

Abstract
The transforming growth factor-beta (TGF-β) superfamily consists of a variety of cytokines expressed in many different cell types including skeletal muscle. Members of this superfamily that are of particular importance in skeletal muscle are TGF-β1, mitogen-activated protein kinases (MAPKs), and myostatin. These signaling molecules play important roles in skeletal muscle homeostasis and in a variety of inherited and acquired neuromuscular disorders. Expression of these molecules is linked to normal processes in skeletal muscle such as growth, differentiation, regeneration, and stress response. However, chronic elevation of TGF-β1, MAPKs, and myostatin is linked to various features of muscle pathology, including impaired regeneration and atrophy. In this review, we focus on the aberrant signaling of TGF-β in various disorders such as Marfan syndrome, muscular dystrophies, sarcopenia, and critical illness myopathy. We also discuss how the inhibition of several members of the TGF-β signaling pathway has been implicated in ameliorating disease phenotypes, opening up novel therapeutic avenues for a large group of neuromuscular disorders.
Introduction
The transforming growth factor-beta (TGF-β) superfamily plays a crucial role in normal physiology and pathogenesis in a number of tissues. It is important to emphasize that downstream effects of this signaling cascade are often tissue-specific, thereby dictating which target genes will be activated in response to the transduction signal. Given its multifaceted effects in different tissues, deregulation of TGF-β signaling cascades can lead to a multitude of developmental defects and/or disease [1]. Several members of the TGF-β family have been shown to play important roles in regulating muscle growth and atrophy. The most extensively characterized ligands, in terms of the effects on skeletal muscle, are TGF-β1, mitogen-activated protein kinases (MAPKs), and myostatin. In this review, we focus on these signaling molecules in normal homeostasis and pathological conditions affecting skeletal muscle and describe the therapeutic avenues that have recently been explored to target the TGF-β signaling cascade.
Overview of the TGF-β superfamily signaling cascade
The TGF-β superfamily of cytokines consists of a variety of signaling molecules including isoforms of TGF-β (1 to 3), bone morphogenic proteins (BMPs 1 to 20), growth and differentiation factors (GDFs), activins (A and B), inhibins (A and B), nodal, leftys (1 and 2), and Mullerian inhibiting substance [1]. They are generally divided into two branches defined by the utilization of receptor Smads (R-Smads): the TGF-β branch, consisting of TGF-β, activin, Nodal, and myostatin (GDF-8), signals through R-Smads 2 and 3 and the BMP branch, consisting of BMPs and other GDFs, signals through R-Smads 1, 5 and 8. This superfamily is known to be involved in embryonic development, adult tissue homeostasis, and disease pathogenesis. Specifically, it has been shown to control proliferation, differentiation, apoptosis, migration, extracellular matrix (ECM) remodeling, immune functions, and tumor invasion/metastasis [2].
TGF-β1 is synthesized as a precursor that is cleaved intracellularly into an inactive complex consisting of the mature TGF-β1 non-covalently bound to the portion of the precursor peptide termed the latency-associated peptide (LAP) [3]. This inactive TGF-β1-LAP complex forms a larger complex with latent transforming growth factor-binding proteins (LTBPs), which directly bind and release TGF-β1 from the ECM. Specifically, LTBP-4 sequesters and regulates the availability of TGF-β1 to bind with its receptor [4]. Cleavage of TGF-β1 from the latent complex is achieved through the action of proteases such as plasmin, thrombin, plasma transglutaminases, or endoglycosylases, or through the physical interaction of LAPs with other proteins [3]. Activation occurs extracellularly [3], and once TGF-β1 is released, it is able to interact with and complex its type I (usually TβR-II) and type II (usually activin receptor-like kinase (ALK) 5) receptors. The constitutively active type II receptor phosphorylates and activates the type I receptor, which in turn directly phosphorylates Smad2 and/or Smad3 (which are recruited by adaptor proteins) to initiate signal transduction through the canonical cascades [5]. Once R-Smad has been phosphorylated, it forms a complex with the common mediator Smad (co-Smad), Smad4, which translocates to the nucleus, where it directly binds defined elements on the DNA [2]. Adding to the regulation are the inhibitory Smads 6 and 7. Smad7 is involved in both branches and competes with R-Smads for interaction with the type I receptor, whereas Smad6 only participates in the BMP pathway and competes with Smad4 for binding to Smad1 [5] (Figure 1).
Figure 1.
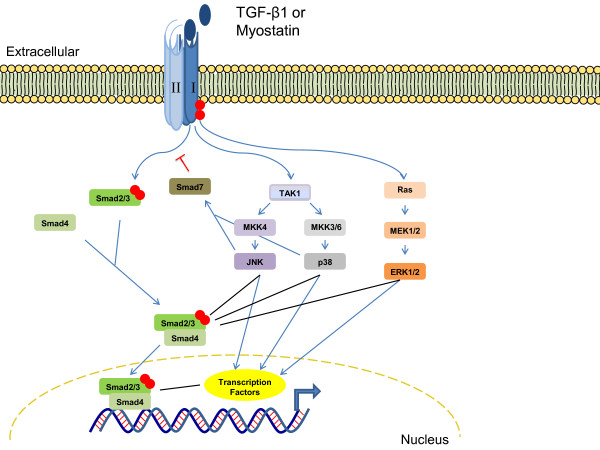
Crosstalk between the canonical and non-canonical transforming growth factor-beta1 (TGF-β1) and myostatin pathways. Once the TGF-β1 or myostatin ligands bind to the appropriate type I and type II receptors, cross-phosphorylation of the type I receptor occurs, leading to the phosphorylation of downstream effectors. In the canonical pathway, the type I receptor phosphorylates Smad2/3, which then binds to Smad4 and translocates into the nucleus to act as transcription factors. In the non-canonical pathway, the type I receptor phosphorylates proteins that are involved in the activation of the mitogen-activated protein kinases (MAPKs). Activated MAPKs can then regulate transcription factors and/or the Smad proteins through direct interactions or via downstream proteins.
TGF-β1 can also signal via induction of non-canonical pathways including MAPK. The MAPK family consists of isoforms of extracellular signal-regulated kinases (ERKs) (1 and 2), c-Jun N-terminal kinase (JNKs) (1to 3), and p38 (α, β, γ and δ). The mechanisms of MAPK activation by TGF-β1 and the subsequent biological consequences are cell-type-specific [6]. Generally in the non-Smad pathway, the type I receptor associates with the adaptor proteins, Shc and tumor necrosis factor receptor-associated factor (TRAF) 6, for the activation of Ras and TGF-β-activated kinase (TAK) 1 and subsequently, the ERK and p38/JNK pathways, respectively [7]. However, MAPK may also modulate TGF-β1-induced Smad signals and phosphorylate Smad proteins independent of TGF-β1, providing evidence for crosstalk between canonical and non-canonical TGF-β pathways [6,7] (Figure 1).
Myostatin (MSTN), predominantly expressed in skeletal muscle, also signals through the TGF-β branch [8]. It is synthesized as a precursor protein that undergoes processing by furin proteases to generate a propeptide. After proteolytic processing, however, the biologically active MSTN remains bound non-covalently to the propeptide, and in this complex, the propeptide maintains its inactive, latent state [9,10]. MSTN also seems to be regulated extracellularly by other binding proteins: follistatin [9,11], follistatin-related gene (FLRG) protein [12], and growth and differentiation factor-associated serum protein (GASP) 1 [13]. When not bound to its propeptide or binding proteins, active MSTN is able to signal to target cells by binding to the activin type II receptors, ActRIIA or ActRIIB [14,15]. The activation of the type I receptor (usually ALK5 and to a lesser extent ALK4) leads to the phosphorylation of R-Smads 2 and 3 [15]. More recently, it has been shown that MSTN is also able to induce the activation of the MAPK signaling pathway in Smad-dependent and -independent mechanisms [16-18], and to inhibit the Akt/TORC1/p70S6K signaling pathway [19] (Figure 1). For a more extensive summary of MSTN, see [20].
Physiological role of TGF-β signaling in skeletal muscle
TGF-β1 is expressed during myogenesis, and its spatial and temporal expression in the developing connective tissue is correlated with the fiber-type composition of the surrounding myotubes. Myotubes formed before the expression of TGF-β1 develop into slow fibers, whereas fast fibers form when myoblasts are adjacent to connective tissue expressing TGF-β1 [21]. TGF-β1 has been shown to inhibit the differentiation of fetal myoblasts but does not affect embryonic myoblasts [22]. In mature adult muscle, TGF-β negatively affects skeletal muscle regeneration by inhibiting satellite cell proliferation, myofiber fusion, and expression of some muscle-specific genes [23]. Furthermore, TGF-β1 induces the transformation of myogenic cells into fibrotic cells after injury [24].
Not much is known about the role of the different MAPKs in embryogenesis [25]; although, they have been shown to play a role in myogenesis and regeneration. p38 is speculated to regulate regeneration through the activation of p21, a cyclin-dependent inhibitor that causes irreversible withdrawal from the cell cycle (necessary for the differentiation of myoblasts) and through interactions with Pax7, myogenic regulatory factors, and myocyte enhancer factors [26,27]. JNK is proposed to inhibit myogenesis [28], and ERK may have multiple roles: preventing the initiation of myogenesis [29], enhancing myoblast proliferation during the acute stages, and repressing muscle-specific gene expression and myoblast differentiation, if expression is sustained [30]. Generally, in mature muscle, MAPKs mediate the transduction of diverse external stress stimuli into intracellular signals that regulate adaptive cellular responses such as proliferation, differentiation, self-renewal, and survival in diseased and healthy states [2,31]. For example, MAPK levels are modulated during exercise and aging as a stress response [31,32].
