

Abstract
Recognition of causes of pulmonary hypertension other than congenital heart disease is increasing in children. Diagnosis and treatment of any underlying cause of pulmonary hypertension is crucial for optimal management of pulmonary hypertension. This article discusses the available knowledge regarding several disorders associated with pulmonary hypertension in children: idiopathic pulmonary arterial hypertension (IPAH), pulmonary capillary hemangiomatosis, pulmonary veno-occlusive disease, hemoglobinopathies, hepatopulmonary syndrome, portopulmonary hypertension and HIV. Three classes of drugs have been extensively studied for the treatment of IPAH in adults: prostanoids (epoprostenol, treprostinil, iloprost, beraprost), endothelin receptor antagonists (bosentan, sitaxsentan, ambrisentan), and phosphodiesterase inhibitors (Sildenafil, tadalafil). These medications have been used in treatment of children with pulmonary arterial hypertension, although randomized clinical trial data is lacking. As pulmonary vasodilator therapy in certain diseases may be associated with adverse outcomes, further study of these medications is needed before widespread use is encouraged.
Keywords: Pulmonary arterial hypertension, Idiopathic pulmonary arterial hypertension, Pulmonary capillary hemangiomatosis, Pulmonary veno-occlusive disease, Sickle cell disease, Hepatopulmonary syndrome, Portopulmonary hypertension, Human immunodeficiency virus
1. Idiopathic pulmonary arterial hypertension
Idiopathic pulmonary arterial hypertension (IPAH), previously called primary pulmonary hypertension, is a diagnosis of exclusion. The natural history if IPAH in children is poor. Data from 1965 revealed that 22 of 35 children diagnosed with IPAH died within 1 year of diagnosis [1]. In the NIH registry, the median untreated survival for children after diagnosis was reported to be 10 months as opposed to 2.8 years for adults [2]. This poor prognosis without targeted therapy was recently confirmed [3]. Advances in the understanding of the pathobiology of idiopathic pulmonary arterial hypertension has led to new treatment therapies and have resulted in an improvement in the prognosis for children with IPAH [4,5]. As IPAH still has no cure, the aim of treatment is to improve quality of life, hemodynamics, exercise capacity, and survival. Medical management of children follows a similar algorithm to that of adults treated with idiopathic pulmonary vascular disease [6]. Children appear to be more reactive to acute vasodilator testing compared with adults [7], and may have a better long-term outcome in the current era than adults (Fig. 1) [8].
Fig. 1.

Kaplan–Meier survival curve of children with idiopathic pulmonary arterial hypertension (IPAH) and associated pulmonary arterial hypertension (APAH) from the UK Pulmonary Hypertension service from 2001–2007.
Haworth and Hislop. Heart 2009;95:312–317.
1.1. Definition
Pulmonary arterial hypertension is defined as a mean pulmonary arterial pressure greater than 25 mmHg at rest, with a normal pulmonary capillary wedge pressure of less than 15 mm Hg and an increased pulmonary vascular resistance greater than 3 Wood units·m2 [9,10]. The Venice classification scheme, established in 2003 at the Third World Symposium on Pulmonary Arterial Hypertension is appropriate for adults and children. This classification has recently been updated at the Dana Point, California Fourth World Symposium on Pulmonary Arterial Hypertension [11]. Exercise criteria have been deleted for the current definition [10]. The diagnosis of IPAH is one of exclusion and therefore requires a complete evaluation of all possible etiologies of associated pulmonary arterial hypertension, left heart disease, and respiratory disease [10,12]
1.2. Heritability
Bone morphogenetic protein receptor type 2 (BMPR2) mutations have been identified in children and adults with IPAH and familial PAH [13–18]. This genetic mutation in the TGF-Beta receptor has been found in patients with familial PAH (50%) [17] and sporadic PAH (15–26%) [18]. BMPR2 mutations are inherited as in autosomal dominant pattern with reduced penetrance and genetic anticipation. In many families, it is the child who presents first with severe disease, and then further evaluation of first degree relatives reveals milder disease in the parents or grandparents [19]. In children, BMPR2 mutations have been evaluated with inconsistent results. Grunig found no BMPR2 mutations or deletions in 13 children with idiopathic pulmonary arterial hypertension [16]. However, in a study by Harrison et al., 22% of children with IPAH or pulmonary hypertension associated with congenital heart disease had activin-like kinase type-1(ALK-1) or BMPR2 mutations [15]. More recently, a study by Rosenzweig et al. evaluated whether children and adults with pulmonary arterial hypertension had a positive response to acute vasodilator testing, and found that BMPR2 mutation positive children appeared less likely to respond to acute vasodilator testing than mutation negative children [20]. These findings are similar to those by Elliott et al. who reported that IPAH and FPAH adult patients with BMPR2 mutations are less likely to respond to acute vasodilator testing than BMPR2 mutation negative patients [21]. A study from Japan suggested that mutations of the activin receptor-like kinase 1 gene in addition to bone morphogenetic protein receptor II gene may be important in the development of IPAH in children [22].
Other genetic loci may also play important roles. Studies have shown mutations of the serotonin transporter gene in some adults with PAH [23], and a study in children found that homozygosity for the long variant of the serotonin transporter gene was highly associated with idiopathic pulmonary hypertension in children [24]. Chung et al. demonstrated an association of a polymorphism in the angiotension II type 1 receptor (AGTR1) with the age at diagnosis of pulmonary hypertension [25]. Future genetic studies may provide further insight regarding disease severity and age of onset in children.
1.3. Evaluation
A complete evaluation for all possible causes of PAH is required before the diagnosis of IPAH is made. Certain diseases, such as connective tissue disease or chronic thromboembolic pulmonary hypertension, are less likely to be discovered in children than adults, but still should be excluded. Cardiac catheterization is required to rule out subtle congenital heart disease, such as pulmonary vein disease, to determine right atrial pressure, exact pulmonary arterial pressure, pulmonary vascular resistance, and to determine vasoreactivity to acute vasodilator testing to target therapy. Lung biopsy is rarely performed but may be helpful to exclude certain diseases, such as pulmonary veno-occlusive disease, pulmonary capillary hemangiomatosis or alveolar capillary dysplasia (see below). Furthermore, in certain forms of interstitial lung disease, such as pulmonary capillaritis or hypersensitivity pneumonitis, lung biopsy may be beneficial as treatment of these disorders varies markedly from the approach used in IPAH.
As in adults, the 6-minute walk test is feasible and has been used to measure sub maximal exercise. Unfortunately, the 6 MW test has not been validated in children with PAH, but normal values in children have recently been described [26]. Children with pulmonary hypertension have significant impairment in aerobic capacity, with a peak oxygen consumption of 20.7±6.9 versus 35.5±7.4 ml/kg/min in healthy controls (p<0.0001) [27].
Interest in biomarkers has grown in the last several years. In adults, brain natriuretic peptide (BNP) is a useful tool to assess mortality risk, progression of the disease and response to therapy [28,29]. Recent studies in children have begun to identify usefulness of BNP or N-terminal pro brain natriuretic peptide (NT-proBNP) [30–32]. BNP correlates positively with functional status in children with PAH and values above 130 pg/ml are associated with increased risk of death or need for transplantation [31]. Furthermore, change in BNP measurements over time correlates with the change in the hemodynamic and echocardiographic parameters of children with PAH; with a BNP value >180 pg/ml predicting a decreased survival rate (Fig. 2). The change in BNP level in a specific patient over time was shown to be more helpful in determining risk or hemodynamic response to therapy than the average BNP value in a pediatric PAH population [30]. NT-proBNP, uric acid, or norepinephrine levels have also been shown to correlate with outcome in children with PAH [32]. When using a cutoff value of NT-proBNP of 1664 pg/ml, sensitivity and specificity for predicting mortality was 100% and 94%, respectively [32].
Fig. 2.

Differences in the last observed brain natriuretic peptide (BNP) distributions between survivors and nonsurvivors for IPAH patients (A). Kaplan–Meier curves estimating cumulative survival for IPAH patients categorized with either high BNP (>180 pg/ml) or normal (<180 pg/ml).
Bernus, A. et al. Chest 2009;135:745–751.
1.4. Conventional therapy
Conventional therapy in patients used to treat right ventricular failure is frequently used in pulmonary arterial hypertension in children. Digoxin is used in the presence of right ventricular failure, although there are no clear-cut data in children [33]. Furthermore, warfarin and other antithrombotic agents are generally recommended to prevent thrombosis in situ, although data specific to the pediatric population are lacking. In adults and children who receive anticoagulation, low dose warfarin is frequently used to target an INR of 1.5 to 2 [34,35]. However, in younger children, maintenance of an adequate level is frequently difficult and toddlers may be treated with aspirin or very low dose warfarin when benefit outweighs risk. Diuretics are used to treat peripheral edema or ascites in the presence of right heart failure, however, excessive diuresis should be avoided. Careful attention to respiratory tract infections is required as this may worsen alveolar hypoxia and routine influenza vaccination is recommended. We advise against the use of decongestants with pseudoephedrine or other stimulant-type medications as these have been associated with PAH [36]. In children who require the use of oral contraceptive agents either for prevention of pregnancy or for regulation of menses, we recommend agents that have no estrogen content. Pulse oximetry and polysomnography is indicated and chronic hypoxemia or nighttime desaturation is aggressively treated. However, oxygen therapy is not used as a mainstay of therapy in children with normal daytime saturations. In the presence of resting hypoxemia, chronic supplemental oxygen may be used. Approximately 5% of children with idiopathic disease may have an elevation of antinuclear antibodies as well as evidence of hypothyroidism or hyperthyroidism suggesting an autoimmune association [37–39]. Furthermore, children should be screened for evidence of a hypercoagulable state as some may have an underlying coagulopathy such as antiphospholipid antibody syndrome. Reversible lower airways obstruction is the most common lung function abnormality among pediatric patients with IPAH [40].
1.5. Vasoreactivity testing
As in adults, cardiac catheterization with acute vasodilator testing is essential prior to selecting targeted therapy in children. Cardiac catheterization carries a greater risk in those children with baseline suprasystemic pulmonary arterial pressure (Odds Ratio = 8.1, p=0.02) [41,42]. As in adults, a short acting vasodilator is used, such as inhaled nitric oxide [43–45]. Previously, patients were considered responsive to acute vasodilator testing if there was a 20% fall in mean pulmonary artery pressure and pulmonary vascular resistance to a vasodilator agent with no change or an increase in cardiac output [44]. Based on studies by Sitbon, more stringent criteria for defining acute vasoreactivity in adults have been established, with the fall in mean pulmonary artery pressure greater than 10 mmHg to less than 40 mmHg to predict long-term response to calcium channel blocker therapy [46]. Although the more strict criteria are frequently used in children, this has not been adequately studied in this population. Recent data have shown that 7% of children with IPAH and 6% of those with APAH are responders to acute vasodilator testing [8,47].
1.6. Targeted pharmacological therapy of PAH
Based on known mechanisms of action, three classes of drugs have been extensively studied for the treatment of IPAH in adults: prostanoids (epoprostenol, treprostinil, iloprost, beraprost), endothelin receptor antagonists (bosentan, sitaxsentan, ambrisentan), and phosphodiesterase inhibitors (Sildenafil, tadalafil).
1.7. Calcium channel blockers
Unless a patient is acutely robustly responsive to inhaled nitric oxide or other selective pulmonary vasodilators, the use of calcium channel antagonists to evaluate vasoreactivity is dangerous, as these drugs can cause a decrease in cardiac output or a marked drop in systemic blood pressure [5]. Consequently, elevated right atrial pressure and low cardiac output are contraindications to acute or chronic calcium channel blockade.
