

Abstract
A1 adenosine receptor (AR) agonists are neuroprotective, cardioprotective, and anxiolytic. (N)-Methanocarba adenine nucleosides designed to bind to human A1AR were truncated to eliminate 5′-CH2OH. This modification previously converted A3AR agonists into antagonists, but the comparable effect at A1AR is unknown. In comparison to ribosides, affinity at the A1AR was less well preserved than that at the A3AR, although a few derivatives were moderately A1AR selective, notably full agonist 21 (N6-dicyclopropylmethyl, Ki 47.9 nM). Thus, at the A1AR, recognition elements for nucleoside binding depend more on 5′ region interactions, and in their absence, A3AR selectivity predominates. Based on the recently reported agonist-bound AR structure, this difference between subtypes likely correlates with an essential His residue in transmembrane domain 6 of A1 but not A3AR. The derivatives ranged from partial to full agonists in A1AR-mediated adenylate cyclase inhibition. Truncated derivatives have more druglike physical properties than other A1AR agonists; this approach is appealing for preclinical development.
Keywords: G protein-coupled receptor, purines, molecular modeling, radioligand binding, adenylate cyclase
Adenosine modulates many physiological processes by activating one or more of four subtypes of G protein-coupled receptors (GPCRs).1 The medicinal chemistry of adenosine receptors (ARs) is now well advanced in comparison to the cases of many other GPCRs, with the existence of numerous selective agonists and antagonists, allosteric modulators, prodrugs, radioligands for imaging, fluorescent probes, and macromolecular ligand conjugates.2 A1AR ligands have been considered clinically for a variety of conditions: agonists (diabetes, pain), partial agonists (arrhythmias), and antagonists (heart failure, renal protection).3
The adenosine structure has been extensively modified, on both the nucleobase and ribose, for pharmacological optimization to selectively activate ARs. One means of achieving AR subtype selectivity has been the replacement of ribose with a sterically constrained methanocarba ([3.1.0]-bicyclohexane) ring system. This bicyclic system adopts a North (N)-envelope conformation in MRS3558 (1) (Chart 1) to maintain a receptor-preferred conformation and enhanced affinity at the A3AR.6 The (N)-methanocarba modification is also tolerated at A1AR, but without affinity enhancement. Consequently, the cardioprotective MRS3630 (2) containing the A1AR-favoring N6-cyclopentyl substituent is a mixed A1/A3AR agonist.7
Chart 1. Structures of Representative Ribosides and Ring-Constrained Methanocarba Nucleoside Derivatives That Have Been Characterized as Agonist, Partial Agonists, and Antagonists at the A3AR (Ki, nM, in Binding to the hA3AR in Italics)6−10.

Another useful modification is truncation of the ribose at the 4′ position, that is, removing the 5′-CH2OH moiety, while retaining all other features of the ribose-like moiety and its stereochemistry. Thus, 4′-thionucleoside antagonist LJ-1251 (3) and 4′-oxo antagonist 4 preserve affinity and selectivity at the A3AR, while removing the ability to induce the required conformational change for receptor activation.8,9 Truncation of (N)-methanocarba nucleosides originally was reported to convert A3AR agonists into selective antagonists in a guanine nucleotide binding assay.10 Subsequently, partial agonism in a functional assay of adenosine 3′,5′-cyclic phosphate (cyclic AMP) at the Gi-coupled A3AR was shown for MRS5127 (5) and congeners.11
In contrast to the reduced A3AR efficacy of 5′-truncated nucleosides, at the Gs-coupled A2AAR, full agonism is retained, as shown recently for the 4′-thio series.12 The effects of truncation on A1AR efficacy are unknown. Our major objective was to probe the effects of truncated (N)-methanocarba nucleosides at the human (h) A1AR, both pharmacologically and with insight into the structural basis for receptor recognition. Therefore, we have incorporated N6 substituents that are expected to promote A1AR affinity. For example, nucleoside 6, previously characterized at the A3AR,12 contains N6-cyclopentyl, which generally produces A1AR selectivity in the riboside series. The Gi-coupled A1AR is more homologous in primary sequence and effector coupling to the Gi-coupled A3AR than to the Gs-coupled A2AAR. If it more closely resembles A3AR in ligand binding and activation mechanism, the truncated analogues such as 6 will be A1AR antagonists or partial agonists. However, if its activation more closely resembles the A2AAR, then these truncated derivatives will be full A1AR agonists. With the recent structural elucidation of an A2AAR active state,15 it is feasible to relate these findings to specific binding site interactions.