Myostatin is expressed in developing skeletal muscle throughout embryogenesis and has been shown to be a negative regulator of adult skeletal muscle mass by acting on different mechanisms [20]. Genetic studies in mice, cattle, sheep, dogs, chickens, and humans have all shown that myostatin normally functions to limit muscle mass [33-40]. In mice, targeted ablation of the Mstn gene causes a doubling of skeletal muscle mass throughout the body, as a result of a combination of muscle fiber hyperplasia and hypertrophy [33]. Moreover, postnatal inhibition of myostatin signaling through the delivery of propeptides, neutralizing antibodies, antisense RNA, inhibitory proteins, and soluble ActRIIB has been shown to induce significant muscle growth when administered to mice of different ages, demonstrating the importance of this signaling pathway in regulating muscle homeostasis [10,14,41-50].
TGF-β signaling and skeletal-muscle repair
After skeletal muscle injury, a well-coordinated repair process occurs. This process includes the release of growth factors and cytokines and the migration and proliferation of macrophages and fibroblasts that increase the production of ECM components; these components are degraded as normal regeneration occurs. The inflammatory response serves to clear myofiber debris and modulate regeneration. The formation of new myofibers begins with the activation of satellite cells, followed by proliferation, differentiation, and fusion of myocytes [51] (Figure 2).
Figure 2.

Regulated and dysregulated muscle regeneration. In regulated muscle regeneration, a transient inflammatory response occurs upon injury, which includes the chemotaxis of growth factors, cytokines, macrophages, and fibroblasts. This is followed by the activation and proliferation of satellite cells. Once activated, myoblasts differentiate into myocytes, and then fuse together to form myofibers, which exhibit central nuclei. This process is primarily orchestrated by the expression of the myogenic regulatory factors. In dysregulated muscle regeneration, there is a persistent inflammatory response and overexpression of proteins such as transforming growth factor-beta1 (TGF-β1) and myostatin, which promote the formation of fibrotic tissue to replace damaged myofibers.
TGF-β1, a potent regulator of tissue wound healing and fibrosis, is physiologically upregulated in regenerating skeletal muscle after injury and exercise and is thought to participate in a transient inflammatory response to muscle damage [51,52]. Persistent exposure to the inflammatory response leads to an altered ECM and increased levels of growth factors and cytokines, including TGF-β1, which contribute to the formation of fibrotic tissue [51,52]. Therefore, TGF-β1 is one of the major factors promoting the transformation of myoblasts into fibrotic tissue after injury. Furthermore, increased levels of TGF-β1 inhibit satellite cell activation and impair myocyte differentiation [23,53] (Figure 2). Interestingly, reducing the levels of TGF-β1 in various physiological and pathological conditions associated with muscle homeostasis and regeneration has proven to be beneficial for several myopathic conditions [54-70] (Table 1).
Table 1.
Comprehensive overview of studies using agents to blunt transforming growth factor (TGF)-β signaling
| Compound | Mechanism of action | Clinical condition | Model organism | Phenotypic findings | Ref |
|---|---|---|---|---|---|
| FDA-approved medications | |||||
| Losartan | AT1a receptor antagonist (mostly used for hypertension, cardiomyopathies) | MFSb | Fbn1C1039G/+ mice | Improved muscle architecture, function and regeneration | [54] |
| DMDc | mdx mice | Improved skeletal, diaphragmatic and cardiac muscle architecture, function and regeneration | [54,55] | ||
| Muscle Injury | Younge mice | Decreased fibrosis and improved regeneration | [56] | ||
| Suramin | TGF-β1 receptor antagonist (anti-parasitic, anti-neoplasic) | DMD | mdx mice | Decreased fibrosis and prevented decrease in grip strength | [57] |
| Muscle injury | Adultf mice | Decreased fibrosis, improved regeneration and function | [58-60] | ||
| Anti-fibrotic agents | |||||
| Decorin | Binds to TGF-β1 ligands | Muscle injury | Young mice | Decreased fibrosis, improved regeneration and functional recovery | [61] |
| DMD | mdx mice | Decreased collagen type I levels in diaphragm | [62] | ||
| γ-Interferon | Induces Smad7 expression | Muscle injury | Young mice | Decreased fibrosis, improved regeneration and functional recovery | [63] |
| Pirfenidone | TGF-β1 antagonist | DMD | mdx mice | Improved cardiac function, minor alterations on the development of fibrosis, and no improvement in diaphragmatic function | [64,65] |
| Halofuginone | Inhibits TGF-β-dependent phosphorylation of Smad3 | DMD | mdx mice | Decreased fibrosis and improved function of the heart,diaphragm and limb muscles | [66,67] |
| CMDd | dy2J/dy2J | Decreased fibrosis and improved functional performance but did not improve strength | [68] | ||
| TGF-β neutralizing antibody | |||||
| Neutralizes TGF-β (1 and/or 2) ligands | MFS | Fbn1C1039G/+ mice | Prevented muscle atrophy and improved regeneration | [54] | |
| DMD | mdx mice | Decreased fibrosis and improved regeneration | [54,69] | ||
| Sarcopenia | Agedg mice | Failed to improve regeneration | [70] | ||
| TGF-β receptor kinase inhibitor | |||||
| Decoy receptor composed of extracellular portion of TGF-β receptor II | Sarcopenia | Aged mice | Improved regeneration after direct intramuscular injection | [70] | |
aAngiotensin II type 1 receptor.
bMarfan syndrome.
cDuchenne muscular dystrophy.
dCongenital muscular dystrophy.
eAge ≤ 3 months.
fAge 3-15 months.
gAge ≥ 15 months.
Myostatin also impairs skeletal muscle regeneration. It is proposed to hinder the chemotaxis of macrophages and myoblasts [71], while simultaneously activating and attracting fibroblasts to the site of injury. Once fibroblasts are within the environment of the injured muscle, they express MSTN and differentiate into myofibroblasts, a process that in turn accelerates the deposition of collagen and connective tissue, ultimately promoting the formation of tissue fibrosis [72,73]. Furthermore, myostatin inhibits the activation, differentiation, and self-renewal of satellite cells [71,74,75] and the expression of the muscle regulatory factors crucial for the regeneration and differentiation process of myofibers [76,77] (Figure 2). Inhibiting MSTN in various myopathic conditions has yielded mixed results, depending on the disease model and mechanism of inhibition [43,44,48,50,78-100] (Table 2).
Table 2.
Comprehensive overview of studies using post-natal inhibition of myostatin
| Disease | Model organism | Phenotypic findings | Ref |
|---|---|---|---|
| Neutralizing antibody: binds to active myostatin and prevents receptor binding | |||
| DMDa | mdx mice | Improved regeneration and function, induced hypertrophy, decreased degeneration (diaphragm) and fibrosis | [78,79] |
| LGMD2Cb | sgcg-/- mice | Improved function, induced hypertrophy but no histopathological improvement | [80] |
| LGMD2F | sgcd-/- mice | Increased muscle mass, regeneration (young) and fibrosis (aged) | [81] |
| ALSc | SOD1G93A mice and rats |
Delayed onset of muscle atrophy and functional decline without extending survival | [82] |
| Sarcopenia | Agedf mice | Prevented loss of body weight, muscle mass and function, and decline in physical activity, reduced apoptosis, no change in fibrosis | [44,83] |
| Disuse atrophy | Adultg mice | Partially protected against but did not prevent atrophy | [99] |
| ActRIIB-Fcd: soluble, decoy receptor binding active myostatin | |||
| DMD | mdx mice | Increased body weight and function, induced hypertrophy | [84,100] |
| LGMD1C | CAV-3P104L mice | Induced muscle hypertrophy | [85] |
| SMAe | SMAΔ7 mice | Modestly increased muscle weight and strength, decreased survival | [86] |
| ALS | SOD1G93A mice | Delayed onset of disease but did not extend survival, reduced weakness after onset | [87] |
| Cachexia | Lewis-lung carcinoma | Protected against loss of body weight and muscle mass | [88] |
| Cachexia | Colon-26 carcinoma | Protected against or restored loss of body weight, muscle mass and grip strength, and increased survival | [88,89] |
| MSTN Propeptide: binds to myostatin and prevents release of active form | |||
| DMD | mdx mice | Induced hypertrophy, increased strength, improved histopathological features of limb and diaphragm, decreased endurance, produced adverse effects on cardiomyopathy | [48,50,90] |
| LGMD2A | Capn3-/- mice | Increased muscle mass and force, no improvement in histopathological features | [91] |
| LGMD2D | sgca-/- mice | Insufficient delivery of vector resulted in no hypertrophy or any change in necrosis | [91] |
| Muscle Injury | Adult mice | Increased muscle mass, improved regeneration, decreased fibrosis | [92] |
| Follistatin: inhibitory protein that binds to myostatin | |||
| SMA | SMAΔ7 mice | Improved muscle mass (during early stages of disease), motor function and extended survival | [93] |
| ALS | SOD1G93A mice | Increased muscle mass (hyperplasia) and strength (not performance) but no survival extension | [94] |
| HDAC Inhibitors: induce expression of follistatin | |||
| DMD | mdx mice | Induced hypertrophy, decreased fibrosis and necrosis, restored muscle architecture, increased strength and performance | [95] |
| LGMD2D | sgca-/- mice | Induced hypertrophy and reduced fibrosis | [95] |
| Cachexia | Colon-26 carcinoma | Did not protect against loss of body weight, muscle mass or function | [88,96] |
| Muscle injury | Youngh mice | Improved regeneration | [97] |
| MSTN peptide: dominant negative truncated myostatin peptide that binds ActRIIB | |||
| Sarcopenia | Aged mice | Improved grip strength and enhanced inflammatory response after injury | [98] |
| Muscle injury | Adult mice | Improved regeneration, decrease in necrosis | [98] |
| Antisense RNA: binds myostatin messenger RNA and inactivates it | |||
| Cachexia | S-180 ascitic tumor |
Increased muscle mass | [43] |
aDuchenne muscular dystrophy.
bLimb-girdle muscular dystrophy.
cAmyotrophic lateral sclerosis.
dActivin type IIB receptor.
eSpinal muscular atrophy.
fAge≥ 15 months.
gAge 3-15 months.
hAge ≤ 3 months.