Our preference is to perform an acute trial of calcium channel blocker therapy only in those patients who are acutely responsive to either nitric oxide, prostacyclin or inhaled prostanoids. Likewise, patients who do not have an acute vasodilatory response to short acting agents and who are then placed on calcium channel blocker therapy are unlikely to benefit from this form of therapy [44]. Recent study examined a previously identified cohort of 77 children diagnosed between 1982 and 1995 with IPAH and followed up through 2002. For acute responders treated with CCB (n=31), survival at 1, 5, and 10 years was 97%, 97%, and 81%, respectively; treatment success was 84%, 68%, and 47%, respectively (Fig. 3) [48]. Sixty to eighty percent of children with severe pulmonary hypertension are non-responsive to acute vasodilator testing, and therefore require therapy other than calcium channel antagonists. Children and adults treated with calcium channel blockers may lose this response over time and must be monitored carefully for sustained efficacy [44,48].
Fig. 3.

Kaplan–Meier curves for survival and treatment success in acute responders with IPAH on calcium channel blockers (n=31). Survival rates at 1, 5, and 10 years were 97%, 97%, and 81% respectively; treatment success rates at 1, 3, 5, and 10 years were 84%, 71%, 68%, and 47%, respectively.
Yung D, et al. Circulation 2004; 110:497–503.
1.8. Prostacyclins
Adults with IPAH and children with congenital heart disease demonstrate an imbalance in the biosynthesis of thromboxane A2 and prostacyclin favoring vasoconstriction and platelet aggregation [49,50]. Likewise, adults and children with severe pulmonary hypertension show diminished prostacyclin synthase expression in the lung vasculature [51]. Prostacyclin administered over the long term, utilizing intravenous epoprostenol, has shown to improve survival and quality of life in adults and children with idiopathic pulmonary arterial hypertension, even if not acutely reactive to postacyclin or other pulmonary vasodilators [44,48,52–54].
Prostacyclin and prostacyclin analogues enhance the cyclic-AMP pathway to increase pulmonary vasodilation. Intravenous epoprostenol-prostacyclin was first used in the 1980s and continues to be the gold standard for treatment of severe disease. The treatment of patients with epoprostenol is beneficial; however the therapy is cumbersome due to the delivery system and short half-life. Epoprostenol must be infused 24 h/day via a central venous catheter and kept cold with ice packs; the half-life of the drug is 2–5 min placing the patient at risk for an acute pulmonary hypertensive crisis if there is an accidental discontinuation of the medication. In addition, the side effects of the drug include nausea, diarrhea, jaw pain, bone pain and headaches. Epoprostenol was FDA approved in 1995. In a recent study, 77 children diagnosed between 1982 and 1995 with idiopathic pulmonary arterial hypertension were followed up through 2002. Survival for all children treated with epoprostenol (n=35) at 1, 5, and 10 years was 94%, 81%, and 61%, respectively, while treatment success was 83%, 57%, and 37%, respectively (Fig. 4) [48]. Due to the development of tolerance, the dose of intravenous epoprostenol is incrementally increased and is usually higher in children than adults. The mean dose in young children ranges anywhere from 30 to 90 ng/kg/min but may be higher.
Fig. 4.

Kaplan–Meier curves for survival and treatment success in all patients with IPAH who received epoprostenol (n=37). Survival rates at 1, 3, 5, and 10 years were 94, 88%, 81%, and 61% respectively; treatment success rates at 1, 3, 5, and 10 years were 83%, 65%, 57%, and 37%, respectively.
Yung D, et al. Circulation 2004; 110:497–503.
The prostacyclin analogue, treprostinil was approved by the FDA, initially for subcutaneous use (2002), and more recently for intravenous (2004) and inhaled (2009) administration. While subcutaneous treprostinil allows patients to remain free of central venous catheters, it can cause severe pain at the infusion site. Recent data has shown long-term efficacy in adults with PAH [55], but it has not been well studied in children. Treprostinil has also been given in the intravenous form [56,57]. Intravenous treprostinil still requires central line access and continuous infusion, but is easier for families to mix, may be used at room temperature, and has a half-life of four hours. Intravenous treprostinil has fewer side effects than intravenous epoprostenol, but there are no studies comparing efficacy [57]. Some studies have suggested a higher rate of bacteremia in children and adults treated with intravenous treprostinil [58], but this may be decreased by protecting catheter connections and avoiding water on any connection [59,60]. Treprostinil is also being studied in an inhaled form [61] and was FDA approved for adults in 2009.
An inhaled prostacyclin analogue, iloprost, received approval for the treatment of PAH in adults in the United States in December 2004. This medication is administered by nebulization 6–9 times a day. Iloprost has been shown to improve hemodynamics and exercise capacity. Iloprost requires patient cooperation with the treatment administration lasting 10–15 min [62–64], which is difficult for young children [65]. The advantage of an inhaled prostacyclin is that it can cause selective pulmonary vasodilation without affecting systemic blood pressure. In the acute setting, inhaled iloprost lowers mean pulmonary artery pressure and improves systemic oxygen saturation [66]. In a select patient population, inhaled iloprost may benefit children as add on therapy or to replace intravenous therapy in children with repeated central line infections. However, some children may develop reactive airways obstruction limiting usefulness of this therapy. Inhaled iloprost has also been studied in combination with bosentan and Sildenafil, among others [63]. In some cases, intravenous iloprost has been studied with beneficial effects but this form has not received US authority approval [67–69].
Beraprost is an orally active prostacyclin analogue with a half-life of 35–40 min. While beneficial effects have been noted in short term trials [69], these may be attenuated with prolonged treatment [6,70]. Beraprost is not currently available in North America.
1.9. Endothelin receptor antagonists
Another target for treatment of pulmonary hypertension is the vasoconstrictor peptide endothelin (ET) [71,72]. The endothelins are a family of isopeptides consisting of ET-1, ET-2, and ET-3. ET-1 is a potent vasoactive peptide produced primarily in the vascular endothelial cell, but also may be produced by smooth muscle cells. Two receptor subtypes, ETA and ETB, mediate the activity of ET-1. ETA and ETB receptors on vascular smooth muscle mediate vasoconstriction, whereas ETB receptors on endothelial cells cause release of nitric oxide (NO) and prostacyclin (PGI2), and act as clearance receptors for circulating ET-1. ET-1 expression is increased in the pulmonary arteries of patients with pulmonary hypertension.
Bosentan, a dual ET receptor antagonist, lowers pulmonary artery pressure and resistance, and improves exercise tolerance in adults with pulmonary arterial hypertension [71]. Bosentan has been approved since 2001 for the treatment of WHO functional Class III and IV patients over 12 years of age, and has recently shown beneficial effects in Class II patients [73]. These results have been extrapolated to children [53,74–77]. In BREATHE-3, treatment with bosentan resulted in an increase in cardiac index of 0.5 l/min/m2, a decrease in mean pulmonary artery pressure of 8 mmHg and a decrease in pulmonary vascular resistance index of 3.8 Wood units·m2 after 12 weeks [74]. In this study, the dosing regimen after up titration of 31.25 mg bid was used for patients weighing 10–20 kg, 62.5 mg bid for those weighing 20–40 kg and 125 mg bid for those weighing >40 kg. Risks associated with endothelin receptor antagonist therapy include dose-related hepatotoxicity, teratogenicity and perhaps male infertility [74]. Bosentan therapy added on to epoprostenol in children allowed for a decrease in epoprostenol dose and its associated side effects [53]. A more recent retrospective study of 86 children on bosentan for a median exposure of 14 months with and without concomitant therapy found that bosentan as part of an overall treatment strategy provided a sustained clinical and hemodynamic improvement was overall well tolerated, and two year survival estimates were 91%. In this study, 90% improved or remained unchanged in WHO FC after median treatment duration of 14 months [75]. Comparable results were reported by Maiya et al., except that in IPAH stabilization was achieved in 95% but combined therapy with epoprostenol was necessary in 60% [76]. Elevated hepatic aminotransferase levels occur in approximately 12% of adults treated with bosentan but were only 3.5% in children [75]. Recently, a European, prospective, noninterventional, internet-based post marketing surveillance database was evaluated. Pediatric patients (aged 2–11 years) were compared with patients over 12 years of age. Over a 30-month period, 4994 patients, including 146 bosentan-naive pediatric patients, were captured in the database. PAH was idiopathic in 40% and related to congenital heart disease in 45%. The median exposure to bosentan was 29.1 weeks, and elevated aminotransferases were reported in 2.7% of children fewer than 12 years of age versus 7.8% in older patients. The discontinuation rate was 14.4% in children versus 28.1% in patients over 12 years [77].
Selective ETA receptor blockade has also been studied using ambrisentan or sitaxsentan, ET receptor antagonists with high oral bioavailability and a long duration of action, and high specificity for the ETA receptor. Selective ETA receptor blockade may benefit patients with pulmonary arterial hypertension by blocking the vasoconstrictor effects of ETA receptors while maintaining the vasodilator/clearance functions of ETB receptors. Sitaxsentan given orally for 12 weeks improved exercise capacity and cardiopulmonary hemodynamics in patients with pulmonary arterial hypertension that was idiopathic, or related to connective tissue or congenital heart disease [78,79]. Ambrisentan, an endothelin receptor antagonist that is selective for the endothelin type-A (ETA) receptor was approved by the U.S. FDA in June 2007. Adults showed significant improvements in 6-minute walk distance and significant delay in clinical worsening on ambrisentan. The incidence of elevated hepatic aminotransferase levels was 2.8% [80]. There is no data on use of selective ETA receptor blockade in children.
1.10. Phosphodiesterase-5 inhibitors
In models of PAH, phosphodiesterase-5 activity is increased and protein is localized to vascular smooth muscle [81]. Specific phosphodiesterase-5 inhibitors, such as Sildenafil [8,73,82–88], promote an increase in cGMP levels and thus promote pulmonary vasodilation and remodeling. Sildenafil is as effective a pulmonary vasodilator as inhaled NO [85] and may potentiate the effects of inhaled NO [89–92]. In certain settings, intravenous Sildenafil may worsen oxygenation [92,93]. However, Sildenafil has been shown to prevent rebound PAH on withdrawal from inhaled NO [94,95]. Addition of Sildenafil to long-term intravenous epoprostenol therapy in adults with PAH has been shown to be beneficial [73].
Sildenafil has been approved for the treatment of WHO functional class II–IV PAH adult patients [84–87]; the data in children remain limited. Studies examining the use of oral phosphodiesterase-5 inhibitors in children are ongoing [8,82,83,96,97]. Although no formal dose-response study has been performed, the doses generally used are 0.5–1 mg/kg/dose given three to four times a day with some children requiring even higher doses. The principle side effects include hyperactivity, muscle pain, rare erections and systemic hypotension. Results of a large randomized placebo controlled pediatric trial are expected in 2009.
Tadalalfil, another selective phosphodiesterase type 5 inhibitor, has a longer duration of action, and was recently approved by the FDA in 2009 for adults with PAH. In adults with severe PAH, tadalafil has been used in addition to prostacyclin with some improvement [98–101]. No data are available in children.
1.11. Atrial septostomy and potts shunt for refractory PAH
The general indications for atrial septostomy include pulmonary hypertension with syncope, intractable heart failure refractory to chronic vasodilator treatment [102–105] and symptomatic low cardiac output states. Risks associated with this procedure include worsening of hypoxemia with resultant right ventricular ischemia, worsening right ventricular failure, increased left atrial pressure, and pulmonary edema. We favor a graded balloon dilation approach utilizing intracardiac echo and saturation monitoring to determine adequacy of shunt. We have frequently used a cutting balloon with the initial inflation followed by static balloon dilations. Recently, a Potts anastamosis with connection of the left pulmonary artery to descending aorta, has been attempted to allow a direct shunt allowing an immediate reduction in right ventricular afterload, but this has rarely been performed [106].