Results
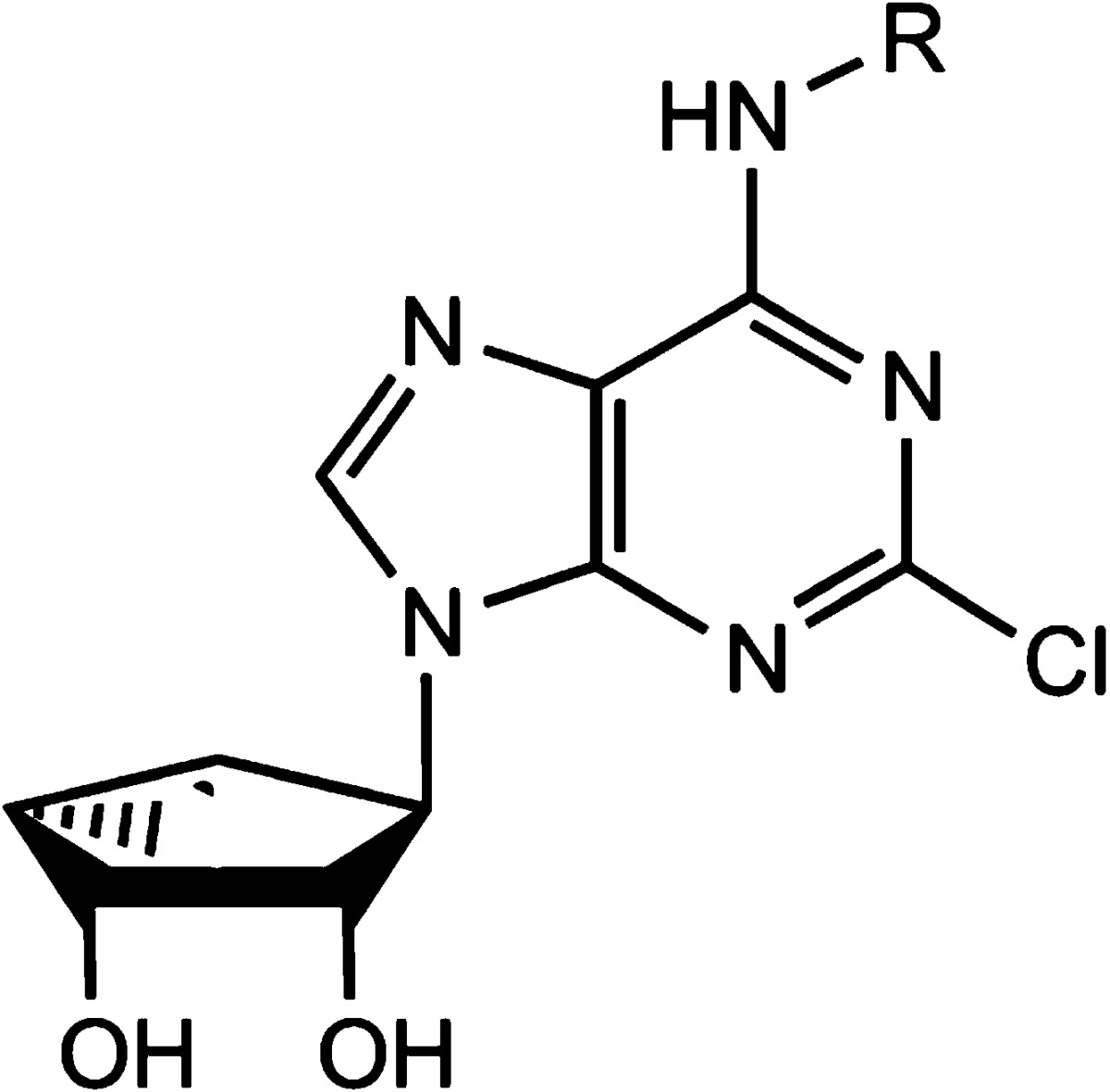
With the objective of increasing A1AR affinity, we explored N6 substitution of 4′-truncated (N)-methanocarba nucleoside derivatives, which were shown previously to be selective A3AR antagonists/partial agonists.12 Therefore, the series included N6-cycloalkyl (8–12) and N6-bicycloalkyl (13,14), and N6-acyclic alkyl and N6-cyclopropylalkyl (15–21) substitutions associated previously with A1AR selectivity of ribosides (Table 1).13 Finally, certain substituted N6-benzyladenosine derivatives, such as 2-fluorobenzyl, were reported to have enhanced affinity at the A1AR.23 Therefore, a variety of fluorinated and nonfluorinated N6-benzyl derivatives (22–28) were prepared.
