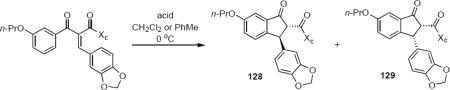
| starting material | acid (equiv) | yield (%) | product ratio (128a–c:129a–c:other) | |
|---|---|---|---|---|
|
127a | SnCI4 (1.1) | 85 | 88:12:0 |
| 127b | SnCI4 (1.1) | 74 | 71:16:14 | |
|
127c | SnCI4 (1.1) | 90 | 92:4:4 |
| 127a | MeSO3H (2.0) | 88 | 85:15:0 | |
| 127c Xc = 8-phenylmenthoxy | 127a | TiCI4 (1.1) | 60 | 70:30:0 |
Acknowledgment
We thank the National Institutes of Health (GM57873) for generous support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Nazarov IN, Zaretskaya II. Izv. Akad. Nauk SSSR, Ser. Khim. 1941:211–224. [Google Scholar]; (b) Nazarov IN, Zaretskaya II. Zh. Obshch. Khim. 1957;27:693–713. [Google Scholar]; (c) Nazarov IN, Zaretskaya II, Sorkina TI. Zh. Obshch. Khim. 1960;30:746–754. [Google Scholar]
- 2.Reviews of the Nazarov reaction: Nakanishi W, West FG. Curr. Op. Drug Disc. Dev. 2009;12:732–751.. Grant TN, Rieder CJ, West FG. Chem. Commun. 2009;38:5676–5688. doi: 10.1039/b908515g.. Frontier AJ, Collison C. Tetrahedron. 2005;61:7577–7606.. Pellissier H. Tetrahedron. 2005;61:6479–6517.. Tius MA. Eur. J. Org. Chem. 2005:2193–2206.. Harmata M. Chemtracts. 2004;17:416–435.. Tius MA. Acc. Chem. Res. 2003;36:284–290. doi: 10.1021/ar0200394.. Habermas KL, Denmark S, Jones TK. In: Organic Reactions. Paquette LA, editor. Vol. 45. John Wiley & Sons, Inc.; New York: 1994. pp. 1–158.. Krohn K. Org. Synth. Highlights. 1991:137–144.. Santelli-Rouvier C, Santelli M. Synthesis. 1983:429–442..
- 3.There are some examples of anionic, disrotatory 6 p electrocyclizations: Bates RB, McCombs DA. Tetrahedron Lett. 1969;10:977–978.. Shoppee CW, Henderson GN. Chem. Commun. 1974:561–562.. Williams DR, Reeves JT, Nag PP, Pitcock WH, Jr., Baik M-H. J. Am. Chem. Soc. 2006;128:12339–12348. doi: 10.1021/ja063243l..
