

Abstract
Gaucher's disease is the most frequent sphingolipid storage disease. We present a case of type 1 non-neuropathic type of adult Gaucher's disease patient with atypical presentation.
Keywords: Asymptomatic, Gaucher's disease, non-neuropathic
INTRODUCTION
Gaucher's disease, the most frequent sphingolipid storage disease, is an autosomal recessive disorder caused by mutations in the beta-glucocerebrosidase gene leading to deficient activity of this lysosomal enzyme. It is most prevalent in the Ashkenazi Jewish (AJ) population (1/450). The clinical features of type 1 Gaucher's disease are inconsistent. We report a case of type 1 adult Gaucher's disease patient with unusual presentation.
CASE REPORT
A 30-year-old male, Bora Muslim by religion and born to parents with nonconsanguineous marriage, came to us with a history of recurrent mucosal and skin infections since last 5 years. He said that every 30–45 days he gets infection, which is associated with fever and subsides after taking antibiotics and antipyretics. The frequency of infections had increased in the last 1 year. He never had any history of weight loss, bleeding tendencies, lymphadenopathy, infections of bone, or recurrent fractures.
When he presented to us, he was febrile (38.2 C), with tachycardia (pulse= 102/bpm) and normal blood pressure. He had furuncle over his left thigh with localized raise in temperature, tenderness, and induration. He was anicteric, had no lymphadenopathy, no hepatosplenomegaly, and no bone tenderness. His other systems examination was essentially normal.
His complete blood counts (CBCs) in the past 5 years (during infections) had been normal with a good neutrophilic response (hemoglobin [Hb], 13.4 gm/dL; total leukocyte count [TLC] 11,900 cubic mm with neutrophils 78%; and platelets = 320,000 cubic mm). Since the last year his total leukocyte count and neutrophil count were decreasing and there was an increase in the lymphocyte count. At the time of presentation his CBC was done. His CBC showed normal hemoglobin, low total leukocyte count with decreased neutrophils (Hb, 12.9 gm/dL; TLC, 3300 cubic mm with neutrophils of 42%; and platelets, 290,000 cubic mm). Peripheral smear examination showed no abnormal cells. He was again given antipyretics and antibiotics and was asked to follow-up. He was followed over a period of next 6–8 weeks both clinically and hematologically. Although clinically his furuncle subsided and he became afebrile, there was no change in his total leukocyte and neutrophilic count (Hb, 13.2 gm/dL, TLC, 2800 cubic mm with neutrophils 45%; platelets, 420,000 cubic mm). In view of a persistent low total leukocyte and low neutrophilic count a bone marrow (BM) examination was done. BM smear showed normoblastic marrow with normal megakaryocytic and leukocyte lineage. Immunohistochemistry of bone marrow was normal.
The patient came to us again in the next month with another skin infection and a further decline in neutrophilic and total leukocyte response (TLC, 1890 cubic mm with neutrophils 38%; platelets, 375,000 cubic mm). A repeat BM examination was done, which showed an abnormal cell, possibly a Gaucher's cell. His enzyme levels for GM1 Gangloisidase were positive. A diagnosis of adult Gaucher's was confirmed.
In retrospect, we again interviewed and examined the patient for any evidence of hepatosplenomegaly, avascular necrosis of bone, pathological fractures, anemia, or thrombocytopenia. But the patient did not have any of these features.
DISCUSSION
Gaucher's disease (GD), the most frequent sphingolipid storage disease, is an autosomal recessive disorder caused by mutations in the beta-glucocerebrosidase gene leading to deficient activity of this lysosomal enzyme.[1] The resulting accumulation of glucocerebroside is primarily in macrophages, giving rise to foam cells or “Gaucher cells.”[1] The glucocerebrosidase gene is located on chromosome 1q21.[2]
The disease has conventionally been classified into three clinical phenotypes: type I—adult, non-neuronopathic; type II—infantile or acute neuronopathic form (rapidly progressive neurovisceral storage disease, almost always leading to death during infancy); and type III—juvenile or chronic neuronopathic (less rapidly progressive neurovisceral storage disease).[3]
Type I is the most common form of Gaucher's disease with an incidence of approximately 1/40–60,000 in the general population.[3] The incidence varies between 1/40,000 in Central Europe and 1/2,000 in some non-European countries, such as Israel.[2] It is most prevalent in the Ashkenazi Jewish (AJ) population (1/450).[3] The clinical features of type 1 Gaucher's disease are inconsistent. Spleen, liver, and bone marrow are usually infiltrated by Gaucher's cells.[4] This type has a varied spectrum of presentation ranging from splenomegaly and hypersplenism to asymptomatic or with trivial hematologic abnormalities in old age.[4] The patient may present with apparently disconnected signs and symptoms, such as splenomegaly, hepatomegaly, pancytopenias, bone manifestations, and pulmonary disease.[3] Table 1 describes and classifies type 1 Gaucher's according to its severity.[2] The postulation that the nature of the mutation in the glucocerebrosidase gene would determine disease severity has provoked the search for genotype/phenotype correlations to explain this wide variation in the clinical course of the disease.[4]
Table 1.