Role of TGF-β signaling in disease pathogenesis of inherited myopathies
Dysregulation of TGF-β signaling has been implicated in various pathological conditions affecting skeletal muscle, both inherited and acquired [51]. Inherited conditions can be progressive, and therefore, there are unique phenotypic characteristics that may require different modes of intervention. Indeed, increased levels of TGF-β, MAPK, and/or MSTN have been associated with spinal muscular atrophy and Kennedy disease [101,102], and inhibition of MSTN improves familial amyotrophic lateral sclerosis (ALS) [82,87], but this review focuses on altered signaling in the pathogenesis of Marfan syndrome (MFS) and the muscular dystrophies.
MFS is an autosomal dominant systemic disorder of connective tissue, caused by mutations in FBN1, the gene encoding the ECM protein, fibrillin-1 [103]. A large subset of patients exhibit a significant decrease in muscle mass, often associated with hypotonia, particularly during early childhood, and experience a life-long inability to increase muscle mass despite physical exercise. Histological analyses of skeletal muscle from fibrillin-1-deficient mice and patients with MFS demonstrated a decrease in the number and size of myofibers, accompanied by an increase in fibrosis, fat deposition, and the number of split fibers. Further molecular analyses revealed that an increase in TGF-β signaling was indeed responsible for the abnormal muscle phenotype and the impaired ability to regenerate muscle in response to injury. Interestingly, when TGF-β signaling was blunted via treatment with a TGF-β neutralizing antibody or losartan, mice deficient in fibrillin-1 exhibited normal muscle architecture and regeneration capabilities [54].
'Muscular dystrophy' (MD) is a term used to describe a group of over 30 inherited disorders characterized by variable progressive muscle weakness and wasting [104,105]. Genetic mutations in genes encoding proteins spanning every subcellular aspect of the myofiber have been described [105]. There are currently no unifying hypotheses integrating all forms of MDs, but various lines of evidence suggest that repeated cycles of degeneration and regeneration may eventually impair the ability of satellite cells to repopulate damaged muscle [104]. Once muscle regeneration declines, there is often an accumulation of inflammation and fibrosis, which results in an abundance of growth factors and cytokines including TGF-β1, as stated above [51,62,106-108]. Similarly, the levels of decorin and biglycan, components of the ECM that interact with cytokines such as TGF-β1, as well as modulations of MAPK and myostatin signaling, are altered in various forms of muscular dystrophies including congenital MD, Emery-Dreifuss MD (EDMD) and Becker MD [109-115]. This review will elaborate on findings in Duchenne and limb-girdle MDs.
Duchenne muscular dystrophy (DMD) is an X-linked disorder characterized by a complete lack of dystrophin, which renders the myofiber membrane unstable. Inflammation is thought to precede the overexpression of TGF-β1 and actual muscle wasting [116,117]. Additionally, other factors including ECM components, immune system components, osteopontin, and fibrinogen are increased and have been linked to fibrosis in patients and animal models of DMD [52,108-110,116,118-120]. Furthermore, increased levels of the MAPKs ERK1/2, JNK1, and p38 have also been suggested to play a role in the pathogenesis of the skeletal and cardiac muscle phenotype in animal models of DMD [121-123]. Insight into the different factors contributing to the fibrosis accompanying DMD has led to various mechanisms to improve the phenotype observed in cardiac and skeletal (diaphragm and limb) muscles (Table 1 and 2) [52,54,64,124,125].
Limb-girdle muscular dystrophy (LGMD) describes a group of disorders primarily affecting the shoulder and pelvic girdle muscles, which have both autosomal dominant and recessive inheritance, and involve a variety of proteins including sarcoglycans, dysferlin, and caveolin [105]. Studies in a Drosophila model of LGMD with γ/δ-sarcoglycan deficiency have shown that partial reduction of the Drosophila genes homologous to Smads 2/3, Smads 1/5/7, and Smad4 improved muscle function, as shown by increased climbing ability of the flies. Similarly, reducing the levels of the homologue for Smads 2/3 and Smad4 improved the heart tube phenotype [125]. Not only has this research provided a novel animal model for studying dystrophic disease processes, but the results also indicate that targeting the R-Smads and co-Smad may be of therapeutic interest. Furthermore, genetic manipulation of LTBP-4, a latent TGF-β binding protein discussed above, affected the severity of a mouse model of sarcoglycan-deficient LGMD2C, providing evidence for an important genetic modifier of MD. An insertion in the LTBP4 gene reduced proteolysis and Smad signaling [4]. Thus, targeting the latent complex of TGF-β1 opens up yet another therapeutic avenue for inhibiting this cytokine in various conditions affecting skeletal muscle.
Acquired myopathies implicating aberrant TGF-β signaling in disease progression
Alterations in the expression of the TGF-β signaling cascades have also been linked to acquired forms of myopathies. Muscle atrophy caused by hypoxia [126], microgravity exposure [127], starvation [128], acute daily psychological stress [129], various models of disuse [130-137], cancer [138,139], sporadic ALS [140], HIV [141], and glucocorticoid steroids [142] is associated with increased activation of MAPK, TGF-β1, and/or MSTN. However, in this paper, we focus on sarcopenia and critical illness myopathy (CIM).
Sarcopenia refers to the physiological age-related loss of skeletal muscle mass and function [143]. Several changes occur with age, including a decrease in myofiber size and number and diminished ability of satellite cells to activate and proliferate in response to injury, leading to impaired muscle remodeling [144,145]. The molecular mechanisms underlying sarcopenia are largely unknown. However, alterations in the canonical and non-canonical TGF-β signaling pathways have been shown to play a role in the pathogenesis of sarcopenia. Studies in elderly men have demonstrated an increase in MAPK at baseline, suggesting that aging skeletal muscle is functioning under 'stress-like' conditions at rest [32]. However, a different study conducted in mice and humans found an age-related decrease in ERK signaling in skeletal muscle and satellite cells, suggesting a contribution to the impaired regeneration [146]. Clearly, more in-depth studies are necessary to characterize the role of MAPK signaling in aging. Additionally, alterations in the canonical TGF-β pathway include an increase in circulating TGF-β1 levels and pSmad3, which contributes to the enrichment of connective tissue within the ECM, creating an environment that interferes with satellite cell activation and proliferation and subsequent remodeling [70,145]. Other studies have shown an upregulation of MSTN [147] and that inhibition of MSTN results in an increase in muscle mass, function, and regeneration in sarcopenic mice, suggesting an important role for this protein in the process of age-related loss of muscle mass [44,98] (Table 2).
CIM is characterized by generalized progressive muscle weakness and atrophy, occurring in critically ill patients who are hospitalized in the intensive care unit [148,149]. There are several factors thought to contribute to the loss of muscle mass in CIM, including immobilization, systemic inflammation, high dose steroids, and other toxins [148]. The precise molecular mechanisms underlying CIM are unknown [148]; however, constitutively active members of the canonical and non-canonical TGF-β signaling pathways may contribute to the muscle phenotype. In fact, atrophic fibers with apoptotic features express TGF-β ligands and receptors, p38, and downstream effectors of pJNK [149]. It is therefore tempting to speculate that TGF-β inhibition may slow or halt the progression of CIM.
Therapeutic inhibition of TGF-β signaling
Aberrant TGF-β signaling has an important role in inherited and acquired myopathies. Therefore, research has been aimed at identifying compounds that can attenuate the increased signaling of TGF-β1, MSTN, and/or MAPK levels in order to improve disease phenotypes.
Several compounds have been shown to reduce the levels of TGF-β1 in myopathies. These include FDA-approved medications with other primary clinical uses, anti-fibrotic agents, TGF-β neutralizing antibodies, and TGF-β receptor blockers. Most yielded favorable results, but there are some conditions in which blunting TGF-β signaling was not beneficial. Table 1 provides a comprehensive overview of existing agents targeting TGF-β signaling in specific disease models.
Losartan is a widely studied, FDA-approved drug commonly used in the treatment of hypertension. Its ability to attenuate TGF-β signaling in chronic renal disease, cardiomyopathies, and MFS [54,150,151] made it an appealing molecule in the treatment of myopathies associated with increased TGF-β signaling. Long-term administration of losartan to dystrophin-negative mdx mice attenuated TGF-β signaling, decreased skeletal muscle fibrosis, and improved muscle regeneration and in vitro and in vivo function [54]. Furthermore, long-term administration of losartan, in conjunction with exercise, in mdx mice improved the cardiac muscle function and decreased fibrosis in the cardiac, diaphragm, and limb muscles but did not improve limb muscle function [55]. Other mechanisms of TGF-β inhibition have also yielded favorable results in the treatment of DMD and other conditions (Table 1).
Furthermore, recent experimental evidence has identified novel therapeutic targets in the TGF-β pathway. Molecules involved upstream (LTBP-4) and downstream (R-Smads and co-Smad) have been shown to modulate disease severity [4,125]. It is important to emphasize that LTBP-4 is a specific target for TGF-β1 [4], whereas Smad molecules incorporate a variety of different pathways, which could potentially lead to a number of adverse effects if they were therapeutically modified [125,152] (Figure 1). Moreover, osteopontin, an inflammatory regulator that also modulates TGF-β1, has recently been shown to be upregulated during muscle regeneration and in DMD [118,153-155]. Lack of osteopontin in mdx mice improved fibrotic tissue formation and muscle function, making osteopontin a potential therapeutic target [118].