Table 1. Potency of a Series of Truncated (N)-Methanocarba Adenosine Derivatives at Three Subtypes of hARs and Relative Efficacy at hA1AR.
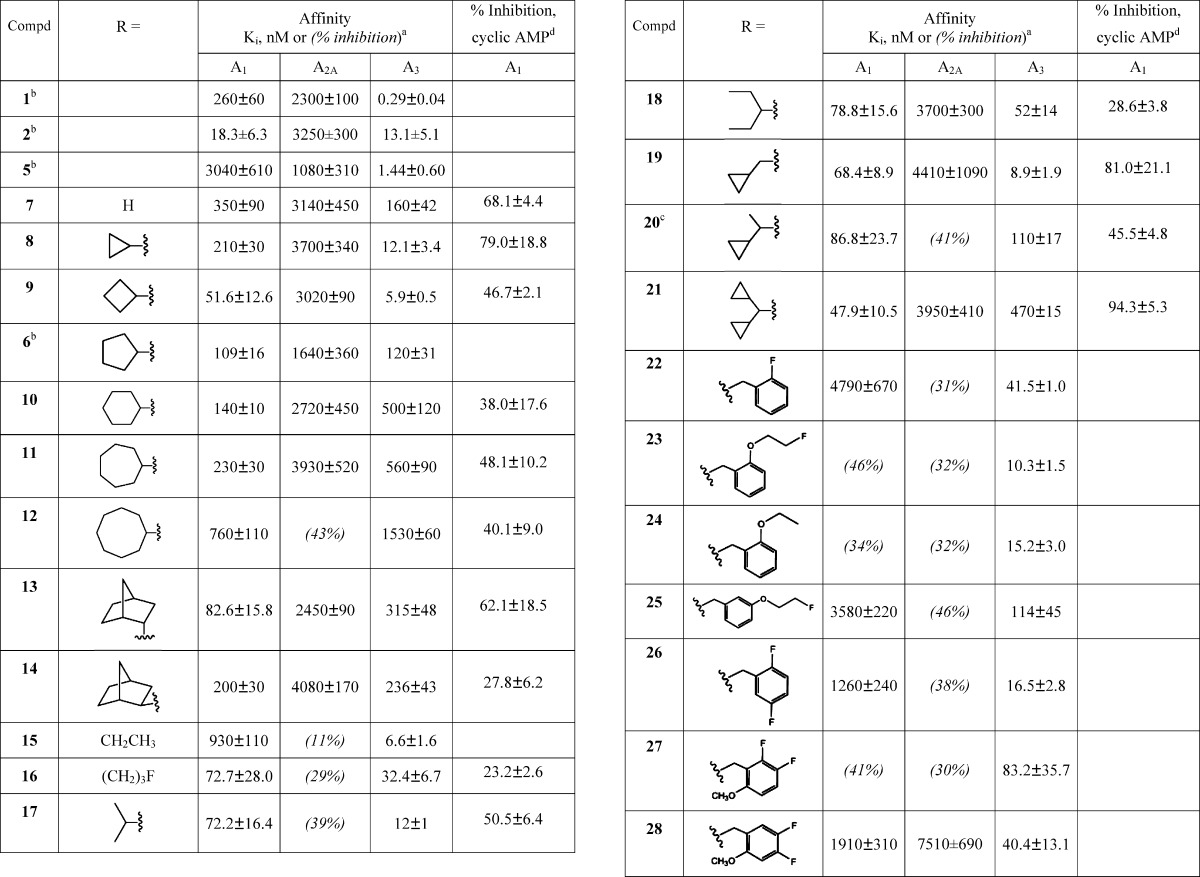
Using CHO or HEK293 (A2A only) cells stably expressing a hAR (Supporting Information); affinity was expressed as Ki value (n = 3–5) or percent inhibition of radioligand binding at 10 μM.
20 is a diastereomeric mixture.