2. Pulmonary capillary hemangiomatosis and pulmonary veno-occlusive disease
Pulmonary capillary hemangiomatosis (PCH) and pulmonary veno-occlusive disease (PVOD) both are characterized by precapillary pulmonary arterial hypertension and are included in WHO group 1 [107–111]. However, these disorders differ in that treatment of these disorders may differ from many of the other WHO group 1 diagnoses, and thus are in a separate category in the new WHO classification. In particular, vasodilators may worsen pulmonary edema in PCH and PVOD [112,113]. Furthermore, these two disorders may be difficult to diagnose and are frequently found at autopsy [114,115]. Both PVOD and PCH are characterized by widespread vascular obstruction at either the alveolar capillary bed (PCH) or the pulmonary venules and small veins (PVOD) [116–118]. Treatment of either PVOD or PCH has been frequently unsuccessful and may require lung or heart lung transplantation. In both disorders the definitive diagnosis is usually made by surgical lung biopsy. Clues to the diagnosis of PVOD and PCH may lie in underlying pulmonary edema on chest X-ray and these findings may worsen with the initiation of either oxygen or inhaled nitric oxide [111,119]. Characteristic findings may be seen on a chest CT scan and include central pulmonary artery prominence with widespread interlobular septa in the case of PVOD as well as ground-glass opacities (Fig. 5). While, in PCH, the chest X-ray demonstrates enlarged central pulmonary arteries as well as a reticulonodular or micronodular areas of opacity which may be seen on chest X-ray but are more prominent on the chest CT [116,117,120] (Fig. 6). Likewise, in both of these disorders pulmonary arteriograms may appear normal, particularly if the PVOD is microvascular in nature and not associated with larger vessel pulmonary vein disease. However, PVOD is rare in the spectrum of pulmonary vein abnormalities usually seen in children. In both disorders, pulmonary artery pressure is elevated and may be suprasystemic in patients with PVOD. Likewise, the pulmonary capillary wedge pressure is frequently normal in both disorders.
Fig. 5.
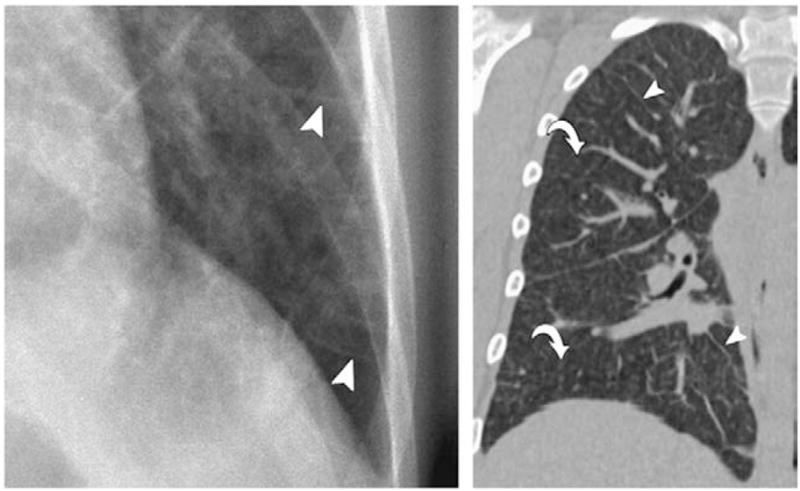
Chest X-ray of a patient with pulmonary veno-occlusive disease showing the presence of Kerley B lines (A). Axial computed tomographic (CT) image reveals widespread septal lines (arrowheads) and diffuse, ill-defined ground-glass nodules (arrows) and geographic ground-glass opacities.
Frazier, A. A. et al. Radiographics 2007; 27:867–882.
Fig. 6.

Chest X-ray of a patient with pulmonary capillary hemangiomatosis showing the widespread nodular opacities (A). Axial computed tomographic (CT) image reveals well-circumscribed ground-glass nodules (arrowheads) and no septal lines.
Frazier, A. A. et al. Radiographics 2007; 27:867–882.
Clinical experience has shown that potent vasodilators (including continuous intravenous prostacyclin and calcium channel blockers) induce florid and even fatal pulmonary edema in patients with either PVOD or PCH [115]. If the pulmonary muscular arteries and arterioles are dilated and yet the pulmonary vein resistance remains fixed, the increased transcapillary hydrostatic pressure leads to transudation of fluid into the lung parenchyma [115]. To exclude unsuspected radiologic evidence of PVOD or PCH, it is currently suggested that patients with presumed IPAH should undergo a high-resolution CT examination before initiation of vasodilator therapy [115]. Therapy for these disorders is limited and is not uniformly effective. Lung transplantation is the only curative option. However, trials of anticoagulation and steroids have been attempted in children with PVOD with mixed results. Due to the findings of myofibroblasts seen in patients with pulmonary vein disorders investigators have attempted use of chemotherapeutic agents with mixed results [121]. In general vasodilator therapy is unsuccessful, frequently contra-indicated, and may worsen the patient status [111]. In the case of PCH experimental therapies have included alpha interferon [122], doxycycline [123], or possibly PDGF antagonists [124]. However, there are no large scale reports of successful treatment.
3. Hemoglobinopathies
Pulmonary hypertension is a known complication of many hereditary hemolytic anemias including sickle cell disease, thalassemia, and spherocytosis [125–128]. Echocardiographic studies have estimated an incidence of PAH in 20–30% of patients with sickle cell disease screened [127,129]. Furthermore, recent studies have shown that upto 75% of sickle cell disease patients may have physiological evidence of pulmonary hypertension at the time of death. Gladwin et al. studied 195 patients with pulmonary arterial hypertension and found a 32% prevalence of pulmonary hypertension [127]. The mortality of those with pulmonary hypertension was 40% at 45 months whereas mortality was only 2% at 45 months in those without pulmonary arterial hypertension. Pulmonary hypertension was defined as a tricuspid regurgitation velocity of greater than 2.5 m/s. Thus a mild elevation of right ventricular systolic pressure carried a greater than tenfold risk of death in adults with pulmonary hypertension. Data in children are lacking but recent studies have begun to evaluate the prevalence and risk factors for pulmonary hypertension [5,129–137].
In a study by Pashshankar, screening echocardiograms were performed in children over 6 years of age with homozygous ss or thalassemia [129]. When using a definition of pulmonary hypertension as a pulmonary artery systolic pressure greater than 30 mm Hg, corresponding to a peak of tricuspid regurgitation velocity greater than or equal to 2.5 m/s, 30% of patients had elevated tricuspid regurgitation jet velocity greater than or equal to 2.5 m/s. Furthermore, 1/3 of these patients had tricuspid regurgitation greater than 3 m/s. Almost all patients with an elevated tricuspid regurgitation jet velocity had hemoglobin homozygous ss disease. Factors associated with elevated pulmonary artery pressure included: a high reticulocyte count, low oxygen saturation, and a high platelet count. Interestingly, acute chest syndrome, hydroxyurea therapy, blood transfusion, stroke, hemoglobin and bilirubin levels between patients with and without pulmonary arterial pressures were not different. Furthermore, in patients with an elevated tricuspid regurgitation velocity, transcranial Doppler examinations were normal [129]. Further study by Pashankar et al. followed patients with echocardiographic evidence of pulmonary arterial hypertension [132]. Those with a tricuspid regurgitation velocity greater than 3 m/s were offered treatment with hydroxyurea; a normalization of tricuspid regurgitation velocity was seen in 8 of 19 patients treated with hydroxyurea.
In children as well as adults with sickle cell disease, pulmonary hypertension is likely due to multifactorial causes such as left ventricular diastolic dysfunction or thromboembolic disease due to hypercoagulability [138,139]. Importantly, intravascular hemolysis may lead to release of hemoglobin which reacts with and destroys nitric oxide [140] (Fig. 7). Likewise, arginase released from the erythrocyte degrades arginine and thus further reduces nitric oxide formation through arginine [134,141]. Xanthine oxidase may also lead to a decrease in nitric oxide. This reduced nitric oxide bioavailability leads to a cascade of other events which further exacerbates pulmonary hypertension and includes an increase in endothelin, an increase in activation of adhesion molecules, and platelet activation [142,143]. Due to abnormalities of nitric oxide, Sildenafil is being studied as a potential therapy for treatment of PAH associated with sickle cell disease [133,144,145]. However, caution is needed. The National Heart, Lung, and Blood Institute of the National Institutes of Health stopped a recent clinical trial testing Sildenafil treatment for pulmonary hypertension in adults with sickle cell disease nearly 1 year early due to safety concerns. Compared to participants on placebo, participants taking Sildenafil were significantly more likely to have serious medical problems, such as sickle cell crises (NIH News, July 28, 2009). Further studies in children are required to determine risk factors, an appropriate screening regimen, and therapy.
Fig. 7.

Pathogenesis and therapeutic targets in hemolysis-associated pulmonary hypertension and vasculopathy. Intravascular hemolysis releases hemoglobin into plasma, which reacts with and destroys endothelial-derived nitric oxide (NO). Arginase is also released from the red cell into plasma during hemolysis and degrades arginine, further reducing NO formation from arginine. Xanthine oxidase bound to endothelium produces superoxide, which also inhibits NO. Reduced NO bioavailability promotes vasoconstriction, activation of adhesion molecules (VCAMs), activation of endothlin-1, a potent vasoconstrictor, and activation of platelets and thrombosis. A number of therapies that target these pathways are shown on the outside of the blood vessel. Lin, E. E. et al. Haematologica 2005; 90:441–444. Obtained from Haematologica/the Hematology Journal website http://www.haematologica.org.
4. Hepatopulmonary syndrome and portopulmonary hypertension
Hepatopulmonary syndrome (HPS) and portopulmonary hypertension (PPHTN) are distinct pulmonary vascular complications of hepatic and extrahepatic portal hypertension [146]. HPS is defined as dilated pulmonary capillaries and precapillary arteriovenous malformations resulting in intrapulmonary vascular shunting, ventilation–perfusion mismatching and chronic hypoxemia in the setting of liver disease or portal hypertension. PPHTN is defined as pulmonary artery hypertension with an elevated mean pulmonary artery pressure and increased pulmonary vascular resistance caused by a pulmonary arteriopathy in the setting of portal hypertension and in the absence of underlying cardiopulmonary disease [146–149]. PPHTN and HPS are estimated to occur in 2–10% and 4–29%, respectively, of adults with cirrhosis [147]. Both PPHTN and HPS are associated with increased morbidity and mortality and are significant risk factors for orthotopic liver transplantation (OLT) [150–153]. However, resolution of both disorders is possible after successful OLT [152,154]. PPHTN and HPS have been reported in children with portal hypertension from both cirrhosis and from congenital or acquired portal vein abnormalities; however, the incidence of these complications in children is unknown [155–159].
The pathogenesis of PPHTN and HPS remains unknown. Proposed theories suggest these disorders result from a combination of hyper dynamic circulation, increased cardiac output, sheer injury to vascular walls and an imbalance of circulating vasoactive peptides [160]. Abnormal hepatic synthesis of vasoactive peptides, such asendothelin-1, or impaired hepatic metabolism of intestinally derived endotoxins, cytokines and neurohormones may result in these substances reaching the pulmonary vascular bed via portosystemic shunting, directly altering vessel tone or leading to pulmonary vascular inflammation and remodeling. The resulting pathology is strikingly different in these two disorders with vasodilation of pulmonary arterioles and capillaries causing arteriovenous shunting in HPS and intimal fibrosis with endothelial and smooth muscle cell proliferation leading to increased pulmonary vascular resistance in PPHTN (Fig. 8).