Classification of type 1 Gaucher's disease according to its severity[2]
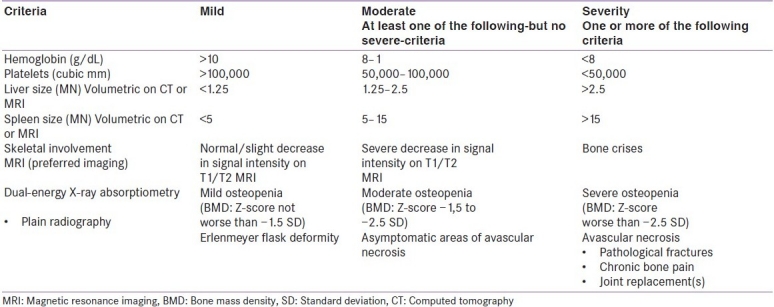
Various studies have documented that up to two-thirds (70%) of AJ homozygotes for the common mutation (N370S) are asymptomatic throughout life and never come to medical attention.[5] Also 43% of non-Jewish Gaucher's patients with N370S generally give rise to a mild disease phenotype. But some patients with this genotype do develop disabling Gaucher's disease.[4] In a study it was found that about 65% GD patients reported no GD medical complaints. However, 49% had anemia and/or thrombocytopenia. Ninety-seven percent of the patients had mild to moderate splenomegaly and 55% had hepatomegaly; skeletal imaging revealed marrow infiltration (100%), Erlenmeyer flask deformities (43%), lucencies (22%), and bone infarcts (14%).[5] Another mutation, L444P has been seen in about 12% of non-neuropathic patients and more than 50% of neuropathic patients.[4] The diagnosis of Gaucher's disease can be confirmed by the measurement of the activity of the enzyme glucocerebrosidase in leukocytes or fibroblasts.[2]
The patient in our case did not have any hepatosplenomegaly or any manifestations of bone or pulmonary disease. The patient had been having persistent cytopenias and increased propensity for infections. In absence of any overt clinical signs or symptoms of GD, bone marrow examination showing classical Gaucher's cells clinched the diagnosis.
Enzyme replacement therapy (ERT) has been available since about 2 decades and has revolutionized the treatment for Gaucher's disease. Imiglucerase is the accepted standard of care for the treatment of patients with Gaucher types I and type III disease.[3] It consists of life-long, intravenous replacement of the deficient enzyme, glucocerebrosidase. This is given at regular intervals, once every 2 weeks. If enzyme replacement therapy is begun early enough, with a sufficiently high dose, it usually leads to a significant improvement of hepatosplenomegaly, hematological parameters, and bone disease and various laboratory-chemical changes. As a result there is a considerable improvement of the patient's general condition and quality of life. While this therapy is highly effective, such chronic treatment places a burden on the individual patient, and is costly for society.[2] Substrate reduction therapy with Miglustat has recently been accepted as the treatment of symptomatic type I patients with mild to moderate disease for whom ERT is unsuitable or not a therapeutic option. Future may open better avenues like gene therapy for the treatment of Gaucher's disease.[3]
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Beutler E, Grabowski GA. Glucosylceramide lipidoses. In: Scriver CR, Beudet AL, Sly WS, Valle D, editors. The Metabolic Basis of Inherited Diseases. 7th ed. New York: McGraw-Hill; 1995. pp. 2641–70. [Google Scholar]
- 2.Abramowicz MJ, Carton D, De Meirleir L, Eyskens FJ, Goyens P, Louwagie AC. The Belgian working group on Gaucher's disease. Guidelines for diagnosis, treatment and monitoring of Gaucher's disease. 2004:1–12. [Google Scholar]
- 3.Di Rocco M, Giona F, Carubbi F, Linari S, Minichilli F, Brady RO, et al. A new severity score index for phenotypic classification and evaluation of responses to treatment in type I Gaucher disease. Haematologica. 2008;93:1211–8. doi: 10.3324/haematol.12379. [DOI] [PubMed] [Google Scholar]
- 4.Lachmann RH, Grant IR, Halsall D, Cox TM. Twin pairs showing discordance of phenotype in adult Gaucher's disease. QJM. 2004;97:199–204. doi: 10.1093/qjmed/hch036. [DOI] [PubMed] [Google Scholar]
- 5.Balwani M, Fuerstman L, Kornreich R, Edelmann L, Desnick RJ. Type 1 Gaucher disease: Significant disease manifestations in “asymptomatic” homozygotes. Arch Intern Med. 2010;170:1463–9. doi: 10.1001/archinternmed.2010.302. [DOI] [PMC free article] [PubMed] [Google Scholar]