- 4.Rueping M, Ieawsuwan W, Antonchick AP, Nachtsheim BJ. Angew. Chem., Int. Ed. 2007;46:2097–2100. doi: 10.1002/anie.200604809. [DOI] [PubMed] [Google Scholar]
- 5.Casson S, Kocienski P. J. Chem. Soc., Perkin Trans. 1. 1994:1187–1191. [Google Scholar]
- 6.He W, Sun X, Frontier AJ. J. Am. Chem. Soc. 2003;125:14278–14279. doi: 10.1021/ja037910b. [DOI] [PubMed] [Google Scholar]
- 7.Tius MA, Kwok C-K, Gu X.-q., Zhao C. Synth. Commun. 1994;24:871–885. [Google Scholar]
- 8.Rueping M, Ieawsuwan W. Adv. Synth. Catal. 2009;351:78–84. [Google Scholar]
- 9.Pousse G, Devineau A, Dalla V, Humphreys L, Lasne M-C, Rouden J, Blanchet J. Tetrahedron. 2009;65:10617–10622. [Google Scholar]
- 10.Basak AK, Shimada N, Bow WF, Vicic DA, Tius M,A. J. Am. Chem. Soc. 2010;132:8266–8267. doi: 10.1021/ja103028r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang G, Trauner D. J. Am. Chem.Soc. 2004;126:9544–9545. doi: 10.1021/ja0476664. [DOI] [PubMed] [Google Scholar]
- 12.Liang G, Gradl SN, Trauner D. Org. Lett. 2003;5:4931–4934. doi: 10.1021/ol036019z. [DOI] [PubMed] [Google Scholar]
- 13.Nie J, Zhu H-W, Cui H-F, Hua M-Q, Ma J-A. Org. Lett. 2007;9:3053–3056. doi: 10.1021/ol071114j. [DOI] [PubMed] [Google Scholar]
- 14.Yaji K, Shindo M. Synlett. 2009:2524–2528. [Google Scholar]
- 15.Kawatsura M, Kajita K, Hayase S, Itoh T. Synlett. 2010:1243–1246. [Google Scholar]
- 16.Cao P, Deng C, Zhou Y-Y, Sun X-L, Zheng J-C, Xie Z, Tang Y. Angew. Chem., Int. Ed. 2010;49:4463–4466. doi: 10.1002/anie.200907266. [DOI] [PubMed] [Google Scholar]
- 17.Huang J, Frontier AJ. J. Am. Chem. Soc. 2007;129:8060–8061. doi: 10.1021/ja0716148. [DOI] [PubMed] [Google Scholar]
- 18.Kokubo M, Kobayashi S. Chem. Asian J. 2009;4:526–528. doi: 10.1002/asia.200800461. [DOI] [PubMed] [Google Scholar]
- 19.Walz I, Togni A. Chem. Commun. 2008:4315–4317. doi: 10.1039/b806870d. [DOI] [PubMed] [Google Scholar]
- 20.Aggarwal VK, Belfield AJ. Org. Lett. 2003;5:5075–5078. doi: 10.1021/ol036133h. [DOI] [PubMed] [Google Scholar]
- 21.Bow WF, Basak AK, Jolit A, Vicic DA, Tius MA. Org. Lett. 2010;12:440–443. doi: 10.1021/ol9025765.. See also, Shimada N, Ashburn BO, Basak AK, Bow WF, Vicic DA, Tius MA. Chem. Commun. 2010;46:3774–3775. doi: 10.1039/b927564a..
- 22.Denmark SE, Wallace MA, Walker CB., Jr. J. Org. Chem. 1990;55:5543–5545. [Google Scholar]
- 23.Hu H, Smith D, Cramer RE, Tius MA. J. Am. Chem. Soc. 1999;121:9895–9896. [Google Scholar]
- 24.Schultz-Fademrecht C, Tius MA, Grimme S, Wibbeling B, Hoppe D. Angew. Chem., Int. Ed. 2002;41:1532–1535. doi: 10.1002/1521-3773(20020503)41:9<1532::aid-anie1532>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 25.Zimmermann M, Wibbeling B, Hoppe D. Synthesis. 2004:765–774. [Google Scholar]
- 26.Williams DR, Robinson LA, Nevill CR, Reddy JP. Angew. Chem., Int. Ed. 2007;46:915–918. doi: 10.1002/anie.200603853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.α-Ketoenones can undergo Nazarov cyclization under much milder reaction conditions: Forest J, Bee C, Cordaro F, Tius MA. Org. Lett. 2003;5:4069–4072. doi: 10.1021/ol035498z.. Batson WA, Sethumadhavan D, Tius MA. Org. Lett. 2005;7:2771–2774. doi: 10.1021/ol050970x..