Fig. 8.
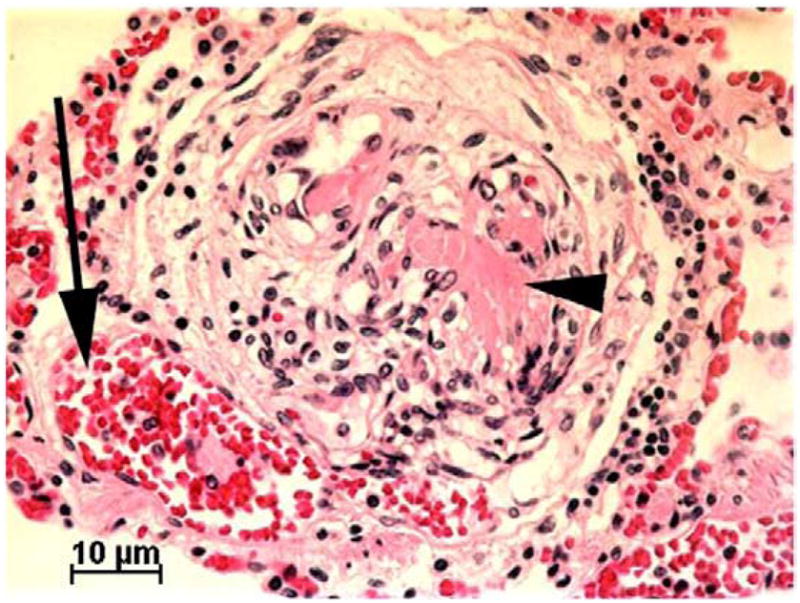
Lung histology of a child who died with portopulmonary hypertension while on intravenous prostacyclin therapy, showing a plexiform lesion. A dilated arteriole (arrow) sits adjacent to a developing plexiform lesion. Partial organization of thrombotic material has occurred leading to formation of slit like vascular spaces. Focal unorganized thrombus remains (arrowhead).
Condino, A. A. et al. J Pediatr 2005; 147:20–26.
Patients with elevated right ventricular systolic pressure by ECHO should undergo confirmatory right heart catheterization [151,152]. In children, a dilated pulmonary artery on CXR and loud second heart sound are suggestive of pulmonary hypertension, whereas the EKG is not uniformly abnormal [161]. In children the initial clinical presentation is subtle with the initial presentation rarely being death from a pulmonary hypertensive crisis [155].
PPHTN is frequently resistant to many first line medications. If the diagnosis of PPHTN is made early prior to the development of irreversible pulmonary vasculopathy, then liver transplantation can be successfully performed and may reverse the process of PPHTN. Similar to adults, few patients with PPHTN respond to calcium channel blocker therapy. In more severe cases treatment with epoprostenol is used, in addition to other targeted PAH agents [162–167].
5. Human immunodeficiency virus (HIV)
Since the first description by Kim et al. [168] the association between pulmonary arterial hypertension and HIV infection has been well established; there are multiple reports for adult patients [169–171] whereas little is known about the incidence, clinical outcome and therapy options for children [172,173]. The average age of patients of patients presenting with HIV-PAH is 33 years (range 2 to 56 years) [174] with a male-to-female ratio of about 1.5:1 [175]. The incidence of PAH in adults with HIV is approximately 0.5% [170] whereas the results from the National Registry in France suggested that HIV was the cause for PAH in about 6.2% [176]. Most children with HIV are infected in the perinatal period [177–179]. Pongprot et al. detected pulmonary hypertension in 41% of their highly selected cohort of HIV-infected children [173].
Changes in the lung are characterized by the typical plexiform lesions (78%) intimal fibrosis without plexiform lesions (7%) and medial hypertrophy (11%) [174] in addition to inflammation surrounding the hypertrophic vasa vasorum in the pulmonary arteries of HIV-infected children [180].
The pathogenesis of HIV-PAH is not completely understood. HIV-PAH has been reported without a history of thromboembolic disease, intravenous drug use, or pulmonary infections [181,182] and HIV or its proteins have not been detected the vascular endothelium of the lungs. It has been hypothesized that the HIV infection may damage the cells by stimulating the host to release proinflammatory cytokines, growth factors or endothelin 1 that would result in PAH [181,183,184].
The prognosis of HIV-PAH is limited but seems to improve in adults with highly active antiretroviral therapy (HAART). However, the efficacy of HAART in patients with HIV related PAH in general and, in the pediatric population more specifically, is still uncertain [171,185].
Treatment options are similar to other forms of PAH. Opravil et al. reported disappointing results for patients with HIV-PAH treated with calcium channel blockers [186]. In addition, co administration of the antiretroviral agents ritonavir and indinavir may increase the plasma concentrations of calcium channel blockers [187]. There are a few case reports about treatment with the phosphodiesterase V inhibitor Sildenafil [188–190]. Recently Degano et al. reported on the long-term benefits of bosentan therapy with improvements in symptoms, exercise capacity, hemodynamics and favorable overall survival [191]. Similar results were seen with the use of intravenous epoprostenol [192].
Currently there is no consensus for routine PAH screening of HIV-infected individuals. Hsue et al. advised against the screening as the clinical implications of “preclinical” PAH amongst HIV-infected persons are unknown [193]
6. Summary
Recognition of causes of pulmonary hypertension other than congenital heart disease is increasing in children. A complete evaluation for all possible causes of PAH is required before the diagnosis of IPAH is made. Cardiac catheterization is required to rule out subtle congenital heart disease and to guide therapy. Although lung biopsy is rarely indicated, in select diseases, such as pulmonary veno-occlusive disease or pulmonary capillary hemangiomatosis, biopsy is important to determine the etiology of PH as use of vasodilator therapy may be detrimental in these disorders. Three classes of drugs have been extensively studied for the treatment of IPAH in adults: prostanoids (epoprostenol, treprostinil, iloprost, beraprost), endothelin receptor antagonists (bosentan, sitaxsentan, ambrisentan), and phosphodiesterase inhibitors (Sildenafil, tadalafil). These medications have been used in treatment of pulmonary hypertension in children with pulmonary arterial hypertension caused by various disorders, although randomized clinical trial data is lacking. Use of these medications requires caution as treatment may lead to other morbidities, as seen in more frequent sickle cell crises in a recent NIH trial of Sildenafil in adult sickle cell disease. Further study of these medications in PAH associated with other disorders is needed before widespread use is encouraged.
References
- 1.Thilenius OG, Nadas AS, Jockin H. Primary pulmonary vascular obstruction in children. Pediatrics. 1965;36:75–87. [PubMed] [Google Scholar]
- 2.D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115:343–9. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- 3.Sandoval J, Bauerle O, Gomez A, Palomar A, Martinez Guerra ML, Furuya ME. Primary pulmonary hypertension in children: clinical characterization and survival. J Am Coll Cardiol. 1995;25:466–74. doi: 10.1016/0735-1097(94)00391-3. [DOI] [PubMed] [Google Scholar]
- 4.Rashid A, Ivy D. Severe paediatric pulmonary hypertension: new management strategies. Arch Dis Child. 2005;90:92–8. doi: 10.1136/adc.2003.048744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenzweig EB, Barst RJ. Pulmonary arterial hypertension in children: a medical update. Curr Opin Pediatr. 2008;20:288–93. doi: 10.1097/MOP.0b013e3282ff5fdc. [DOI] [PubMed] [Google Scholar]
- 6.Badesch DB, Abman SH, Ahearn GS, et al. Medical therapy for pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126:35S–62S. doi: 10.1378/chest.126.1_suppl.35S. [DOI] [PubMed] [Google Scholar]
- 7.Barst RJ. Pharmacologically induced pulmonary vasodilatation in children and young adults with primary pulmonary hypertension. Chest. 1986;89:497–503. doi: 10.1378/chest.89.4.497. [DOI] [PubMed] [Google Scholar]
- 8.Haworth SG, Hislop AA. Treatment and survival in children with pulmonary arterial hypertension: the UK Pulmonary Hypertension Service for Children 2001–2006. Heart. 2009;95:312–7. doi: 10.1136/hrt.2008.150086. [DOI] [PubMed] [Google Scholar]
- 9.Rich S. Executive Summary from the World Symposium. Primary Pulmonary Hypertension World Health Orgnization; 1998. Primary Pulmonary Hypertension. [Google Scholar]
- 10.Badesch DB, Champion HC, Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S55–66. doi: 10.1016/j.jacc.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 11.Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 12.Rosenzweig EB, Barst RJ. Idiopathic pulmonary arterial hypertension in children. Curr Opin Pediatr. 2005;17:372–80. doi: 10.1097/01.mop.0000163356.51027.c1. [DOI] [PubMed] [Google Scholar]
- 13.Austin ED, Loyd JE, Phillips JA., III Genetics of pulmonary arterial hypertension. Semin Respir Crit Care Med. 2009;30:386–98. doi: 10.1055/s-0029-1233308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trembath RC, Harrison R. Insights into the genetic and molecular basis of primary pulmonary hypertension. Pediatr Res. 2003;53:883–8. doi: 10.1203/01.PDR.0000061565.22500.E7. [DOI] [PubMed] [Google Scholar]
- 15.Harrison RE, Berger R, Haworth SG, et al. Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation. 2005;111:435–41. doi: 10.1161/01.CIR.0000153798.78540.87. [DOI] [PubMed] [Google Scholar]
- 16.Grunig E, Koehler R, Miltenberger-Miltenyi G, et al. Primary pulmonary hypertension in children may have a different genetic background than in adults. Pediatr Res. 2004;56:571–8. doi: 10.1203/01.PDR.0000139481.20847.D0. [DOI] [PubMed] [Google Scholar]
- 17.Newman JH, Wheeler L, Lane KB, et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med. 2001;345:319–24. doi: 10.1056/NEJM200108023450502. [DOI] [PubMed] [Google Scholar]
- 18.Thomson JR, Machado RD, Pauciulo MW, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet. 2000;37:741–5. doi: 10.1136/jmg.37.10.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loyd JE, Slovis B, Phillips JA, III, et al. The presence of genetic anticipation suggests that the molecular basis of familial primary pulmonary hypertension may be trinucleotide repeat expansion. Chest. 1997;111:82S–3S. doi: 10.1378/chest.111.6_supplement.82s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenzweig EB, Morse JH, Knowles JA, et al. Clinical implications of determining BMPR2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J Heart Lung Transplant. 2008;27:668–74. doi: 10.1016/j.healun.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 21.Elliott CG, Glissmeyer EW, Havlena GT, et al. Relationship of BMPR2 mutations to vasoreactivity in pulmonary arterial hypertension. Circulation. 2006;113:2509–15. doi: 10.1161/CIRCULATIONAHA.105.601930. [DOI] [PubMed] [Google Scholar]
- 22.Fujiwara M, Yagi H, Matsuoka R, et al. Implications of mutations of activin receptor-like kinase 1 gene (ALK1) in addition to bone morphogenetic protein receptor II gene (BMPR2) in children with pulmonary arterial hypertension. Circ J. 2008;72:127–33. doi: 10.1253/circj.72.127. [DOI] [PubMed] [Google Scholar]