Maximal efficacy (at 10 μM) in an A1AR functional assay, determined by inhibition of forskolin-stimulated cyclic AMP production in AR-transfected CHO cells, expressed as percent inhibition (mean ± standard error, n = 3–5) in comparison to effect (100%) of full agonist CPA 30 at 10 μM. The value for NECA was 100 ± 15.
The synthetic route to the truncated derivatives involves nucleophilic displacement by the appropriate amine of a 6-chloroadenine group in a 2′,3′-isopropylidene-protected precursor 29 (Scheme S1 of the Supporting Information). All of the analogues contain a 2-chloro substitution of the adenine ring, which has been shown to increase affinity at either or both A1AR and A3AR. 2-Chloro substitution of the A1AR agonist N6-cyclopentyladenosine (CPA, 30) also was shown to reduce agonist efficacy at the A3 but not A1AR.14
Several previously reported 2-chloro (N)-methanocarba derivatives (1, 2, 5, and 6) were used for comparison in the biological assays (Table 1). Binding assays at three hAR subtypes were carried out using standard radioligands and membrane preparations from Chinese hamster ovary (CHO) cells (A1 and A3) or human embryonic kidney (HEK) 293 cells (A2A) stably expressing a hAR subtype (Table 1).12 Since activity within the class of (N)-methanocarba nucleosides was previously noted to be very weak or absent at the hA2BAR,16 we did not include this receptor in the screening protocol.
Generally, the 5′-truncated (N)-methanocarba-adenosine derivatives, in comparison to the corresponding 9-ribosides, maintained affinity at the A3AR more effectively than at the A1AR. Nevertheless, affinities of <100 nM at the A1AR were achieved for certain N6-alkyl and cycloalkyl members of this series. Among the most potently binding 2-chloro (N)-methanocarba analogues (Ki, in nM) at the hA1AR (with Ki of the corresponding N6 derivatized 9-ribosides at rat A1AR in parentheses13) are the following: cyclobutyl 9, 51.6 (0.7); isopropyl 17, 72.2 (1.9); cyclopropylmethyl 19, 68.4 (0.8); and di(cyclopropyl)methyl 21, 47.9 (0.8). The A1AR affinity of the N6-ethyl analogue 15 was more substantially reduced, by 141-fold, with Ki values of 930 and 6.6 nM for the truncated and 9-riboside analogues, respectively. Other N6 analogues that were more significantly reduced in their hA1AR affinity in comparison to the corresponding riboside at rat A1AR (Ki, nM) are as follows: cyclohexyl 10, 131-fold (0.9); endo-norbornyl 13, 113-fold (0.34); exo-norbornyl 14, 231-fold (0.7); and 2-fluorobenzyl 22, 800-fold (6). Because of this reduced affinity, the selectivity for the A1AR was diminished overall. Only a few derivatives tended toward A1AR selectivity in comparison to the A3AR: di(cyclopropyl)-methyl 21, 10-fold; endo-norbornyl 13, 4-fold; and large cycloalkyl derivatives 10–12, 2–3-fold. The degree of selectivity vs A2AAR was higher: 21, 74-fold; 13, 30-fold. Many of the other derivatives were equipotent in binding to A1 and A3ARs, and the substituted N6-benzyladenosine derivatives (22–28) were generally selective for the A3AR, similar to previous observations with truncated N6-benzyl derivatives.10,12
Functional data determined at a single concentration (10 μM) in an assay of adenylate cyclase (A1AR-induced inhibition of cyclic AMP) are reported in Table 1. The potent and selective agonist CPA was used as the standard full agonist, and the nonselective AR agonist 5′-N-ethylcarboxamidoadenosine (NECA) was also a full agonist in this assay. Most of the analogues were partial agonists of the A1AR. However, concentration–response curve for dicyclopropylmethyl analogue 21 in A1AR-mediated inhibition of cyclic AMP indicated full agonism compared to NECA (Figure S1 of the Supporting Information). The EC50 values for 21 and NECA were 40.7 ± 19.7 and 10.2 ± 3.3 nM, respectively, in close agreement with the A1AR binding affinities.