In addition to altering TGF-β1 signaling, a number of compounds have been shown to inhibit myostatin signaling. Numerous studies breeding myostatin-null mice to several mouse models of inherited and acquired myopathies have shown various beneficial and non-beneficial effects [81,85,88,98,156-159]. Interestingly, several studies on myostatin-null mice alone have shown that despite an increase in muscle fiber size, there is no increase in specific force, which is probably due to a disturbance in mitochondrial metabolism [160,161]. Furthermore, myostatin-null mice have also been reported to have brittle tendons, which may contribute to the decrease in specific force [162]. However, it is important to emphasize that these studies were performed in mice with a complete lack of myostatin during the development of skeletal muscle. Thus, caution is needed when extrapolating the findings obtained from myostatin-null mice to the various compounds targeting myostatin signaling postnatally.
There are several pharmacological compounds that inhibit MSTN postnatally: MSTN propeptide, MSTN peptide, inhibitory proteins (follistatin), MSTN neutralizing antibodies, histone deacetylase inhibitors, and soluble ActRIIB. These techniques have been used in various disease models, and a detailed overview is presented in Table 2.
The soluble receptor, ActRIIB, is currently being used in multiple clinical trials and has been explored in various animal models including a model of DMD. It is important to note that targeting ActRIIB could lead to adverse side effects, because its expression is not limited to skeletal muscle [84] and because other members of the TGF-β superfamily besides myostatin bind to it [14]. Furthermore, myostatin can also signal through another receptor, ActRIIA, but with lower affinity [15].
Preclinical trials with soluble ActRIIB in mdx mice have shown that short-term (3 months) intraperitoneal administration increased skeletal muscle mass and in vitro function and caused a decrease in creatine kinase levels [100]. Adeno-associated virus (AAV)-mediated gene transfer of a soluble form of the extracellular domain of the ActRIIB to the liver provided similar results after 3.5 months, but no changes to cardiac muscle mass were seen [84]. However, long-term (11 months) myostatin inhibition using a recombinant AAV to overexpress myostatin propeptide in mdx mice did not reduce the amount of fibrosis in the diaphragm, but caused cardiac hypertrophy and impaired function in a dose-dependent manner [50]. These results indicate that all modes of myostatin inhibition may not be beneficial.
Similar to TGF-β1 and MSTN, perturbations of MAPK signaling have been documented in several myopathies, but not many studies exist examining the effects of inhibition on disease progression. Some evidence suggest that a reduction in JNK and ERK signaling might be beneficial in DMD [122] and cachexia [163], respectively, but further studies are needed to elaborate on these initial findings. Furthermore, a number of studies have shown that inhibition of the MAPK pathway is beneficial for the cardiomyopathic phenotype of various muscular dystrophies. Specifically, it has been shown that blunting ERK or JNK before and after onset of EDMD results in less cardiac fibrosis and an overall improved function [164-166]. Thus, this is a potential area of interest in designing future pharmacological compounds, because of the potential benefits and current lack of FDA-approved MAPK inhibitors [166].
Conclusions
Increased activity of the TGF-β superfamily plays an important role in the pathogenesis of both inherited and acquired forms of neuromuscular disorders. These alterations cause an unfavorable environment for muscle regeneration and promote an increase in fibrotic tissue formation (Figure 2). Future studies will need to address the precise timeline of alterations in TGF-β signaling in various disease processes in order to establish the optimal therapeutic intervention. A number of drugs (Table 1; Table 2) are close to or currently in clinical trials. These and future clinical trials will need to establish the safety and efficacy of these drugs and address whether certain clinical conditions may benefit from a multi-targeted approach.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
TNB designed the study and drafted the manuscript. RDC drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Tyesha N Burks, Email: tburks1@jhmi.edu.
Ronald D Cohn, Email: rcohn2@jhmi.edu.
Acknowledgements
This project was funded by the National Institute on Aging, Claude D. Pepper Older Americans Independence Center, parent grant P30AG021334 to RDC and supplemental grant P30AG021334-08S1 to TNB. RDC is also supported by the NIH Director's New Innovator Award DP2 OD004515, by NIH 5K08NS055879 award, and MDA #101938.
References
- Gordon KJ, Blobe GC. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta. 2008;1782:197–228. doi: 10.1016/j.bbadis.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Guo X, Wang XF. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009;19:71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos-Hoff MH. Latency and activation in the control of TGF-beta. J Mammary Gland Biol Neoplasia. 1996;1:353–363. doi: 10.1007/BF02017391. [DOI] [PubMed] [Google Scholar]
- Heydemann A, Ceco E, Lim JE, Hadhazy M, Ryder P, Moran JL, Beier DR, Palmer AA, McNally EM. Latent TGF-beta-binding protein 4 modifies muscular dystrophy in mice. J Clin Invest. 2009;119:3703–3712. doi: 10.1172/JCI39845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi RA, Leof EB. TGF-beta signaling: a tale of two responses. J Cell Biochem. 2007;102:593–608. doi: 10.1002/jcb.21501. [DOI] [PubMed] [Google Scholar]
- Javelaud D, Mauviel A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene. 2005;24:5742–5750. doi: 10.1038/sj.onc.1208928. [DOI] [PubMed] [Google Scholar]
- Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- Thies RS, Chen T, Davies MV, Tomkinson KN, Pearson AA, Shakey QA, Wolfman NM. GDF-8 propeptide binds to GDF-8 and antagonizes biological activity by inhibiting GDF-8 receptor binding. Growth Factors. 2001;18:251–259. doi: 10.3109/08977190109029114. [DOI] [PubMed] [Google Scholar]
- Wolfman NM, McPherron AC, Pappano WN, Davies MV, Song K, Tomkinson KN, Wright JF, Zhao L, Sebald SM, Greenspan DS, Lee SJ. Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc Natl Acad Sci USA. 2003;100:15842–15846. doi: 10.1073/pnas.2534946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, McPherron AC, Wolfman NM, Lee SJ. Induction of cachexia in mice by systemically administered myostatin. Science. 2002;296:1486–1488. doi: 10.1126/science.1069525. [DOI] [PubMed] [Google Scholar]
- Hill JJ, Davies MV, Pearson AA, Wang JH, Hewick RM, Wolfman NM, Qiu Y. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J Biol Chem. 2002;277:40735–40741. doi: 10.1074/jbc.M206379200. [DOI] [PubMed] [Google Scholar]
- Hill JJ, Qiu Y, Hewick RM, Wolfman NM. Regulation of myostatin in vivo by growth and differentiation factor-associated serum protein-1: a novel protein with protease inhibitor and follistatin domains. Mol Endocrinol. 2003;17:1144–1154. doi: 10.1210/me.2002-0366. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Reed LA, Davies MV, Girgenrath S, Goad ME, Tomkinson KN, Wright JF, Barker C, Ehrmantraut G, Holmstrom J. et al. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci USA. 2005;102:18117–18122. doi: 10.1073/pnas.0505996102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L. Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Mol Cell Biol. 2003;23:7230–7242. doi: 10.1128/MCB.23.20.7230-7242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Chen Y, Zhang Y, Wang X, Yang N, Zhu D. Extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase pathway is involved in myostatin-regulated differentiation repression. Cancer Res. 2006;66:1320–1326. doi: 10.1158/0008-5472.CAN-05-3060. [DOI] [PubMed] [Google Scholar]
- Huang Z, Chen D, Zhang K, Yu B, Chen X, Meng J. Regulation of myostatin signaling by c-Jun N-terminal kinase in C2C12 cells. Cell Signal. 2007;19:2286–2295. doi: 10.1016/j.cellsig.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Philip B, Lu Z, Gao Y. Regulation of GDF-8 signaling by the p38 MAPK. Cell Signal. 2005;17:365–375. doi: 10.1016/j.cellsig.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009;296:C1258–1270. doi: 10.1152/ajpcell.00105.2009. [DOI] [PubMed] [Google Scholar]
- Lee SJ. Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol. 2004;20:61–86. doi: 10.1146/annurev.cellbio.20.012103.135836. [DOI] [PubMed] [Google Scholar]
- McLennan IS. Localisation of transforming growth factor beta 1 in developing muscles: implications for connective tissue and fiber type pattern formation. Dev Dyn. 1993;197:281–290. doi: 10.1002/aja.1001970406. [DOI] [PubMed] [Google Scholar]