- 23.Eddahibi S, Adnot S. The serotonin pathway in pulmonary hypertension. Arch Mal Coeur Vaiss. 2006;99:621–5. [PubMed] [Google Scholar]
- 24.Willers ED, Newman JH, Loyd JE, et al. Serotonin transporter polymorphisms in familial and idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;173:798–802. doi: 10.1164/rccm.200509-1361OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung WK, Deng L, Carroll JS, et al. Polymorphism in the angiotensin II type 1 receptor (AGTR1) is associated with age at diagnosis in pulmonary arterial hypertension. J Heart Lung Transplant. 2009;28:373–9. doi: 10.1016/j.healun.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lammers AE, Hislop AA, Flynn Y, Haworth SG. The 6-minute walk test: normal values for children of 4–11 years of age. Arch Dis Child. 2008;93:464–8. doi: 10.1136/adc.2007.123653. [DOI] [PubMed] [Google Scholar]
- 27.Yetman AT, Taylor AL, Doran A, Ivy DD. Utility of cardiopulmonary stress testing in assessing disease severity in children with pulmonary arterial hypertension. Am J Cardiol. 2005;95:697–9. doi: 10.1016/j.amjcard.2004.10.056. [DOI] [PubMed] [Google Scholar]
- 28.Nagaya N, Nishikimi T, Uematsu M, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation. 2000;102:865–70. doi: 10.1161/01.cir.102.8.865. [DOI] [PubMed] [Google Scholar]
- 29.Nagaya N, Nishikimi T, Okano Y, et al. Plasma brain natriuretic peptide levels increase in proportion to the extent of right ventricular dysfunction in pulmonary hypertension. J Am Coll Cardiol. 1998;31:202–8. doi: 10.1016/s0735-1097(97)00452-x. [DOI] [PubMed] [Google Scholar]
- 30.Bernus A, Wagner BD, Accurso F, Doran A, Kaess H, Ivy DD. Brain natriuretic peptide levels in managing pediatric patients with pulmonary arterial hypertension. Chest. 2009;135:745–51. doi: 10.1378/chest.08-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lammers AE, Hislop AA, Haworth SG. Prognostic value of B-type natriuretic peptide in children with pulmonary hypertension. Int J Cardiol. 2009;135:21–6. doi: 10.1016/j.ijcard.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 32.Van Albada ME, Loot FG, Fokkema R, Roofthooft MT, Berger RM. Biological serum markers in the management of pediatric pulmonary arterial hypertension. Pediatr Res. 2008;63:321–7. doi: 10.1203/PDR.0b013e318163a2e7. [DOI] [PubMed] [Google Scholar]
- 33.Rich SSM, Dodin E, et al. The short-term effects of digoxin in patients with right ventricular dysfunction from pulmonary hypertension. Chest. 1998;114:787–92. doi: 10.1378/chest.114.3.787. [DOI] [PubMed] [Google Scholar]
- 34.Frank H, Mlczoch J, Huber K, Schuster E, Gurtner HP, Kneussl M. The effect of anticoagulant therapy in primary and anorectic drug-induced pulmonary hypertension. Chest. 1997;112:714–21. doi: 10.1378/chest.112.3.714. [DOI] [PubMed] [Google Scholar]
- 35.Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327:76–81. doi: 10.1056/NEJM199207093270203. [DOI] [PubMed] [Google Scholar]
- 36.Barst RJ, Abenhaim L. Fatal pulmonary arterial hypertension associated with phenylpropanolamine exposure. Heart. 2004;90:e42. doi: 10.1136/hrt.2004.036491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barst RJ, Flaster ER, Menon A, Fotino M, Morse JH. Evidence for the association of unexplained pulmonary hypertension in children with the major histocompatibility complex. Circulation. 1992;85:249–58. doi: 10.1161/01.cir.85.1.249. [DOI] [PubMed] [Google Scholar]
- 38.Ferris A, Jacobs T, Widlitz A, Barst RJ, Morse JH. Pulmonary arterial hypertension and thyroid disease. Chest. 2001;119:1980–1. doi: 10.1378/chest.119.6.1980. [DOI] [PubMed] [Google Scholar]
- 39.Roberts KE, Barst RJ, McElroy JJ, et al. Bone morphogenetic protein receptor 2 mutations in adults and children with idiopathic pulmonary arterial hypertension: association with thyroid disease. Chest. 2005;128:618S. doi: 10.1378/chest.128.6_suppl.618S. [DOI] [PubMed] [Google Scholar]
- 40.Rastogi D, Ngai P, Barst RJ, Koumbourlis AC. Lower airway obstruction, bronchial hyperresponsiveness, and primary pulmonary hypertension in children. Pediatr Pulmonol. 2004;37:50–5. doi: 10.1002/ppul.10363. [DOI] [PubMed] [Google Scholar]
- 41.Carmosino MJ, Friesen RH, Doran A, Ivy DD. Perioperative complications in children with pulmonary hypertension undergoing noncardiac surgery or cardiac catheterization. Anesth Analg. 2007;104:521–7. doi: 10.1213/01.ane.0000255732.16057.1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Friesen RH, Williams GD. Anesthetic management of children with pulmonary arterial hypertension. Paediatr Anaesth. 2008;18:208–16. doi: 10.1111/j.1460-9592.2008.02419.x. [DOI] [PubMed] [Google Scholar]
- 43.Berman Rosenzweig E, Barst RJ. Pulmonary arterial hypertension: a comprehensive review of pharmacological treatment. Treat Respir Med. 2006;5:117–27. doi: 10.2165/00151829-200605020-00005. [DOI] [PubMed] [Google Scholar]
- 44.Barst RJ, Maislin G, Fishman AP. Vasodilator therapy for primary pulmonary hypertension in children. Circulation. 1999;99:1197–208. doi: 10.1161/01.cir.99.9.1197. [DOI] [PubMed] [Google Scholar]
- 45.Beghetti M. Current treatment options in children with pulmonary arterial hypertension and experiences with oral bosentan. Eur J Clin Invest. 2006;36 (Suppl 3):16–24. doi: 10.1111/j.1365-2362.2006.01681.x. [DOI] [PubMed] [Google Scholar]
- 46.Sitbon O, Humbert M, Jais X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111:3105–11. doi: 10.1161/CIRCULATIONAHA.104.488486. [DOI] [PubMed] [Google Scholar]
- 47.Haworth SG. The management of pulmonary hypertension in children. Arch Dis Child. 2008;93:620–5. doi: 10.1136/adc.2007.120493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yung D, Widlitz AC, Rosenzweig EB, Kerstein D, Maislin G, Barst RJ. Outcomes in children with idiopathic pulmonary arterial hypertension. Circulation. 2004;110:660–5. doi: 10.1161/01.CIR.0000138104.83366.E9. [DOI] [PubMed] [Google Scholar]
- 49.Christman BW, McPherson CD, Newman JH, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327:70–5. doi: 10.1056/NEJM199207093270202. [DOI] [PubMed] [Google Scholar]
- 50.Adatia I, Barrow SE, Stratton PD, Miall-Allen VM, Ritter JM, Haworth SG. Thromboxane A2 and prostacyclin biosynthesis in children and adolescents with pulmonary vascular disease. Circulation. 1993;88:2117–22. doi: 10.1161/01.cir.88.5.2117. [DOI] [PubMed] [Google Scholar]
- 51.Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:1925–32. doi: 10.1164/ajrccm.159.6.9804054. [DOI] [PubMed] [Google Scholar]
- 52.Lammers AE, Hislop AA, Flynn Y, Haworth SG. Epoprostenol treatment in children with severe pulmonary hypertension. Heart. 2007;93:739–43. doi: 10.1136/hrt.2006.096412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ivy DD, Doran A, Claussen L, Bingaman D, Yetman A. Weaning and discontinuation of epoprostenol in children with idiopathic pulmonary arterial hypertension receiving concomitant bosentan. Am J Cardiol. 2004;93:943–6. doi: 10.1016/j.amjcard.2003.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barst RJ. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996:296–302. doi: 10.1056/NEJM199602013340504. [DOI] [PubMed] [Google Scholar]
- 55.Barst RJ, Galie N, Naeije R, et al. Long-term outcome in pulmonary arterial hypertension patients treated with subcutaneous treprostinil. Eur Respir J. 2006;28:1195–203. doi: 10.1183/09031936.06.00044406. [DOI] [PubMed] [Google Scholar]
- 56.Tapson VF, Gomberg-Maitland M, McLaughlin VV, et al. Safety and efficacy of IV treprostinil for pulmonary arterial hypertension: a prospective, multicenter, open-label, 12-week trial. Chest. 2006;129:683–8. doi: 10.1378/chest.129.3.683. [DOI] [PubMed] [Google Scholar]
- 57.Ivy DD, Claussen L, Doran A. Transition of stable pediatric patients with pulmonary arterial hypertension from intravenous epoprostenol to intravenous treprostinil. Am J Cardiol. 2007;99:696–8. doi: 10.1016/j.amjcard.2006.09.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bloodstream infections among patients treated with intravenous epoprostenol or intravenous treprostinil for pulmonary arterial hypertension—seven sites, United States, 2003–2006. MMWR Morb Mortal Wkly Rep. 2007;56:170–2. [PubMed] [Google Scholar]
- 59.Doran AK, Ivy DD, Barst RJ, Hill N, Murali S, Benza RL. Guidelines for the prevention of central venous catheter-related blood stream infections with prostanoid therapy for pulmonary arterial hypertension. Int J Clin Pract Suppl. 2008:5–9. doi: 10.1111/j.1742-1241.2008.01811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ivy DD, Calderbank M, Wagner BD, et al. Closed-hub systems with protected connections and the reduction of risk of catheter-related bloodstream infection in pediatric patients receiving intravenous prostanoid therapy for pulmonary hypertension. Infect Control Hosp Epidemiol. 2009;30:823–9. doi: 10.1086/605320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Channick RN, Olschewski H, Seeger W, Staub T, Voswinckel R, Rubin LJ. Safety and efficacy of inhaled treprostinil as add-on therapy to bosentan in pulmonary arterial hypertension. J Am Coll Cardiol. 2006;48:1433–7. doi: 10.1016/j.jacc.2006.05.070. [DOI] [PubMed] [Google Scholar]
- 62.Hoeper MM, Schwarze M, Ehlerding S, et al. Long-term treatment of primary pulmonary hypertension with aerosolized iloprost, a prostacyclin analogue. N Engl J Med. 2000;342:1866–70. doi: 10.1056/NEJM200006223422503. [DOI] [PubMed] [Google Scholar]
- 63.McLaughlin VV, Oudiz RJ, Frost A, et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174:1257–63. doi: 10.1164/rccm.200603-358OC. [DOI] [PubMed] [Google Scholar]
- 64.Olschewski H, Simonneau G, Galie N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347:322–9. doi: 10.1056/NEJMoa020204. [DOI] [PubMed] [Google Scholar]