A correlation plot of the hA3AR affinity of truncated 2-chloro-(N)-methanocarba analogues in the present study (x-axis) vs the hA3AR affinity of 2-unsubstituted adenine-7-riboside analogues (y-axis)13 demonstrated a parallel in these parameters (Figure 1B). The affinity at the A3AR subtype was relatively well maintained (correlation coefficient of 0.55). The enhanced A3AR affinity of a 5′-truncated ribonucleoside derivative was first noted for the 2-chloro-N6-(3-iodobenzyl) derivative16 and has been validated consistently in SAR studies that also showed lowered efficacy in this series.8−10 However, a similar plot of A1AR affinity comparing ribonucleosides and truncated ring-constrained nucleosides illustrated the trend of consistently lower affinity with truncation (Figure 1A), but without correlation of Ki values (correlation coefficient of 0.33). Therefore, in comparison to the case of ribosides, the affinity of the truncated ring-constrained analogues at the A1AR was less well preserved than that at the A3AR.
demonstrated a parallel in these parameters (Figure 1B). The affinity at the A3AR subtype was relatively well maintained (correlation coefficient of 0.55). The enhanced A3AR affinity of a 5′-truncated ribonucleoside derivative was first noted for the 2-chloro-N6-(3-iodobenzyl) derivative16 and has been validated consistently in SAR studies that also showed lowered efficacy in this series.8−10 However, a similar plot of A1AR affinity comparing ribonucleosides and truncated ring-constrained nucleosides illustrated the trend of consistently lower affinity with truncation (Figure 1A), but without correlation of Ki values (correlation coefficient of 0.33). Therefore, in comparison to the case of ribosides, the affinity of the truncated ring-constrained analogues at the A1AR was less well preserved than that at the A3AR.
Figure 1.

(A) Correlation of the affinity of truncated 2-chloro-(N)-methanocarba analogues (x-axis, at the hA1AR) and 2-unsubstituted adenine-7-riboside analogues (y-axis, at the rat A1AR).13 (B) Correlation of the hA3AR affinity of truncated 2-chloro-(N)-methanocarba analogues (x-axis) and 2-unsubstituted adenine-7-riboside analogues (y-axis).13
One derivative that tended toward A1AR-selectivity, the full agonist 21, contained a N6-dicyclopropylmethyl group. Curiously, truncated analogues of N6-cyclopentyl and N6-benzyl derivatives were notably reduced in A1AR affinity, even in the case of a 2-fluoro analogue 22, a modification previously found to enhance A1AR selectivity in the riboside series.13 Compound 22 was 115-fold selective for the A3AR in comparison to the A1AR. A 2-fluoroethyl ether 23 was a potent and selective ligand at the A3AR (Ki 10.3 nM), which might be sufficient affinity for use in radiofluorination for positron emission tomography.
There is a general phenomenon of reduced AR efficacy by removing H-bonding groups from the ribose moiety, especially at the A3AR but also at the A1AR.5,16 A precedent for reduced A1AR efficacy in nucleoside derivatives is the series of xanthine 7-ribosides. The 5′-uronamide analogue 5′-N-methyl 1,3-dibutylxanthine 7-β-d-ribofuronamide (DBXRM) was a full agonist at both A1 and A3ARs. However, removal of the 3′-hydroxyl group produced antagonism at the rat adipocytes A1AR and partial agonism at the rat A3AR. In examining the question of whether 5′-truncation in the present series of rigid (N)-methanocarba analogues provides agonists or antagonists at the A1AR, we found that the answer lies in the middle; that is, the agonist efficacy is partially maintained for many of the derivatives, as shown in Table 1, but the pattern is not uniform.
To examine docking of truncated (N)-methanocarba nucleosides, homology models of the hA1AR and hA3AR were produced from the agonist-bound A2AAR X-ray structure (3QAK).15 It was noted that key interactions of the nucleoside ligand with His251 in transmembrane domain (TM) 6, conserved between A1AR and A2AAR, and with Thr91 in TM3 are necessarily missing in the truncated derivatives. In the A2AAR structure, this essential His residue, which is absent in the A3AR, and Thr91 both H-bonded to a 5′-CO-NH-alkyl moiety of a cocrystallized NECA-like agonist.15 We predict that the difference in this key His residue is related to the dramatically altered binding of the truncated ring-constrained nucleosides at A1 and A3ARs. Thus, in general, A3AR recognition would depend more on interactions of groups other than the nucleoside 5′ substituent; A1AR recognition depends more on 5′ region interactions, and in their absence, A3AR selectivity predominates. This is consistent with the truncated derivatives maintaining A3AR, but not A1AR, affinity.