- 28.Gao S, Wang Q, Chen C. J. Am. Chem. Soc. 2009;131:1410–1412. doi: 10.1021/ja808110d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao S, Wang Q, Huang LJ-S, Lum L, Chen C. J. Am. Chem. Soc. 2010;132:371–383. doi: 10.1021/ja908626k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Churruca F, Fousteris M, Ishikawa Y, Rekowski MW, Hounsou C, Surrey T, Giannis A. Org. Lett. 2010;12:2096–2099. doi: 10.1021/ol100579w. [DOI] [PubMed] [Google Scholar]
- 31.Noyori R, Ohnishi Y, Kato M. Tetrahedron Lett. 1971;19:1515–1518. [Google Scholar]
- 32.Crandall JK, Haseltine RP. J. Am. Chem. Soc. 1968;90:6251–6253. [Google Scholar]
- 33.Leitich J, Heise I, Werner S, Krueger C, Schaffner K. J. Photochem. Photobiol. A: Chem. 1991;57:127–151.. See also: Matlin AR, Jin K. Tetrahedron Lett. 1989;30:637–640..
- 34.Leitich J, Heise I, Rust J, Schaffner K. Eur. J. Org. Chem. 2001:2719–2726.. See also: Smith AB, III, Agosta WC. J. Am. Chem. Soc. 1973;95:1961–1968..
- 35.Mazzola RD, Jr., White TD, Vollmer-Snarr HR, West FG. Org. Lett. 2005;7:2799–2801. doi: 10.1021/ol051169q.. For related studies see: Liu C, Sowa JR., Jr. Tetrahedron Lett. 1996;37:7241–7244..
- 36.Wu Y-K, West FG. J. Org. Chem. 2010;75:5410–5413. doi: 10.1021/jo101112t. [DOI] [PubMed] [Google Scholar]
- 37.Prandi C, Ferrali A, Guarna A, Venturello P, Occhiato EG. J. Org. Chem. 2004;69:7705–7709. doi: 10.1021/jo0489263.. For related studies see: Srikrishna A, Neetu G. Tetrahedron: Asymmetry. 2010;21:2067–2071..
- 38.Occhiato EG, Prandi C, Ferrali A, Guarna A. J. Org. Chem. 2005;70:4542–4545. doi: 10.1021/jo0504058. [DOI] [PubMed] [Google Scholar]
- 39.Pridgen LN, Huang K, Shilcrat S, Tickner-Eldridge A, DeBrosse C, Haltiwanger RC. Synlett. 1999:1612–1614. [Google Scholar]
- 40.Kerr DJ, Metje C, Flynn BL. Chem. Commun. 2003:1380–1381. [PubMed] [Google Scholar]
- 41.Kerr DJ, White JM, Flynn BL. J. Org. Chem. 2010;75:7073–7084. doi: 10.1021/jo100736p. [DOI] [PubMed] [Google Scholar]
- 42.Harrington PE, Tius MA. Org. Lett. 2000;2:2447–2450. doi: 10.1021/ol0001362. [DOI] [PubMed] [Google Scholar]
- 43.delos Santos DB, Banaag AR, Tius MA. Org. Lett. 2006;8:2579–2582. doi: 10.1021/ol060816q. [DOI] [PubMed] [Google Scholar]
- 44.(a) Banaag AR, Tius MA. J. Am. Chem. Soc. 2007;129:5328–5329. doi: 10.1021/ja069342g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Banaag AR, Tius MA. J. Org. Chem. 2008;73:8133–8141. doi: 10.1021/jo801503c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harrington PE, Tius MA. J. Am. Chem. Soc. 2001;123:8509–8514. doi: 10.1021/ja011242h. [DOI] [PubMed] [Google Scholar]
- 46.Harrington PE, Murai T, Chu C, Tius MA. J. Am. Chem. Soc. 2002;124:10091–10100. doi: 10.1021/ja020591o. [DOI] [PubMed] [Google Scholar]
- 47.Dhoro F, Kristensen TE, Stockmann V, Yap GPA, Tius MA. J. Am. Chem. Soc. 2007;129:7256–7257. doi: 10.1021/ja0718873. [DOI] [PMC free article] [PubMed] [Google Scholar]