- 65.Ivy DD, Doran AK, Smith KJ, et al. Short- and long-term effects of inhaled iloprost therapy in children with pulmonary arterial hypertension. J Am Coll Cardiol. 2008;51:161–9. doi: 10.1016/j.jacc.2007.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Limsuwan A, Wanitkul S, Khosithset A, Attanavanich S, Samankatiwat P. Aerosolized iloprost for postoperative pulmonary hypertensive crisis in children with congenital heart disease. Int J Cardiol. 2008;129:333–8. doi: 10.1016/j.ijcard.2007.08.084. [DOI] [PubMed] [Google Scholar]
- 67.Ewert R, Opitz CF, Wensel R, Winkler J, Halank M, Felix SB. Continuous intravenous iloprost to revert treatment failure of first-line inhaled iloprost therapy in patients with idiopathic pulmonary arterial hypertension. Clin Res Cardiol. 2007;96:211–7. doi: 10.1007/s00392-007-0490-3. [DOI] [PubMed] [Google Scholar]
- 68.Hallioglu O, Dilber E, Celiker A. Comparison of acute hemodynamic effects of aerosolized and intravenous iloprost in secondary pulmonary hypertension in children with congenital heart disease. Am J Cardiol. 2003;92:1007–9. doi: 10.1016/s0002-9149(03)00991-3. [DOI] [PubMed] [Google Scholar]
- 69.Galie N, Humbert M, Vachiery JL, et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2002;39:1496–502. doi: 10.1016/s0735-1097(02)01786-2. [DOI] [PubMed] [Google Scholar]
- 70.Barst RJ, McGoon M, McLaughlin V, et al. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2003;41:2119–25. doi: 10.1016/s0735-1097(03)00463-7. [DOI] [PubMed] [Google Scholar]
- 71.Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]
- 72.Abman SH. Role of endothelin receptor antagonists in the treatment of pulmonary arterial hypertension. Annu Rev Med. 2009;60:13–23. doi: 10.1146/annurev.med.59.110106.212434. [DOI] [PubMed] [Google Scholar]
- 73.Simonneau G, Rubin LJ, Galie N, et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med. 2008;149:521–30. doi: 10.7326/0003-4819-149-8-200810210-00004. [DOI] [PubMed] [Google Scholar]
- 74.Barst RJ, Ivy D, Dingemanse J, et al. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin Pharmacol Ther. 2003;73:372–82. doi: 10.1016/s0009-9236(03)00005-5. [DOI] [PubMed] [Google Scholar]
- 75.Rosenzweig EB, Ivy DD, Widlitz A, et al. Effects of long-term bosentan in children with pulmonary arterial hypertension. J Am Coll Cardiol. 2005;46:697–704. doi: 10.1016/j.jacc.2005.01.066. [DOI] [PubMed] [Google Scholar]
- 76.Maiya S, Hislop AA, Flynn Y, Haworth SG. Response to bosentan in children with pulmonary hypertension. Heart. 2006;92:664–70. doi: 10.1136/hrt.2005.072314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beghetti M, Hoeper MM, Kiely DG, et al. Safety experience with bosentan in 146 children 2–11 years old with pulmonary arterial hypertension: results from the European Postmarketing Surveillance program. Pediatr Res. 2008;64:200–4. doi: 10.1203/PDR.0b013e318179954c. [DOI] [PubMed] [Google Scholar]
- 78.Barst RJ, Langleben D, Badesch D, et al. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol. 2006;47:2049–56. doi: 10.1016/j.jacc.2006.01.057. [DOI] [PubMed] [Google Scholar]
- 79.Barst RJ, Langleben D, Frost A, et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;169:441–7. doi: 10.1164/rccm.200307-957OC. [DOI] [PubMed] [Google Scholar]
- 80.Galie N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117:3010–9. doi: 10.1161/CIRCULATIONAHA.107.742510. [DOI] [PubMed] [Google Scholar]
- 81.Hanson KA, Ziegler JW, Rybalkin SD, Miller JW, Abman SH, Clarke WR. Chronic pulmonary hypertension increases fetal lung cGMP phosphodiesterase activity. Am J Physiol. 1998;275:L931–41. doi: 10.1152/ajplung.1998.275.5.L931. [DOI] [PubMed] [Google Scholar]
- 82.Humpl T, Reyes JT, Holtby H, Stephens D, Adatia I. Beneficial effect of oral sildenafil therapy on childhood pulmonary arterial hypertension: twelve-month clinical trial of a single-drug, open-label, pilot study. Circulation. 2005;111:3274–80. doi: 10.1161/CIRCULATIONAHA.104.473371. [DOI] [PubMed] [Google Scholar]
- 83.Karatza AA, Bush A, Magee AG. Safety and efficacy of sildenafil therapy in children with pulmonary hypertension. Int J Cardiol. 2005;100:267–73. doi: 10.1016/j.ijcard.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 84.Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–57. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 85.Michelakis ETW, Lien D, Webster L, Hasimoto K, Archer S. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation. 2002;105:2398–403. doi: 10.1161/01.cir.0000016641.12984.dc. [DOI] [PubMed] [Google Scholar]
- 86.Michelakis ED, Tymchak W, Noga M, et al. Long-term treatment with oral sildenafil is safe and improves functional capacity and hemodynamics in patients with pulmonary arterial hypertension. Circulation. 2003;108:2066–9. doi: 10.1161/01.CIR.0000099502.17776.C2. [DOI] [PubMed] [Google Scholar]
- 87.Sastry BK, Narasimhan C, Reddy NK, Raju BS. Clinical efficacy of sildenafil in primary pulmonary hypertension: a randomized, placebo-controlled, double-blind, crossover study. J Am Coll Cardiol. 2004;43:1149–53. doi: 10.1016/j.jacc.2003.10.056. [DOI] [PubMed] [Google Scholar]
- 88.Wilkins MR, Paul GA, Strange JW, et al. Sildenafil versus endothelin receptor antagonist for pulmonary hypertension (SERAPH) study. Am J Respir Crit Care Med. 2005;171:1292–7. doi: 10.1164/rccm.200410-1411OC. [DOI] [PubMed] [Google Scholar]
- 89.Abrams D, Schulze-Neick I, Magee AG. Sildenafil as a selective pulmonary vasodilator in childhood primary pulmonary hypertension. Heart. 2000;84:E4. doi: 10.1136/heart.84.2.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Prasad S, Wilkinson J, Gatzoulis MA. Sildenafil in primary pulmonary hypertension. N Engl J Med. 2000;343:1342. doi: 10.1056/NEJM200011023431814. [DOI] [PubMed] [Google Scholar]
- 91.Atz Am LA, Fairbrother DL, Uber WE, Bradley SM. Sildenafil augments the effect of inhaled nitric oxide for postoperative pulmonary hypertensive crisis. J Thorac Cardiovasc Surg. 2002;124:628–9. doi: 10.1067/mtc.2002.125265. [DOI] [PubMed] [Google Scholar]
- 92.Schulze-Neick I, Hartenstein P, Li J, et al. Intravenous Sildenafil is a potent pulmonary vasodilator in children with congenital heart disease. Circulation. 2003;108(Suppl 1):II167–73. doi: 10.1161/01.cir.0000087384.76615.60. [DOI] [PubMed] [Google Scholar]
- 93.Stocker C, Penny DJ, Brizard CP, Cochrane AD, Soto R, Shekerdemian LS. Intravenous Sildenafil and inhaled nitric oxide: a randomised trial in infants after cardiac surgery. Intensive Care Med. 2003;29:1996–2003. doi: 10.1007/s00134-003-2016-4. [DOI] [PubMed] [Google Scholar]
- 94.Atz AMWD. Sildenafil ameliorates effects of inhaled nitric oxide withdrawal. Anesthesiology. 1999;91:307–10. doi: 10.1097/00000542-199907000-00041. [DOI] [PubMed] [Google Scholar]
- 95.Namachivayam P, Theilen U, Butt WW, Cooper SM, Penny DJ, Shekerdemian LS. Sildenafil prevents rebound pulmonary hypertension after withdrawal of nitric oxide in children. Am J Respir Crit Care Med. 2006;174:1042–7. doi: 10.1164/rccm.200605-694OC. [DOI] [PubMed] [Google Scholar]
- 96.Fasnacht MS, Tolsa JF, Beghetti M. The Swiss registry for pulmonary arterial hypertension: the paediatric experience. Swiss Med Wkly. 2007;137:510–3. doi: 10.4414/smw.2007.11895. [DOI] [PubMed] [Google Scholar]
- 97.Mourani PM, Sontag MK, Ivy DD, Abman SH. Effects of long-term Sildenafil treatment for pulmonary hypertension in infants with chronic lung disease. J Pediatr. 2009;154:379–84. doi: 10.1016/j.jpeds.2008.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bendayan D, Shitrit D, Kramer MR. Combination therapy with prostacyclin and tadalafil for severe pulmonary arterial hypertension: a pilot study. Respirology. 2008;13:916–8. doi: 10.1111/j.1440-1843.2007.01176.x. [DOI] [PubMed] [Google Scholar]
- 99.Wrishko RE, Dingemanse J, Yu A, Darstein C, Phillips DL, Mitchell MI. Pharmacokinetic interaction between tadalafil and bosentan in healthy male subjects. J Clin Pharmacol. 2008;48:610–8. doi: 10.1177/0091270008315315. [DOI] [PubMed] [Google Scholar]
- 100.Tay EL, Geok-Mui MK, Poh-Hoon MC, Yip J. Sustained benefit of tadalafil in patients with pulmonary arterial hypertension with prior response to Sildenafil: a case series of 12 patients. Int J Cardiol. 2008;125:416–7. doi: 10.1016/j.ijcard.2007.01.046. [DOI] [PubMed] [Google Scholar]
- 101.Palmieri EA, Affuso F, Fazio S, Lembo D. Tadalafil in primary pulmonary arterial hypertension. Ann Intern Med. 2004;141:743–4. doi: 10.7326/0003-4819-141-9-200411020-00033. [DOI] [PubMed] [Google Scholar]
- 102.Barst RJ. Role of atrial septostomy in the treatment of pulmonary vascular disease. Thorax. 2000;55:95–6. doi: 10.1136/thorax.55.2.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kerstein D, Levy PS, Hsu DT, Hordof AJ, Gersony WM, Barst RJ. Blade balloon atrial septostomy in patients with severe primary pulmonary hypertension. Circulation. 1995;91:2028–35. doi: 10.1161/01.cir.91.7.2028. [DOI] [PubMed] [Google Scholar]
- 104.Nihill MROLM, Mullins CE. Effects of atrial septostomy in patients with terminal cor pulmonale due to pulmonary vascular disease. Cathet Cardiovasc Diagn. 1991;24:166–72. doi: 10.1002/ccd.1810240305. [DOI] [PubMed] [Google Scholar]
- 105.Sandoval J, Gaspar J, Pulido T, et al. Graded balloon dilation atrial septostomy in severe primary pulmonary hypertension. A therapeutic alternative for patients nonresponsive to vasodilator treatment. J Am Coll Cardiol. 1998;32:297–304. doi: 10.1016/s0735-1097(98)00238-1. [DOI] [PubMed] [Google Scholar]
- 106.Blanc J, Vouhe P, Bonnet D. Potts shunt in patients with pulmonary hypertension. N Engl J Med. 2004;350:623. doi: 10.1056/NEJM200402053500623. [DOI] [PubMed] [Google Scholar]
- 107.Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43:5S–12S. doi: 10.1016/j.jacc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 108.Barboza CE, Jardim CV, Hovnanian AL, Dias BA, Souza R. Pulmonary veno-occlusive disease: diagnostic and therapeutic alternatives. J Bras Pneumol. 2008;34:749–52. doi: 10.1590/s1806-37132008000900015. [DOI] [PubMed] [Google Scholar]