Furthermore, the hydrophilic region of the receptor associated with ribose binding is critical for activation. This step likely involves essential residues of TM3, TM6, and TM7 throughout the AR family, as in the A2AAR.15 Multiple H-bonding groups in this region promote agonism at the A3AR,14 such as the interactions of the 5′-CH2OH of the ribose moiety or the corresponding 5′-CO-NH-alkyl in NECA-like analogues. Thus, loss of the 5′ substituent is expected to reduce AR efficacy, which it clearly does at the A3AR. However, the effect at the A1AR is highly variable, with relative maximal efficacies of the truncated (N)-methanocarba analogues ranging from low (20–30% in compounds 14, 16, and 18) to high (80–100% in compounds 8, 19, and 21). The structural basis for the full agonism of 21, in contrast to closely related compounds such as the N6-(3-pentyl) derivative 18, which has greatly reduced efficacy at this subtype, must arise from conformational effects of the N6 group.
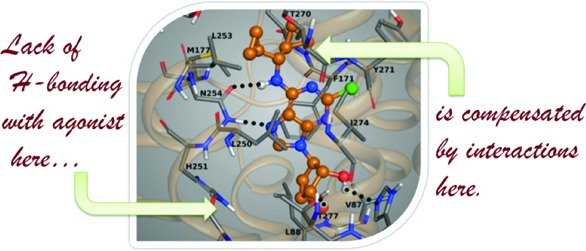
The docking poses of nonselective agonist NECA, full agonist 21, and low efficacy partial agonist 16 in the putative binding site of the A1AR were compared (Figure 2 and Figure S2 of the Supporting Information). The H-bond interactions between the 5′-CO-NH-ethyl group of NECA and the two residues, His251(6.52) and Thr91(3.36), could lock the ribose moiety in an active conformation, with the 2′,3′-hydroxyl groups of the ribose ring correctly directed toward Thr277(7.42) and His278(7.43) in order to pull TM7 toward TM3 to efficiently activate the receptor.15 In the absence of an interaction with His251(6.52) and/or Thr91(3.36) due to the lack of the 5′ substituent, the orientation of the rigid methanocarba moiety could be less effective in forming the H-bond interactions with Thr277(7.42) and His278(7.43) needed to attract TM7. In the case of A3AR, the activation process could be slightly different from that for the A1AR, due to some differences in the key residues of the binding pocket. Position 3.32 in A3AR consists of a nonconserved bulky and hydrophobic leucine residue, while a smaller valine is present in the other AR subtypes. Residue 3.32 was close to the ribose ring of the docked NECA in both A1AR and A3AR binding sites. The longer side chain of Leu90(3.32) in A3AR, in comparison to Val87(3.32) of A1AR, could contribute to a stronger hydrophobic interaction with the methanocarba moiety of the 5′-truncated agonists. At the same time, the different nature of the substituents at the N6 position of the adenine ring could influence the selectivity and the efficacy of agonists at both the A1AR and the A3AR. The N6 groups in the docking poses of the studied agonists lay in a region of the pocket formed by residues in the upper part of TM6 and TM7. The A1AR and A3AR differ in the nature of the residues lining this pocket. The variation of the affinity and potency of this series of 5′ truncated methanocarba analogues at the A1AR indicates that the N6 substituent can greatly affect these parameters, in some cases compensating for the lack of H-bonding interactions in the ribose 4′ region. From the docking pose of full agonist 21 in the A1AR model (Figure 2B), the favorable interactions of the N6-dicyclopropylmethyl substituent and the residues in the upper part of TM6 and TM7 (such as Thr257, Leu253, and Thr270) could maintain the adenine and methanocarba moieties in an efficacious active conformation with strong H-bond and hydrophobic interactions with residues in TM6, TM3, TM7, and EL2. A smaller and more flexible N6 group, such as the 3-fluoropropyl substituent of 16, in addition to the lack of interactions with His251(6.52) and Thr91(3.36), could negatively affect the ability to fully activate the receptor through conformational affects originating at the upper pocket of TM6 and TM7 (Figure S3 of the Supporting Information).
Figure 2.

Docking poses of NECA (A) and truncated nucleoside 21 (B) in the binding site of the hA1AR homology model based on the X-ray structure of an agonist-bound hA2AAR, indicating the main H-bond interactions.