- Cusella-De Angelis MG, Molinari S, Le Donne A, Coletta M, Vivarelli E, Bouche M, Molinaro M, Ferrari S, Cossu G. Differential response of embryonic and fetal myoblasts to TGF beta: a possible regulatory mechanism of skeletal muscle histogenesis. Development. 1994;120:925–933. doi: 10.1242/dev.120.4.925. [DOI] [PubMed] [Google Scholar]
- Allen RE, Boxhorn LK. Inhibition of skeletal muscle satellite cell differentiation by transforming growth factor-beta. J Cell Physiol. 1987;133:567–572. doi: 10.1002/jcp.1041330319. [DOI] [PubMed] [Google Scholar]
- Li Y, Foster W, Deasy BM, Chan Y, Prisk V, Tang Y, Cummins J, Huard J. Transforming growth factor-beta1 induces the differentiation of myogenic cells into fibrotic cells in injured skeletal muscle: a key event in muscle fibrogenesis. Am J Pathol. 2004;164:1007–1019. doi: 10.1016/S0002-9440(10)63188-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson RJ, Williams DJ, Slack DN, Williamson J, Seternes OM, Keyse SM. Characterization of a murine gene encoding a developmentally regulated cytoplasmic dual-specificity mitogen-activated protein kinase phosphatase. Biochem J. 2002;364:145–155. doi: 10.1042/bj3640145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Woodring PJ, Bhakta KS, Tamura K, Wen F, Feramisco JR, Karin M, Wang JY, Puri PL. p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol Cell Biol. 2000;20:3951–3964. doi: 10.1128/MCB.20.11.3951-3964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V, Marquez VE, Valente S, Mai A, Forcales SV. et al. TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell. 2010;7:455–469. doi: 10.1016/j.stem.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriane M, Roux P, Primig M, Fort P, Gauthier-Rouviere C. Critical activities of Rac1 and Cdc42Hs in skeletal myogenesis: antagonistic effects of JNK and p38 pathways. Mol Biol Cell. 2000;11:2513–2528. doi: 10.1091/mbc.11.8.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett AM, Tonks NK. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science. 1997;278:1288–1291. doi: 10.1126/science.278.5341.1288. [DOI] [PubMed] [Google Scholar]
- Jones NC, Fedorov YV, Rosenthal RS, Olwin BB. ERK1/2 is required for myoblast proliferation but is dispensable for muscle gene expression and cell fusion. J Cell Physiol. 2001;186:104–115. doi: 10.1002/1097-4652(200101)186:1<104::AID-JCP1015>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Widegren U, Jiang XJ, Krook A, Chibalin AV, Bjornholm M, Tally M, Roth RA, Henriksson J, Wallberg-henriksson H, Zierath JR. Divergent effects of exercise on metabolic and mitogenic signaling pathways in human skeletal muscle. FASEB J. 1998;12:1379–1389. doi: 10.1096/fasebj.12.13.1379. [DOI] [PubMed] [Google Scholar]
- Williamson D, Gallagher P, Harber M, Hollon C, Trappe S. Mitogen-activated protein kinase (MAPK) pathway activation: effects of age and acute exercise on human skeletal muscle. J Physiol. 2003;547:977–987. doi: 10.1113/jphysiol.2002.036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- Grobet L, Martin LJ, Poncelet D, Pirottin D, Brouwers B, Riquet J, Schoeberlein A, Dunner S, Menissier F, Massabanda J. et al. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat Genet. 1997;17:71–74. doi: 10.1038/ng0997-71. [DOI] [PubMed] [Google Scholar]
- Kambadur R, Sharma M, Smith TP, Bass JJ. Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res. 1997;7:910–916. doi: 10.1101/gr.7.9.910. [DOI] [PubMed] [Google Scholar]
- Zhiliang G, Dahai Z, Ning L, Hui L, Xuemei D, Changxin W. The single nucleotide polymorphisms of the chicken myostatin gene are associated with skeletal muscle and adipose growth. Sci China C Life Sci. 2004;47:25–30. doi: 10.1360/02yc0201. [DOI] [PubMed] [Google Scholar]
- Shelton GD, Engvall E. Gross muscle hypertrophy in whippet dogs is caused by a mutation in the myostatin gene. Neuromuscul Disord. 2007;17:721–722. doi: 10.1016/j.nmd.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Mosher DS, Quignon P, Bustamante CD, Sutter NB, Mellersh CS, Parker HG, Ostrander EA. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 2007;3:e79. doi: 10.1371/journal.pgen.0030079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibe B, Bouix J, Caiment F, Elsen JM, Eychenne F. et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006;38:813–818. doi: 10.1038/ng1810. [DOI] [PubMed] [Google Scholar]
- Whittemore LA, Song K, Li X, Aghajanian J, Davies M, Girgenrath S, Hill JJ, Jalenak M, Kelley P, Knight A. et al. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem Biophys Res Commun. 2003;300:965–971. doi: 10.1016/S0006-291X(02)02953-4. [DOI] [PubMed] [Google Scholar]
- Haidet AM, Rizo L, Handy C, Umapathi P, Eagle A, Shilling C, Boue D, Martin PT, Sahenk Z, Mendell JR, Kaspar BK. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci USA. 2008;105:4318–4322. doi: 10.1073/pnas.0709144105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CM, Yang Z, Liu CW, Wang R, Tien P, Dale R, Sun LQ. Myostatin antisense RNA-mediated muscle growth in normal and cancer cachexia mice. Gene Ther. 2008;15:155–160. doi: 10.1038/sj.gt.3303016. [DOI] [PubMed] [Google Scholar]
- Murphy KT, Koopman R, Naim T, Leger B, Trieu J, Ibebunjo C, Lynch GS. Antibody-directed myostatin inhibition in 21-mo-old mice reveals novel roles for myostatin signaling in skeletal muscle structure and function. FASEB J. 2010;24:4433–4442. doi: 10.1096/fj.10-159608. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhao B, Kim YS, Hu CY, Yang J. Administration of a mutated myostatin propeptide to neonatal mice significantly enhances skeletal muscle growth. Mol Reprod Dev. 2010;77:76–82. doi: 10.1002/mrd.21111. [DOI] [PubMed] [Google Scholar]
- Cadena SM, Tomkinson KN, Monnell TE, Spaits MS, Kumar R, Underwood KW, Pearsall RS, Lachey JL. Administration of a soluble activin type IIB receptor promotes skeletal muscle growth independent of fiber type. J Appl Physiol. 2010;109:635–642. doi: 10.1152/japplphysiol.00866.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsakas A, Foster K, Otto A, Macharia R, Elashry MI, Feist S, Graham I, Foster H, Yaworsky P, Walsh F. et al. Molecular, cellular and physiological investigation of myostatin propeptide-mediated muscle growth in adult mice. Neuromuscul Disord. 2009;19:489–499. doi: 10.1016/j.nmd.2009.06.367. [DOI] [PubMed] [Google Scholar]
- Qiao C, Li J, Jiang J, Zhu X, Wang B, Xiao X. Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum Gene Ther. 2008;19:241–254. doi: 10.1089/hum.2007.159. [DOI] [PubMed] [Google Scholar]
- Hu S, Chen C, Sheng J, Sun Y, Cao X, Qiao J. Enhanced muscle growth by plasmid-mediated delivery of myostatin propeptide. J Biomed Biotechnol. 2010;2010:862591. doi: 10.1155/2010/862591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morine KJ, Bish LT, Pendrak K, Sleeper MM, Barton ER, Sweeney HL. Systemic myostatin inhibition via liver-targeted gene transfer in normal and dystrophic mice. PLoS One. 2010;5:e9176. doi: 10.1371/journal.pone.0009176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano AL, Munoz-Canoves P. Regulation and dysregulation of fibrosis in skeletal muscle. Exp Cell Res. 2010;316:3050–3058. doi: 10.1016/j.yexcr.2010.05.035. [DOI] [PubMed] [Google Scholar]
- Gosselin LE, McCormick KM. Targeting the immune system to improve ventilatory function in muscular dystrophy. Med Sci Sports Exerc. 2004;36:44–51. doi: 10.1249/01.MSS.0000106185.22349.2C. [DOI] [PubMed] [Google Scholar]
- Allen RE, Boxhorn LK. Regulation of skeletal muscle satellite cell proliferation and differentiation by transforming growth factor-beta, insulin-like growth factor I, and fibroblast growth factor. J Cell Physiol. 1989;138:311–315. doi: 10.1002/jcp.1041380213. [DOI] [PubMed] [Google Scholar]
- Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, Gamradt M, ap Rhys CM, Holm TM, Loeys BL. et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13:204–210. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurney CF, Sali A, Guerron AD, Iantorno M, Yu Q, Gordish-Dressman H, Rayavarapu S, van der Meulen J, Hoffman EP, Nagaraju K. Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx mice. J Cardiovasc Pharmacol Ther. 2011;16:87–95. doi: 10.1177/1074248410381757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedair HS, Karthikeyan T, Quintero A, Li Y, Huard J. Angiotensin II receptor blockade administered after injury improves muscle regeneration and decreases fibrosis in normal skeletal muscle. Am J Sports Med. 2008;36:1548–1554. doi: 10.1177/0363546508315470. [DOI] [PubMed] [Google Scholar]
- Taniguti AP, Pertille A, Matsumura CY, Santo Neto H, Marques MJ. Prevention of muscle fibrosis and myonecrosis in mdx mice by suramin, a TGF-beta1 blocker. Muscle Nerve. 2011;43:82–87. doi: 10.1002/mus.21869. [DOI] [PubMed] [Google Scholar]