- 109.Halank M, Kolditz M, Langner S, Hoffken G. Pulmonary veno-occlusive disease —a rare form of pulmonary arterial hypertension. Dtsch Med Wochenschr. 2008;133(Suppl 6):S212–4. doi: 10.1055/s-0028-1091242. [DOI] [PubMed] [Google Scholar]
- 110.Creagh-Brown BC, Nicholson AG, Showkathali R, Gibbs JS, Howard LS. Pulmonary veno-occlusive disease presenting with recurrent pulmonary oedema and the use of nitric oxide to predict response to Sildenafil. Thorax. 2008;63:933–4. doi: 10.1136/thx.2007.088831. [DOI] [PubMed] [Google Scholar]
- 111.Montani D, Achouh L, Dorfmuller P, et al. Pulmonary veno-occlusive disease: clinical, functional, radiologic, and hemodynamic characteristics and outcome of 24 cases confirmed by histology. Medicine (Baltimore) 2008;87:220–33. doi: 10.1097/MD.0b013e31818193bb. [DOI] [PubMed] [Google Scholar]
- 112.Gugnani MK, Pierson C, Vanderheide R, Girgis RE. Pulmonary edema complicating prostacyclin therapy in pulmonary hypertension associated with scleroderma: a case of pulmonary capillary hemangiomatosis. Arthritis Rheum. 2000;43:699–703. doi: 10.1002/1529-0131(200003)43:3<699::AID-ANR28>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 113.Humbert M, Maitre S, Capron F, Rain B, Musset D, Simonneau G. Pulmonary edema complicating continuous intravenous prostacyclin in pulmonary capillary hemangiomatosis. Am J Respir Crit Care Med. 1998;157:1681–5. doi: 10.1164/ajrccm.157.5.9708065. [DOI] [PubMed] [Google Scholar]
- 114.Harch S, Whitford H, McLean C. Failure of medical therapy in pulmonary arterial hypertension: is there an alternative diagnosis? Chest. 2009;135:1462–9. doi: 10.1378/chest.08-2006. [DOI] [PubMed] [Google Scholar]
- 115.Matsumoto JS, Hoffman AD. Pediatric case of the day. Pulmonary venoocclusive disease. AJR Am J Roentgenol. 1993;160:1331–2. doi: 10.2214/ajr.160.6.8498252. [DOI] [PubMed] [Google Scholar]
- 116.El-Gabaly M, Farver CF, Budev MA, Mohammed TL. Pulmonary capillary hemangiomatosis imaging findings and literature update. J Comput Assist Tomogr. 2007;31:608–10. doi: 10.1097/01.rct.0000284393.76073.87. [DOI] [PubMed] [Google Scholar]
- 117.Frazier AA, Franks TJ, Mohammed TL, Ozbudak IH, Galvin JR. From the archives of the AFIP: pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis. Radiographics. 2007;27:867–82. doi: 10.1148/rg.273065194. [DOI] [PubMed] [Google Scholar]
- 118.Lantuejoul S, Sheppard MN, Corrin B, Burke MM, Nicholson AG. Pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis: a clinicopathologic study of 35 cases. Am J Surg Pathol. 2006;30:850–7. doi: 10.1097/01.pas.0000209834.69972.e5. [DOI] [PubMed] [Google Scholar]
- 119.Van Mieghem W, Verleden G, Demedts M. Acute pulmonary oedema in patients with primary pulmonary hypertension and normal pulmonary capillary wedge pressure. Acta Cardiol. 1994;49:483–8. [PubMed] [Google Scholar]
- 120.Lawler LP, Askin FB. Pulmonary capillary hemangiomatosis: multidetector row CT findings and clinico-pathologic correlation. J Thorac Imaging. 2005;20:61–3. doi: 10.1097/01.rti.0000139390.26138.94. [DOI] [PubMed] [Google Scholar]
- 121.Overbeek MJ, van Nieuw Amerongen GP, Boonstra A, Boonstra A, Smit EF, Vonk-Noordegraaf A. Possible role of imatinib in clinical pulmonary veno-occlusive disease. Eur Respir J. 2008;32:232–5. doi: 10.1183/09031936.00054407. [DOI] [PubMed] [Google Scholar]
- 122.White CW, Wolf SJ, Korones DN, Sondheimer HM, Tosi MF, Yu A. Treatment of childhood angiomatous diseases with recombinant interferon alfa-2a. J Pediatr. 1991;118:59–66. doi: 10.1016/s0022-3476(05)81844-x. [DOI] [PubMed] [Google Scholar]
- 123.Ginns LC, Roberts DH, Mark EJ, Brusch JL, Marler JJ. Pulmonary capillary hemangiomatosis with atypical endotheliomatosis: successful antiangiogenic therapy with doxycycline. Chest. 2003;124:2017–22. doi: 10.1378/chest.124.5.2017. [DOI] [PubMed] [Google Scholar]
- 124.Assaad AM, Kawut SM, Arcasoy SM, et al. Platelet-derived growth factor is increased in pulmonary capillary hemangiomatosis. Chest. 2007;131:850–5. doi: 10.1378/chest.06-1680. [DOI] [PubMed] [Google Scholar]
- 125.Aliyu ZY, Kato GJ, Taylor Jt, et al. Sickle cell disease and pulmonary hypertension in Africa: a global perspective and review of epidemiology, pathophysiology, and management. Am J Hematol. 2008;83:63–70. doi: 10.1002/ajh.21057. [DOI] [PubMed] [Google Scholar]
- 126.Castro O, Gladwin MT. Pulmonary hypertension in sickle cell disease: mechanisms, diagnosis, and management. Hematol Oncol Clin North Am. 2005;19:881–96. vii. doi: 10.1016/j.hoc.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 127.Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350:886–95. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- 128.Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med. 2008;359:2254–65. doi: 10.1056/NEJMra0804411. [DOI] [PubMed] [Google Scholar]
- 129.Pashankar FD, Carbonella J, Bazzy-Asaad A, Friedman A. Prevalence and risk factors of elevated pulmonary artery pressures in children with sickle cell disease. Pediatrics. 2008;121:777–82. doi: 10.1542/peds.2007-0730. [DOI] [PubMed] [Google Scholar]
- 130.Chung EE, Dianzumba SB, Morais P, Serjeant GR. Cardiac performance in children with homozygous sickle cell disease. J Am Coll Cardiol. 1987;9:1038–42. doi: 10.1016/s0735-1097(87)80305-4. [DOI] [PubMed] [Google Scholar]
- 131.Lamers L, Ensing G, Pignatelli R, et al. Evaluation of left ventricular systolic function in pediatric sickle cell anemia patients using the end-systolic wall stress–velocity of circumferential fiber shortening relationship. J Am Coll Cardiol. 2006;47:2283–8. doi: 10.1016/j.jacc.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 132.Pashankar FD, Carbonella J, Bazzy-Asaad A, Friedman A. Longitudinal follow up of elevated pulmonary artery pressures in children with sickle cell disease. Br J Haematol. 2009;144:736–41. doi: 10.1111/j.1365-2141.2008.07501.x. [DOI] [PubMed] [Google Scholar]
- 133.Taylor JGt, Woods GM, Machado R, Kato GJ, Gladwin MT. Severe pulmonary hypertension in an adolescent with sickle cell disease. Am J Hematol. 2008;83:71–2. doi: 10.1002/ajh.21039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Villavicencio K, Ivy D, Cole L, Nuss R. Symptomatic pulmonary hypertension in a child with sickle cell disease. J Pediatr. 2008;152:879–81. doi: 10.1016/j.jpeds.2008.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jing X, Yokoi T, Nakamura Y, et al. Pulmonary capillary hemangiomatosis: a unique feature of congestive vasculopathy associated with hypertrophic cardiomyopathy. Arch Pathol Lab Med. 1998;122:94–6. [PubMed] [Google Scholar]
- 136.Rush C, Langleben D, Schlesinger RD, Stern J, Wang NS, Lamoureux E. Lung scintigraphy in pulmonary capillary hemangiomatosis. A rare disorder causing primary pulmonary hypertension. Clin Nucl Med. 1991;16:913–7. doi: 10.1097/00003072-199112000-00007. [DOI] [PubMed] [Google Scholar]
- 137.Billington DC. Angiogenesis and its inhibition: potential new therapies in oncology and non-neoplastic diseases. Drug Des Discov. 1991;8:3–35. [PubMed] [Google Scholar]
- 138.Adedeji MO, Cespedes J, Allen K, Subramony C, Hughson MD. Pulmonary thrombotic arteriopathy in patients with sickle cell disease. Arch Pathol Lab Med. 2001;125:1436–41. doi: 10.5858/2001-125-1436-PTAIPW. [DOI] [PubMed] [Google Scholar]
- 139.Sachdev V, Machado RF, Shizukuda Y, et al. Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J Am Coll Cardiol. 2007;49:472–9. doi: 10.1016/j.jacc.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Morris CR, Gladwin MT, Kato GJ. Nitric oxide and arginine dysregulation: a novel pathway to pulmonary hypertension in hemolytic disorders. Curr Mol Med. 2008;8:620–32. doi: 10.2174/156652408786241447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Morris CR, Kato GJ, Poljakovic M, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kato GJ, Martyr S, Blackwelder WC, et al. Levels of soluble endothelium-derived adhesion molecules in patients with sickle cell disease are associated with pulmonary hypertension, organ dysfunction, and mortality. Br J Haematol. 2005;130:943–53. doi: 10.1111/j.1365-2141.2005.05701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Villagra J, Shiva S, Hunter LA, Machado RF, Gladwin MT, Kato GJ. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood. 2007;110:2166–72. doi: 10.1182/blood-2006-12-061697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Derchi G, Forni GL, Formisano F, et al. Efficacy and safety of Sildenafil in the treatment of severe pulmonary hypertension in patients with hemoglobinopathies. Haematologica. 2005;90:452–8. [PubMed] [Google Scholar]
- 145.Machado RF, Martyr S, Kato GJ, et al. Sildenafil therapy in patients with sickle cell disease and pulmonary hypertension. Br J Haematol. 2005;130:445–53. doi: 10.1111/j.1365-2141.2005.05625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Krowka MJ, Plevak D. The distinct concepts and implications of hepatopulmonary syndrome and portopulmonary hypertension. Crit Care Med. 2005;33:470. doi: 10.1097/01.ccm.0000153595.76419.f3. [DOI] [PubMed] [Google Scholar]
- 147.Hoeper MM, Krowka MJ, Strassburg CP. Portopulmonary hypertension and hepatopulmonary syndrome. Lancet. 2004;363:1461–8. doi: 10.1016/S0140-6736(04)16107-2. [DOI] [PubMed] [Google Scholar]
- 148.Krowka MJ. Hepatopulmonary syndrome and portopulmonary hypertension: implications for liver transplantation. Clin Chest Med. 2005;26:587–97. vi. doi: 10.1016/j.ccm.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 149.Krowka MJ, Mandell MS, Ramsay MA, et al. Hepatopulmonary syndrome and portopulmonary hypertension: a report of the multicenter liver transplant database. Liver Transplant. 2004;10:174–82. doi: 10.1002/lt.20016. [DOI] [PubMed] [Google Scholar]
- 150.Pilatis ND, Jacobs LE, Rerkpattanapipat P, et al. Clinical predictors of pulmonary hypertension in patients undergoing liver transplant evaluation. Liver Transplant. 2000;6:85–91. doi: 10.1002/lt.500060116. [DOI] [PubMed] [Google Scholar]