The physicochemical properties of nucleosides that act as AR agonists often lead to limited in vivo bioavailability. The C-log p of compound 21 is 1.41, with the optimal for small molecular pharmaceutical substances being typically 2–3.17 The comparable parameter for the related A1AR-selective riboside and prototypical agonist CPA is 0.14, which is less desirable. Also, the polar surface area (PSA) values for 21 and CPA are calculated to be 92.8 and 122 Å2, respectively. Most druglike small molecules have a PSA smaller than 120 Å2. Compound 21 has fewer hydroxyl groups than CPA, which would favor bioavailability. The molecular weight of 21 of 376 is comfortably within the preferred range. Therefore, by several criteria, the full agonist of the A1AR 21 is more druglike than CPA.
The bioavailability of peripherally administered 21 in the brain, i.e. whether its altered physicochemical properties may facilitate its passage across the blood brain barrier, is undetermined. Many of the efforts to develop A1 agonists have attempted to limit central nervous system (CNS) penetration to avoid central mediated side effects, but other envisioned applications of A1 agonists depend on brain entry. In previous studies of the activity of A1AR agonists in the CNS, only a small fraction of a peripherally administered agent crossed the blood brain barrier.4 However, a similar attempt to alter the biodistribution by removing the 2′-hydroxyl group of CPA did not enhance brain uptake.4
In conclusion, truncated (N)-methanocarba adenine nucleosides display highly variable degrees of binding affinity and activation at the hA1AR. Based on the recently reported agonist-bound AR X-ray structure, this difference between subtypes likely correlates with an essential His residue in TM6 of A1 but not A3AR. By overcoming the lack of an important recognition element for receptor binding, i.e., the 5′ substituent, a N6-dicyclopropylmethyl derivative 21 was empirically identified as a moderately A1AR selective, full agonist. This is counterintuitive given that many 5′-modified analogues are partial agonists at the A1AR.18 A1AR agonists hold interest therapeutically for their cardio- and neuroprotective, antiarrhythmic, antiseizure, antilipolytic, antiglaucoma, and anxiolytic actions. It is conceivable that the expanded range of physical properties in the present series of truncated derivatives would offer pharmacokinetic advantages. Therefore, this approach is appealing for preclinical development. This hypothesis will have to be evaluated in the future in vivo studies.
Acknowledgments
We thank Prof. Ray Stevens (Scripps Research Institute, La Jolla, CA) and Dr. Vsevolod Katrich (University of California, San Diego) for helpful discussions and Dr. Noel Whittaker (NIDDK) for mass spectral determinations.
Glossary
Abbreviations
- AR
adenosine receptor
- cyclic AMP
adenosine 3′,5′-cyclic phosphate
- CPA
N6-cyclopentyladenosine
- CHO
Chinese hamster ovary
- GPCR
G protein-coupled receptor
- HEK
human embryonic kidney
- MRS3558
(1′S,2′R,3′S,4′R,5′S)-4′-{2-chloro-6-[(3-chlorophenylmethyl)amino]purin-9-yl}-1-(methylaminocarbonyl)bicyclo[3.1.0]hexane-2,3-diol
- MRS3630
(1′S,2′R,3′S,4′R,5′S)-4-(2-chloro-6-(cyclopentylamino)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide
- MRS5127
(1′S,2′R,3′S,4′R,5′S)-4′-[2-chloro-6-(3-iodobenzylamino)-purine]-2′,3′-O-dihydroxybicyclo[3.1.0]hexane
- NECA
5′-N-ethylcarboxamidoadenosine
- TM
transmembrane domain
Supporting Information Available
Synthetic procedures for compounds 7–28, their characterization and bioassays, and modeling procedures and results. This material is available free of charge via the Internet at http://pubs.acs.org.
The X-ray structure of NECA bound to a thermostabilized A2AAR was recently solved (RCSB ID: 2YDV; Lebon et al. 2011, Nature, doi:10.1038/nature10136) and is very similar to our docked pose of NECA.