- Nozaki M, Li Y, Zhu J, Ambrosio F, Uehara K, Fu FH, Huard J. Improved muscle healing after contusion injury by the inhibitory effect of suramin on myostatin, a negative regulator of muscle growth. Am J Sports Med. 2008;36:2354–2362. doi: 10.1177/0363546508322886. [DOI] [PubMed] [Google Scholar]
- Chan YS, Li Y, Foster W, Horaguchi T, Somogyi G, Fu FH, Huard J. Antifibrotic effects of suramin in injured skeletal muscle after laceration. J Appl Physiol. 2003;95:771–780. doi: 10.1152/japplphysiol.00915.2002. [DOI] [PubMed] [Google Scholar]
- Chan YS, Li Y, Foster W, Fu FH, Huard J. The use of suramin, an antifibrotic agent, to improve muscle recovery after strain injury. Am J Sports Med. 2005;33:43–51. doi: 10.1177/0363546504265190. [DOI] [PubMed] [Google Scholar]
- Fukushima K, Badlani N, Usas A, Riano F, Fu F, Huard J. The use of an antifibrosis agent to improve muscle recovery after laceration. Am J Sports Med. 2001;29:394–402. doi: 10.1177/03635465010290040201. [DOI] [PubMed] [Google Scholar]
- Gosselin LE, Williams JE, Deering M, Brazeau D, Koury S, Martinez DA. Localization and early time course of TGF-beta 1 mRNA expression in dystrophic muscle. Muscle Nerve. 2004;30:645–653. doi: 10.1002/mus.20150. [DOI] [PubMed] [Google Scholar]
- Foster W, Li Y, Usas A, Somogyi G, Huard J. Gamma interferon as an antifibrosis agent in skeletal muscle. J Orthop Res. 2003;21:798–804. doi: 10.1016/S0736-0266(03)00059-7. [DOI] [PubMed] [Google Scholar]
- Van Erp C, Irwin NG, Hoey AJ. Long-term administration of pirfenidone improves cardiac function in mdx mice. Muscle Nerve. 2006;34:327–334. doi: 10.1002/mus.20590. [DOI] [PubMed] [Google Scholar]
- Gosselin LE, Williams JE, Personius K, Farkas GA. A comparison of factors associated with collagen metabolism in different skeletal muscles from dystrophic (mdx) mice: impact of pirfenidone. Muscle Nerve. 2007;35:208–216. doi: 10.1002/mus.20681. [DOI] [PubMed] [Google Scholar]
- Turgeman T, Hagai Y, Huebner K, Jassal DS, Anderson JE, Genin O, Nagler A, Halevy O, Pines M. Prevention of muscle fibrosis and improvement in muscle performance in the mdx mouse by halofuginone. Neuromuscul Disord. 2008;18:857–868. doi: 10.1016/j.nmd.2008.06.386. [DOI] [PubMed] [Google Scholar]
- Huebner KD, Jassal DS, Halevy O, Pines M, Anderson JE. Functional resolution of fibrosis in mdx mouse dystrophic heart and skeletal muscle by halofuginone. Am J Physiol Heart Circ Physiol. 2008;294:H1550–1561. doi: 10.1152/ajpheart.01253.2007. [DOI] [PubMed] [Google Scholar]
- Nevo Y, Halevy O, Genin O, Moshe I, Turgeman T, Harel M, Biton E, Reif S, Pines M. Fibrosis inhibition and muscle histopathology improvement in laminin-alpha2-deficient mice. Muscle Nerve. 2010;42:218–229. doi: 10.1002/mus.21706. [DOI] [PubMed] [Google Scholar]
- Andreetta F, Bernasconi P, Baggi F, Ferro P, Oliva L, Arnoldi E, Cornelio F, Mantegazza R, Confalonieri P. Immunomodulation of TGF-beta 1 in mdx mouse inhibits connective tissue proliferation in diaphragm but increases inflammatory response: implications for antifibrotic therapy. J Neuroimmunol. 2006;175:77–86. doi: 10.1016/j.jneuroim.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Carlson ME, Conboy MJ, Hsu M, Barchas L, Jeong J, Agrawal A, Mikels AJ, Agrawal S, Schaffer DV, Conboy IM. Relative roles of TGF-beta1 and Wnt in the systemic regulation and aging of satellite cell responses. Aging Cell. 2009;8:676–689. doi: 10.1111/j.1474-9726.2009.00517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCroskery S, Thomas M, Platt L, Hennebry A, Nishimura T, McLeay L, Sharma M, Kambadur R. Improved muscle healing through enhanced regeneration and reduced fibrosis in myostatin-null mice. J Cell Sci. 2005;118:3531–3541. doi: 10.1242/jcs.02482. [DOI] [PubMed] [Google Scholar]
- Zhu J, Li Y, Shen W, Qiao C, Ambrosio F, Lavasani M, Nozaki M, Branca MF, Huard J. Relationships between transforming growth factor-beta1, myostatin, and decorin: implications for skeletal muscle fibrosis. J Biol Chem. 2007;282:25852–25863. doi: 10.1074/jbc.M704146200. [DOI] [PubMed] [Google Scholar]
- Li ZB, Kollias HD, Wagner KR. Myostatin directly regulates skeletal muscle fibrosis. J Biol Chem. 2008;283:19371–19378. doi: 10.1074/jbc.M802585200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R. Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol. 2003;162:1135–1147. doi: 10.1083/jcb.200207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnac G, Vernus B, Bonnieu A. Myostatin in the pathophysiology of skeletal muscle. Curr Genomics. 2007;8:415–422. doi: 10.2174/138920207783591672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002;277:49831–49840. doi: 10.1074/jbc.M204291200. [DOI] [PubMed] [Google Scholar]
- Joulia D, Bernardi H, Garandel V, Rabenoelina F, Vernus B, Cabello G. Mechanisms involved in the inhibition of myoblast proliferation and differentiation by myostatin. Exp Cell Res. 2003;286:263–275. doi: 10.1016/S0014-4827(03)00074-0. [DOI] [PubMed] [Google Scholar]
- Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, Ahima RS, Khurana TS. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- Murphy KT, Ryall JG, Snell SM, Nair L, Koopman R, Krasney PA, Ibebunjo C, Holden KS, Loria PM, Salatto CT, Lynch GS. Antibody-directed myostatin inhibition improves diaphragm pathology in young but not adult dystrophic mdx mice. Am J Pathol. 2010;176:2425–2434. doi: 10.2353/ajpath.2010.090932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanovich S, McNally EM, Khurana TS. Myostatin blockade improves function but not histopathology in a murine model of limb-girdle muscular dystrophy 2C. Muscle Nerve. 2008;37:308–316. doi: 10.1002/mus.20920. [DOI] [PubMed] [Google Scholar]
- Parsons SA, Millay DP, Sargent MA, McNally EM, Molkentin JD. Age-dependent effect of myostatin blockade on disease severity in a murine model of limb-girdle muscular dystrophy. Am J Pathol. 2006;168:1975–1985. doi: 10.2353/ajpath.2006.051316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzbaur EL, Howland DS, Weber N, Wallace K, She Y, Kwak S, Tchistiakova LA, Murphy E, Hinson J, Karim R. et al. Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis. Neurobiol Dis. 2006;23:697–707. doi: 10.1016/j.nbd.2006.05.009. [DOI] [PubMed] [Google Scholar]
- LeBrasseur NK, Schelhorn TM, Bernardo BL, Cosgrove PG, Loria PM, Brown TA. Myostatin inhibition enhances the effects of exercise on performance and metabolic outcomes in aged mice. J Gerontol A Biol Sci Med Sci. 2009;64:940–948. doi: 10.1093/gerona/glp068. [DOI] [PubMed] [Google Scholar]
- Morine KJ, Bish LT, Selsby JT, Gazzara JA, Pendrak K, Sleeper MM, Barton ER, Lee SJ, Sweeney HL. Activin IIB receptor blockade attenuates dystrophic pathology in a mouse model of Duchenne muscular dystrophy. Muscle Nerve. 2010;42:722–730. doi: 10.1002/mus.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa Y, Hagiwara H, Nakatani M, Yasue A, Moriyama K, Murakami T, Tsuchida K, Noji S, Sunada Y. Muscular atrophy of caveolin-3-deficient mice is rescued by myostatin inhibition. J Clin Invest. 2006;116:2924–2934. doi: 10.1172/JCI28520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner CJ, Wee CD, Warsing LC, Choe DW, Ng AS, Lutz C, Wagner KR. Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice. Hum Mol Genet. 2009;18:3145–3152. doi: 10.1093/hmg/ddp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BM, Lachey JL, Warsing LC, Ting BL, Pullen AE, Underwood KW, Kumar R, Sako D, Grinberg A, Wong V. et al. A soluble activin type IIB receptor improves function in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2009;217:258–268. doi: 10.1016/j.expneurol.2009.02.017. [DOI] [PubMed] [Google Scholar]
- Benny Klimek ME, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA. Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun. 2010;391:1548–1554. doi: 10.1016/j.bbrc.2009.12.123. [DOI] [PubMed] [Google Scholar]
- Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS. et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142:531–543. doi: 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]
- Bogdanovich S, Perkins KJ, Krag TO, Whittemore LA, Khurana TS. Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J. 2005;19:543–549. doi: 10.1096/fj.04-2796com. [DOI] [PubMed] [Google Scholar]
- Bartoli M, Poupiot J, Vulin A, Fougerousse F, Arandel L, Daniele N, Roudaut C, Noulet F, Garcia L, Danos O, Richard I. AAV-mediated delivery of a mutated myostatin propeptide ameliorates calpain 3 but not alpha-sarcoglycan deficiency. Gene Ther. 2007;14:733–740. doi: 10.1038/sj.gt.3302928. [DOI] [PubMed] [Google Scholar]