- 151.Swanson KL, Krowka MJ. Screen for portopulmonary hypertension, especially in liver transplant candidates. Cleve Clin J Med. 2008;75(121–2):5–30. 33. doi: 10.3949/ccjm.75.2.121. passim. [DOI] [PubMed] [Google Scholar]
- 152.Swanson KL, Wiesner RH, Nyberg SL, Rosen CB, Krowka MJ. Survival in portopulmonary hypertension: mayo clinic experience categorized by treatment subgroups. Am J Transplant. 2008;8:2445–53. doi: 10.1111/j.1600-6143.2008.02384.x. [DOI] [PubMed] [Google Scholar]
- 153.Torregrosa M, Genesca J, Gonzalez A, et al. Role of Doppler echocardiography in the assessment of portopulmonary hypertension in liver transplantation candidates. Transplantation. 2001;71:572–4. doi: 10.1097/00007890-200102270-00015. [DOI] [PubMed] [Google Scholar]
- 154.Iqbal CW, Krowka MJ, Pham TH, Freese DK, El Youssef M, Ishitani MB. Liver transplantation for pulmonary vascular complications of pediatric end-stage liver disease. J Pediatr Surg. 2008;43:1813–20. doi: 10.1016/j.jpedsurg.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 155.Weber MA, Ashworth MT, Sebire NJ. Portopulmonary hypertension in childhood presenting as sudden death. Pediatr Dev Pathol. 2006;9:65–71. doi: 10.2350/08-05-0093.1. [DOI] [PubMed] [Google Scholar]
- 156.Laving A, Khanna A, Rubin L, Ing F, Dohil R, Lavine JE. Successful liver transplantation in a child with severe portopulmonary hypertension treated with epoprostenol. J Pediatr Gastroenterol Nutr. 2005;41:466–8. doi: 10.1097/01.mpg.0000178441.10417.0f. [DOI] [PubMed] [Google Scholar]
- 157.Whitworth JR, Sokol RJ. Hepato-portopulmonary disorders—not just in adults! J Pediatr Gastroenterol Nutr. 2005;41:393–5. [PubMed] [Google Scholar]
- 158.Condino AA, Ivy DD, O’Connor JA, et al. Portopulmonary hypertension in pediatric patients. J Pediatr. 2005;147:20–6. doi: 10.1016/j.jpeds.2005.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Krowka MJ. Many faces of portopulmonary hypertension. J Pediatr. 2005;147:3–4. doi: 10.1016/j.jpeds.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 160.Ling Y, Zhang J, Luo B, et al. The role of endothelin-1 and the endothelin B receptor in the pathogenesis of hepatopulmonary syndrome in the rat. Hepatology. 2004;39:1593–602. doi: 10.1002/hep.20244. [DOI] [PubMed] [Google Scholar]
- 161.Condino AA, Ivy DD, O’Connor JA, et al. Portopulmonary hypertension in pediatric patients. J Pediatr. 2005;147:20–6. doi: 10.1016/j.jpeds.2005.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Barth F, Gerber PJ, Reichen J, Dufour JF, Nicod LP. Efficiency and safety of bosentan in child C cirrhosis with portopulmonary hypertension and renal insufficiency. Eur J Gastroenterol Hepatol. 2006;18:1117–9. doi: 10.1097/01.meg.0000231749.60889.f7. [DOI] [PubMed] [Google Scholar]
- 163.Hoeper MM, Halank M, Marx C, et al. Bosentan therapy for portopulmonary hypertension. Eur Respir J. 2005;25:502–8. doi: 10.1183/09031936.05.00080804. [DOI] [PubMed] [Google Scholar]
- 164.Hoeper MM, Seyfarth HJ, Hoeffken G, et al. Experience with inhaled iloprost and bosentan in portopulmonary hypertension. Eur Respir J. 2007;30:1096–102. doi: 10.1183/09031936.00032407. [DOI] [PubMed] [Google Scholar]
- 165.Kato H, Katori T, Nakamura Y, Kawarasaki H. Moderate-term effect of epoprostenol on severe portopulmonary hypertension. Pediatr Cardiol. 2003;24:50–3. doi: 10.1007/s00246-002-0120-9. [DOI] [PubMed] [Google Scholar]
- 166.Krowka MJ, Swanson KL. How should we treat portopulmonary hypertension? Eur Respir J. 2006;28:466–7. doi: 10.1183/09031936.06.00086506. [DOI] [PubMed] [Google Scholar]
- 167.Tempe DK, Datt V, Datta D. Bosentan for the treatment of portopulmonary hypertension. Ann Card Anaesth. 2008;11:139–40. doi: 10.4103/0971-9784.41595. [author reply 40] [DOI] [PubMed] [Google Scholar]
- 168.Kim KK, Factor SM. Membranoproliferative glomerulonephritis and plexogenic pulmonary arteriopathy in a homosexual man with acquired immunodeficiency syndrome. Hum Pathol. 1987;18:1293–6. doi: 10.1016/s0046-8177(87)80417-3. [DOI] [PubMed] [Google Scholar]
- 169.Polos PG, Wolfe D, Harley RA, Strange C, Sahn SA. Pulmonary hypertension and human immunodeficiency virus infection. Two reports and a review of the literature. Chest. 1992;101:474–8. doi: 10.1378/chest.101.2.474. [DOI] [PubMed] [Google Scholar]
- 170.Speich R, Jenni R, Opravil M, Pfab M, Russi EW. Primary pulmonary hypertension in HIV infection. Chest. 1991;100:1268–71. doi: 10.1378/chest.100.5.1268. [DOI] [PubMed] [Google Scholar]
- 171.Zuber JP, Calmy A, Evison JM, et al. Pulmonary arterial hypertension related to HIV infection: improved hemodynamics and survival associated with antiretroviral therapy. Clin Infect Dis. 2004;38:1178–85. doi: 10.1086/383037. [DOI] [PubMed] [Google Scholar]
- 172.Diogenes MS, Succi RC, Machado DM, Moises VA, Novo NF, Carvalho AC. Cardiac longitudinal study of children perinatally exposed to human immunodeficiency virus type 1. Arq Bras Cardiol. 2005;85:233–40. doi: 10.1590/s0066-782x2005001700002. [DOI] [PubMed] [Google Scholar]
- 173.Pongprot Y, Sittiwangkul R, Silvilairat S, Sirisanthana V. Cardiac manifestations in HIV-infected Thai children. Ann Trop Paediatr. 2004;24:153–9. doi: 10.1179/027249304225013439. [DOI] [PubMed] [Google Scholar]
- 174.Mehta NJ, Khan IA, Mehta RN, Sepkowitz DA. HIV-related pulmonary hypertension: analytic review of 131 cases. Chest. 2000;118:1133–41. doi: 10.1378/chest.118.4.1133. [DOI] [PubMed] [Google Scholar]
- 175.Pellicelli AM, Barbaro G, Palmieri F, et al. Primary pulmonary hypertension in HIV patients: a systematic review. Angiology. 2001;52:31–41. doi: 10.1177/000331970105200105. [DOI] [PubMed] [Google Scholar]
- 176.Humbert M, Sitbon O, Chaouat A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–30. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- 177.Lai WW, Lipshultz SE, Easley KA, et al. Prevalence of congenital cardiovascular malformations in children of human immunodeficiency virus-infected women: the prospective P2C2 HIV Multicenter Study. P2C2 HIV Study Group, National Heart, Lung, and Blood Institute, Bethesda, Maryland. J Am Coll Cardiol. 1998;32:1749–55. doi: 10.1016/s0735-1097(98)00449-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Magder LS, Mofenson L, Paul ME, et al. Risk factors for in utero and intrapartum transmission of HIV. J Acquir Immune Defic Syndr. 2005;38:87–95. doi: 10.1097/00126334-200501010-00016. [DOI] [PubMed] [Google Scholar]
- 179.McDonald AM, Zurynski YA, Wand HC, et al. Perinatal exposure to HIV among children born in Australia, 1982–2006. Med J Aust. 2009;190:416–20. doi: 10.5694/j.1326-5377.2009.tb02488.x. [DOI] [PubMed] [Google Scholar]
- 180.Perez-Atayde AR, Kearney DI, Bricker JT, et al. Cardiac, aortic, and pulmonary arteriopathy in HIV-infected children: the prospective P2C2 HIV multicenter study. Pediatr Dev Pathol. 2004;7:61–70. doi: 10.1007/s10024-003-1001-9. [DOI] [PubMed] [Google Scholar]
- 181.Mette SA, Palevsky HI, Pietra GG, et al. Primary pulmonary hypertension in association with human immunodeficiency virus infection. A possible viral etiology for some forms of hypertensive pulmonary arteriopathy. Am Rev Respir Dis. 1992;145:1196–200. doi: 10.1164/ajrccm/145.5.1196. [DOI] [PubMed] [Google Scholar]
- 182.Seoane L, Shellito J, Welsh D, de Boisblanc BP. Pulmonary hypertension associated with HIV infection. South Med J. 2001;94:635–9. [PubMed] [Google Scholar]
- 183.Humbert M. Mediators involved in HIV-related pulmonary arterial hypertension. AIDS. 2008;22(Suppl 3):S41–7. doi: 10.1097/01.aids.0000327515.55041.da. [DOI] [PubMed] [Google Scholar]
- 184.Humbert M, Monti G, Fartoukh M, et al. Platelet-derived growth factor expression in primary pulmonary hypertension: comparison of HIV seropositive and HIV seronegative patients. Eur Respir J. 1998;11:554–9. [PubMed] [Google Scholar]
- 185.Barbaro G. Cardiovascular monitoring of HIV-infected patients. J Cardiovasc Med (Hagerstown) 2006;7:379. doi: 10.2459/01.JCM.0000223264.85298.81. [author reply 80] [DOI] [PubMed] [Google Scholar]
- 186.Opravil M, Pechere M, Speich R, et al. HIV-associated primary pulmonary hypertension. A case control study. Swiss HIV Cohort Study. Am J Respir Crit Care Med. 1997;155:990–5. doi: 10.1164/ajrccm.155.3.9117037. [DOI] [PubMed] [Google Scholar]
- 187.Glesby MJ, Aberg JA, Kendall MA, et al. Pharmacokinetic interactions between indinavir plus ritonavir and calcium channel blockers. Clin Pharmacol Ther. 2005;78:143–53. doi: 10.1016/j.clpt.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 188.Aschmann YZ, Kummer O, Linka A, et al. Pharmacokinetics and pharmacodynamics of Sildenafil in a patient treated with human immunodeficiency virus protease inhibitors. Ther Drug Monit. 2008;30:130–4. doi: 10.1097/FTD.0b013e318165ba71. [DOI] [PubMed] [Google Scholar]
- 189.Carlsen J, Kjeldsen K, Gerstoft J. Sildenafil as a successful treatment of otherwise fatal HIV-related pulmonary hypertension. AIDS. 2002;16:1568–9. doi: 10.1097/00002030-200207260-00021. [DOI] [PubMed] [Google Scholar]
- 190.Wong AR, Rasool AH, Abidin NZ, Noor AR, Quah BS. Sildenafil as treatment for Human Immunodeficiency Virus-related pulmonary hypertension in a child. J Paediatr Child Health. 2006;42:147–8. doi: 10.1111/j.1440-1754.2006.00816.x. [DOI] [PubMed] [Google Scholar]
- 191.Degano B, Yaici A, Le Pavec J, et al. Long-term effects of bosentan in patients with HIV-associated pulmonary arterial hypertension. Eur Respir J. 2009;33:92–8. doi: 10.1183/09031936.00094808. [DOI] [PubMed] [Google Scholar]
- 192.Aguilar RV, Farber HW. Epoprostenol (prostacyclin) therapy in HIV-associated pulmonary hypertension. Am J Respir Crit Care Med. 2000;162:1846–50. doi: 10.1164/ajrccm.162.5.2004042. [DOI] [PubMed] [Google Scholar]
- 193.Hsue PY, Waters DD. What a cardiologist needs to know about patients with human immunodeficiency virus infection. Circulation. 2005;112:3947–57. doi: 10.1161/CIRCULATIONAHA.105.546465. [DOI] [PubMed] [Google Scholar]