The authors thank the Intramural Research Program of the NIH, NIDDK, for support.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fredholm B. B.; IJzerman A. P.; Jacobson K. A.; Linden J.; Müller C. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller C.; Jacobson K. A. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta, Biomembranes 2011, 1808, 1290–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi I.; Nieri P. Therapeutic potential of A1 adenosine receptor ligands: a survey of recent patent literature. Expert Opin. Therap. Patents 2008, 18, 677–691. [Google Scholar]
- Bueters T. J.; IJzerman A. P.; van Helden H. P.; Danhof M. Characterization of the pharmacokinetics, brain distribution, and therapeutic efficacy of the adenosine A1 receptor partial agonist 2′-deoxy-N6-cyclopentyladenosine in sarin-poisoned rats. Toxicol. Appl. Pharmacol. 2003, 192, 86–94. [DOI] [PubMed] [Google Scholar]
- Park K. S.; Hoffmann C.; Kim H. O.; Padgett W. L.; Daly J. W.; Brambilla R.; Motta C.; Abbracchio M. P.; Jacobson K. A. Activation and desensitization of rat A3-adenosine receptors by selective adenosine derivatives and xanthine-7-ribosides. Drug Dev. Res. 1998, 44, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchilibon S.; Joshi B. V.; Kim S. K.; Duong H. T.; Gao Z. G.; Jacobson K. A. (N)-Methanocarba 2,N6-disubstituted adenine nucleosides as highly potent and selective A3 adenosine receptor agonists. J. Med. Chem. 2005, 48, 1745–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A.; Gao Z. G.; Tchilibon S.; Duong H. T.; Joshi B. V.; Sonin D.; Liang B. T. Semirational design of (N)-methanocarba nucleosides as dual acting A1 and A3 adenosine receptor agonists: Novel prototypes for cardioprotection. J. Med. Chem. 2005, 48, 8103–8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong L. S.; Pal S.; Choe S. A.; Choi W. J.; Jacobson K. A.; Gao Z. G.; Klutz A. M.; Hou X.; Kim H. O.; Lee H. W.; Lee S. K.; Tosh D. K.; Moon H. R. Structure activity relationships of truncated D- and L- 4¢-thioadenosine derivatives as species-independent A3 adenosine receptor antagonists. J. Med. Chem. 2008, 51, 6609–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S.; Choi W. J.; Choe S. A.; Heller C. L.; Gao Z. G.; Chinn M.; Jacobson K. A.; Hou X.; Lee S. K.; Kim H. O.; Jeong L. S. Structure-activity relationships of truncated adenosine derivatives as highly potent and selective human A3 adenosine receptor antagonists. Bioorg. Med. Chem. 2009, 17, 3733–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melman A.; Wang B.; Joshi B. V.; Gao Z. G.; de Castro S.; Heller C. L; Kim S. K.; Jeong L. S.; Jacobson K. A. Selective A3 adenosine receptor antagonists derived from nucleosides containing a bicyclo[3.1.0]hexane ring system. Bioorg. Med. Chem. 2008, 16, 8546–8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X.; Kim H. O.; Alexander V.; Kim K.; Choi S.; Park S. G.; Lee J. H.; Yoo L. S.; Gao Z. G.; Jacobson K. A.; Jeong L. S. Discovery of a new human A2A adenosine receptors agonist, truncated 2-hexynyl 4′-thioadenosine. ACS Med. Chem. Lett. 2010, 1, 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Chinn M.; Ivanov A. A.; Klutz A. M.; Gao Z. G.; Jacobson K. A. Functionalized congeners of A3 adenosine receptor-selective nucleosides containing a bicyclo[3.1.0]hexane ring system. J. Med. Chem. 2009, 52, 7580–7592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z. G.; Blaustein J.; Gross A. S.; Melman N.; Jacobson K. A. N6-Substituted adenosine derivatives: Selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem. Pharmacol. 2003, 65, 1675–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z. G.; Kim S. K.; Biadatti T.; Chen W.; Lee K.; Barak D.; Kim S. G.; Johnson C. R.; Jacobson K. A. Structural determinants of A3 adenosine receptor activation: Nucleoside ligands at the agonist/antagonist boundary. J. Med. Chem. 2002, 45, 4471–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F.; Wu H.; Katritch V.; Han G. W.; Jacobson K. A.; Gao Z. G.; Cherezov V.; Stevens R. C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A.; Siddiqi S. M.; Olah M. E.; Ji X. D.; Melman N.; Bellamkonda K.; Meshulam Y.; Stiles G. L.; Kim H. O. Structure-activity relationships of 9-alkyladenine and ribose-modified adenosine derivatives at rat A3 adenosine receptors. J. Med. Chem. 1995, 38, 1720–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H. Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- Zablocki J. A.; Wu L.; Shryock J.; Belardinelli L. Partial A1 adenosine receptor agonists from a molecular perspective and their potential use as chronic ventricular rate control agents during atrial fibrillation (AF). Curr. Top. Med. Chem. 2004, 4, 839–854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.