- Hamrick MW, Arounleut P, Kellum E, Cain M, Immel D, Liang LF. Recombinant myostatin (GDF-8) propeptide enhances the repair and regeneration of both muscle and bone in a model of deep penetrant musculoskeletal injury. J Trauma. 2010;69:579–583. doi: 10.1097/TA.0b013e3181c451f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose FF Jr, Mattis VB, Rindt H, Lorson CL. Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum Mol Genet. 2009;18:997–1005. doi: 10.1093/hmg/ddn426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, Kim SH, Yamanaka K, Hester M, Umapathi P, Arnson H, Rizo L, Mendell JR, Gage FH, Cleveland DW, Kaspar BK. Gene transfer demonstrates that muscle is not a primary target for non-cell-autonomous toxicity in familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2006;103:19546–19551. doi: 10.1073/pnas.0609411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minetti GC, Colussi C, Adami R, Serra C, Mozzetta C, Parente V, Fortuni S, Straino S, Sampaolesi M, Di Padova M. et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. 2006;12:1147–1150. doi: 10.1038/nm1479. [DOI] [PubMed] [Google Scholar]
- Bonetto A, Penna F, Minero VG, Reffo P, Bonelli G, Baccino FM, Costelli P. Deacetylase inhibitors modulate the myostatin/follistatin axis without improving cachexia in tumor-bearing mice. Curr Cancer Drug Targets. 2009;9:608–616. doi: 10.2174/156800909789057015. [DOI] [PubMed] [Google Scholar]
- Iezzi S, Di Padova M, Serra C, Caretti G, Simone C, Maklan E, Minetti G, Zhao P, Hoffman EP, Puri PL, Sartorelli V. Deacetylase inhibitors increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin. Dev Cell. 2004;6:673–684. doi: 10.1016/S1534-5807(04)00107-8. [DOI] [PubMed] [Google Scholar]
- Siriett V, Platt L, Salerno MS, Ling N, Kambadur R, Sharma M. Prolonged absence of myostatin reduces sarcopenia. J Cell Physiol. 2006;209:866–873. doi: 10.1002/jcp.20778. [DOI] [PubMed] [Google Scholar]
- Murphy KT, Cobani V, Ryall JG, Ibebunjo C, Lynch GS. Acute antibody-directed myostatin inhibition attenuates disuse muscle atrophy and weakness in mice. J Appl Physiol. 2011. [DOI] [PubMed]
- Pistilli EE, Bogdanovich S, Goncalves MD, Ahima RS, Lachey J, Seehra J, Khurana T. Targeting the activin type IIB Receptor to improve muscle mass and function in the mdx mouse model of Duchenne muscular dystrophy. Am J Pathol. 2011;178:1287–1297. doi: 10.1016/j.ajpath.2010.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millino C, Fanin M, Vettori A, Laveder P, Mostacciuolo ML, Angelini C, Lanfranchi G. Different atrophy-hypertrophy transcription pathways in muscles affected by severe and mild spinal muscular atrophy. BMC Med. 2009;7:14. doi: 10.1186/1741-7015-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Minamiyama M, Waza M, Doi H, Kondo N, Mizoguchi H, Nitta A, Yamada K, Banno H. et al. Disrupted transforming growth factor-beta signaling in spinal and bulbar muscular atrophy. J Neurosci. 2010;30:5702–5712. doi: 10.1523/JNEUROSCI.0388-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM. et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- Morgan JE, Zammit PS. Direct effects of the pathogenic mutation on satellite cell function in muscular dystrophy. Exp Cell Res. 2010;316:3100–3108. doi: 10.1016/j.yexcr.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23:1456–1471. doi: 10.1002/1097-4598(200010)23:10<1456::AID-MUS2>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Bernasconi P, Di Blasi C, Mora M, Morandi L, Galbiati S, Confalonieri P, Cornelio F, Mantegazza R. Transforming growth factor-beta1 and fibrosis in congenital muscular dystrophies. Neuromuscul Disord. 1999;9:28–33. doi: 10.1016/S0960-8966(98)00093-5. [DOI] [PubMed] [Google Scholar]
- Ishitobi M, Haginoya K, Zhao Y, Ohnuma A, Minato J, Yanagisawa T, Tanabu M, Kikuchi M, Iinuma K. Elevated plasma levels of transforming growth factor beta1 in patients with muscular dystrophy. Neuroreport. 2000;11:4033–4035. doi: 10.1097/00001756-200012180-00026. [DOI] [PubMed] [Google Scholar]
- Passerini L, Bernasconi P, Baggi F, Confalonieri P, Cozzi F, Cornelio F, Mantegazza R. Fibrogenic cytokines and extent of fibrosis in muscle of dogs with X-linked golden retriever muscular dystrophy. Neuromuscul Disord. 2002;12:828–835. doi: 10.1016/S0960-8966(02)00071-8. [DOI] [PubMed] [Google Scholar]
- Zanotti S, Negri T, Cappelletti C, Bernasconi P, Canioni E, Di Blasi C, Pegoraro E, Angelini C, Ciscato P, Prelle A. et al. Decorin and biglycan expression is differentially altered in several muscular dystrophies. Brain. 2005;128:2546–2555. doi: 10.1093/brain/awh635. [DOI] [PubMed] [Google Scholar]
- Zanotti S, Saredi S, Ruggieri A, Fabbri M, Blasevich F, Romaggi S, Morandi L, Mora M. Altered extracellular matrix transcript expression and protein modulation in primary Duchenne muscular dystrophy myotubes. Matrix Biol. 2007;26:615–624. doi: 10.1016/j.matbio.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Muchir A, Pavlidis P, Bonne G, Hayashi YK, Worman HJ. Activation of MAPK in hearts of EMD null mice: similarities between mouse models of X-linked and autosomal dominant Emery Dreifuss muscular dystrophy. Hum Mol Genet. 2007;16:1884–1895. doi: 10.1093/hmg/ddm137. [DOI] [PubMed] [Google Scholar]
- Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, Worman HJ. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest. 2007;117:1282–1293. doi: 10.1172/JCI29042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin MA, Feng H, Tewari M, Acosta P, Kawana M, Sweeney HL, Discher DE. gamma-Sarcoglycan deficiency increases cell contractility, apoptosis and MAPK pathway activation but does not affect adhesion. J Cell Sci. 2005;118:1405–1416. doi: 10.1242/jcs.01717. [DOI] [PubMed] [Google Scholar]
- Woodman SE, Park DS, Cohen AW, Cheung MW, Chandra M, Shirani J, Tang B, Jelicks LA, Kitsis RN, Christ GJ. et al. Caveolin-3 knock-out mice develop a progressive cardiomyopathy and show hyperactivation of the p42/44 MAPK cascade. J Biol Chem. 2002;277:38988–38997. doi: 10.1074/jbc.M205511200. [DOI] [PubMed] [Google Scholar]
- Markert CD, Meaney MP, Voelker KA, Grange RW, Dalley HW, Cann JK, Ahmed M, Bishwokarma B, Walker SJ, Yu SX. et al. Functional muscle analysis of the Tcap knockout mouse. Hum Mol Genet. 2010;19:2268–2283. doi: 10.1093/hmg/ddq105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YW, Nagaraju K, Bakay M, McIntyre O, Rawat R, Shi R, Hoffman EP. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology. 2005;65:826–834. doi: 10.1212/01.wnl.0000173836.09176.c4. [DOI] [PubMed] [Google Scholar]
- Porter JD, Khanna S, Kaminski HJ, Rao JS, Merriam AP, Richmonds CR, Leahy P, Li J, Guo W, Andrade FH. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum Mol Genet. 2002;11:263–272. doi: 10.1093/hmg/11.3.263. [DOI] [PubMed] [Google Scholar]
- Vetrone SA, Montecino-Rodriguez E, Kudryashova E, Kramerova I, Hoffman EP, Liu SD, Miceli MC, Spencer MJ. Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-beta. J Clin Invest. 2009;119:1583–1594. doi: 10.1172/JCI37662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti S, Gibertini S, Mora M. Altered production of extra-cellular matrix components by muscle-derived Duchenne muscular dystrophy fibroblasts before and after TGF-beta1 treatment. Cell Tissue Res. 2010;339:397–410. doi: 10.1007/s00441-009-0889-4. [DOI] [PubMed] [Google Scholar]
- Sun G, Haginoya K, Wu Y, Chiba Y, Nakanishi T, Onuma A, Sato Y, Takigawa M, Iinuma K, Tsuchiya S. Connective tissue growth factor is overexpressed in muscles of human muscular dystrophy. J Neurol Sci. 2008;267:48–56. doi: 10.1016/j.jns.2007.09.043. [DOI] [PubMed] [Google Scholar]
- Kumar A, Khandelwal N, Malya R, Reid MB, Boriek AM. Loss of dystrophin causes aberrant mechanotransduction in skeletal muscle fibers. FASEB J. 2004;18:102–113. doi: 10.1096/fj.03-0453com. [DOI] [PubMed] [Google Scholar]
- Kolodziejczyk SM, Walsh GS, Balazsi K, Seale P, Sandoz J, Hierlihy AM, Rudnicki MA, Chamberlain JS, Miller FD, Megeney LA. Activation of JNK1 contributes to dystrophic muscle pathogenesis. Curr Biol. 2001;11:1278–1282. doi: 10.1016/S0960-9822(01)00397-9. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Harrod GV, Davies KE. Activation of calcineurin and stress activated protein kinase/p38-mitogen activated protein kinase in hearts of utrophin-dystrophin knockout mice. Neuromuscul Disord. 2001;11:251–259. doi: 10.1016/S0960-8966(00)00201-7. [DOI] [PubMed] [Google Scholar]
- Vidal B, Serrano AL, Tjwa M, Suelves M, Ardite E, De Mori R, Baeza-Raja B, Martinez de Lagran M, Lafuste P, Ruiz-Bonilla V. et al. Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev. 2008;22:1747–1752. doi: 10.1101/gad.465908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JA, Kelly SM, Lopresti PP, Heydemann A, Earley JU, Ferguson EL, Wolf MJ, McNally EM. SMAD signaling drives heart and muscle dysfunction in a Drosophila model of muscular dystrophy. Hum Mol Genet. 2010. [DOI] [PMC free article] [PubMed]
- Hayot M, Rodriguez J, Vernus B, Carnac G, Jean E, Allen D, Goret L, Obert P, Candau R, Bonnieu A. Myostatin up-regulation is associated with the skeletal muscle response to hypoxic stimuli. Mol Cell Endocrinol. 2011;332:38–47. doi: 10.1016/j.mce.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Lalani R, Bhasin S, Byhower F, Tarnuzzer R, Grant M, Shen R, Asa S, Ezzat S, Gonzalez-Cadavid NF. Myostatin and insulin-like growth factor-I and -II expression in the muscle of rats exposed to the microgravity environment of the NeuroLab space shuttle flight. J Endocrinol. 2000;167:417–428. doi: 10.1677/joe.0.1670417. [DOI] [PubMed] [Google Scholar]
- Allen DL, Cleary AS, Lindsay SF, Loh AS, Reed JM. Myostatin expression is increased by food deprivation in a muscle-specific manner and contributes to muscle atrophy during prolonged food deprivation in mice. J Appl Physiol. 2010;109:692–701. doi: 10.1152/japplphysiol.00504.2010. [DOI] [PubMed] [Google Scholar]
- Allen DL, McCall GE, Loh AS, Madden MC, Mehan RS. Acute daily psychological stress causes increased atrophic gene expression and myostatin-dependent muscle atrophy. Am J Physiol Regul Integr Comp Physiol. 2010;299:R889–898. doi: 10.1152/ajpregu.00296.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling M, Cai B, Tidball JG. Modulation of myostatin expression during modified muscle use. FASEB J. 2000;14:103–110. doi: 10.1096/fasebj.14.1.103. [DOI] [PubMed] [Google Scholar]
- Heinemeier KM, Olesen JL, Haddad F, Schjerling P, Baldwin KM, Kjaer M. Effect of unloading followed by reloading on expression of collagen and related growth factors in rat tendon and muscle. J Appl Physiol. 2009;106:178–186. doi: 10.1152/japplphysiol.91092.2008. [DOI] [PubMed] [Google Scholar]
- Jones SW, Hill RJ, Krasney PA, O'Conner B, Peirce N, Greenhaff PL. Disuse atrophy and exercise rehabilitation in humans profoundly affects the expression of genes associated with the regulation of skeletal muscle mass. FASEB J. 2004;18:1025–1027. doi: 10.1096/fj.03-1228fje. [DOI] [PubMed] [Google Scholar]
- Kim J, Won KJ, Lee HM, Hwang BY, Bae YM, Choi WS, Song H, Lim KW, Lee CK, Kim B. p38 MAPK participates in muscle-specific RING finger 1-Mediated atrophy in cast-immobilized rat gastrocnemius muscle. Korean J Physiol Pharmacol. 2009;13:491–496. doi: 10.4196/kjpp.2009.13.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon KA, Davis J, Kapsa RM, Choong P, Byrne E. Myostatin, insulin-like growth factor-1, and leukemia inhibitory factor mRNAs are upregulated in chronic human disuse muscle atrophy. Muscle Nerve. 2001;24:893–899. doi: 10.1002/mus.1086. [DOI] [PubMed] [Google Scholar]
- Hirose T, Nakazato K, Song H, Ishii N. TGF-beta1 and TNF-alpha are involved in the transcription of type I collagen alpha2 gene in soleus muscle atrophied by mechanical unloading. J Appl Physiol. 2008;104:170–177. doi: 10.1152/japplphysiol.00463.2006. [DOI] [PubMed] [Google Scholar]
- Machida S, Booth FW. Changes in signalling molecule levels in 10-day hindlimb immobilized rat muscles. Acta Physiol Scand. 2005;183:171–179. doi: 10.1111/j.1365-201X.2004.01395.x. [DOI] [PubMed] [Google Scholar]
- Carlson CJ, Booth FW, Gordon SE. Skeletal muscle myostatin mRNA expression is fiber-type specific and increases during hindlimb unloading. Am J Physiol. 1999;277:R601–606. doi: 10.1152/ajpregu.1999.277.2.r601. [DOI] [PubMed] [Google Scholar]
- Costelli P, Muscaritoli M, Bonetto A, Penna F, Reffo P, Bossola M, Bonelli G, Doglietto GB, Baccino FM, Rossi Fanelli F. Muscle myostatin signalling is enhanced in experimental cancer cachexia. Eur J Clin Invest. 2008;38:531–538. doi: 10.1111/j.1365-2362.2008.01970.x. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Miyazaki H, Takeda Y, Takeo S. Detection of serum cytokine levels in experimental cancer cachexia of colon 26 adenocarcinoma-bearing mice. Cancer Lett. 1993;72:65–70. doi: 10.1016/0304-3835(93)90012-X. [DOI] [PubMed] [Google Scholar]
- Houi K, Kobayashi T, Kato S, Mochio S, Inoue K. Increased plasma TGF-beta1 in patients with amyotrophic lateral sclerosis. Acta Neurol Scand. 2002;106:299–301. doi: 10.1034/j.1600-0404.2002.01301.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S, Shen R, Lalani R, Asa S, Mamita M. et al. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci USA. 1998;95:14938–14943. doi: 10.1073/pnas.95.25.14938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Mallidis C, Bhasin S, Mahabadi V, Artaza J, Gonzalez-Cadavid N, Arias J, Salehian B. Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am J Physiol Endocrinol Metab. 2003;285:E363–371. doi: 10.1152/ajpendo.00487.2002. [DOI] [PubMed] [Google Scholar]
- Roubenoff R. Sarcopenia and its implications for the elderly. Eur J Clin Nutr. 2000;54(Suppl 3):S40–47. doi: 10.1038/sj.ejcn.1601024. [DOI] [PubMed] [Google Scholar]
- Machida S, Narusawa M. The roles of satellite cells and hematopoietic stem cells in impaired regeneration of skeletal muscle in old rats. Ann N Y Acad Sci. 2006;1067:349–353. doi: 10.1196/annals.1354.049. [DOI] [PubMed] [Google Scholar]
- Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454:528–532. doi: 10.1038/nature07034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson ME, Suetta C, Conboy MJ, Aagaard P, Mackey A, Kjaer M, Conboy I. Molecular aging and rejuvenation of human muscle stem cells. EMBO Mol Med. 2009;1:381–391. doi: 10.1002/emmm.200900045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger B, Derave W, De Bock K, Hespel P, Russell AP. Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res. 2008;11:163–175B. doi: 10.1089/rej.2007.0588. [DOI] [PubMed] [Google Scholar]
- Puthucheary Z, Harridge S, Hart N. Skeletal muscle dysfunction in critical care: wasting, weakness, and rehabilitation strategies. Crit Care Med. 2010;38:S676–682. doi: 10.1097/CCM.0b013e3181f2458d. [DOI] [PubMed] [Google Scholar]
- Di Giovanni S, Molon A, Broccolini A, Melcon G, Mirabella M, Hoffman EP, Servidei S. Constitutive activation of MAPK cascade in acute quadriplegic myopathy. Ann Neurol. 2004;55:195–206. doi: 10.1002/ana.10811. [DOI] [PubMed] [Google Scholar]
- Lavoie P, Robitaille G, Agharazii M, Ledbetter S, Lebel M, Lariviere R. Neutralization of transforming growth factor-beta attenuates hypertension and prevents renal injury in uremic rats. J Hypertens. 2005;23:1895–1903. doi: 10.1097/01.hjh.0000182521.44440.c5. [DOI] [PubMed] [Google Scholar]
- Lim DS, Lutucuta S, Bachireddy P, Youker K, Evans A, Entman M, Roberts R, Marian AJ. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103:789–791. doi: 10.1161/01.cir.103.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Vita J, Sanchez-Lopez E, Esteban V, Ruperez M, Egido J, Ruiz-Ortega M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation. 2005;111:2509–2517. doi: 10.1161/01.CIR.0000165133.84978.E2. [DOI] [PubMed] [Google Scholar]
- Hirata A, Masuda S, Tamura T, Kai K, Ojima K, Fukase A, Motoyoshi K, Kamakura K, Miyagoe-Suzuki Y, Takeda S. Expression profiling of cytokines and related genes in regenerating skeletal muscle after cardiotoxin injection: a role for osteopontin. Am J Pathol. 2003;163:203–215. doi: 10.1016/S0002-9440(10)63644-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira RO, Carvalho SN, Stumbo AC, Rodrigues CA, Porto LC, Moura AS, Carvalho L. Osteopontin expression in coculture of differentiating rat fetal skeletal fibroblasts and myoblasts. In Vitro Cell Dev Biol Anim. 2006;42:4–7. doi: 10.1007/s11626-006-0003-0. [DOI] [PubMed] [Google Scholar]
- Uaesoontrachoon K, Yoo HJ, Tudor EM, Pike RN, Mackie EJ, Pagel CN. Osteopontin and skeletal muscle myoblasts: association with muscle regeneration and regulation of myoblast function in vitro. Int J Biochem Cell Biol. 2008;40:2303–2314. doi: 10.1016/j.biocel.2008.03.020. [DOI] [PubMed] [Google Scholar]
- Wagner KR, McPherron AC, Winik N, Lee SJ. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002;52:832–836. doi: 10.1002/ana.10385. [DOI] [PubMed] [Google Scholar]
- Gilson H, Schakman O, Combaret L, Lause P, Grobet L, Attaix D, Ketelslegers JM, Thissen JP. Myostatin gene deletion prevents glucocorticoid-induced muscle atrophy. Endocrinology. 2007;148:452–460. doi: 10.1210/en.2006-0539. [DOI] [PubMed] [Google Scholar]
- Li ZF, Shelton GD, Engvall E. Elimination of myostatin does not combat muscular dystrophy in dy mice but increases postnatal lethality. Am J Pathol. 2005;166:491–497. doi: 10.1016/S0002-9440(10)62271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon CD, Popovic L, Oldham JM, Jeanplong F, Smith HK, Kambadur R, Sharma M, Maxwell L, Bass JJ. Myostatin-deficient mice lose more skeletal muscle mass than wild-type controls during hindlimb suspension. Am J Physiol Endocrinol Metab. 2003;285:E82–87. doi: 10.1152/ajpendo.00275.2002. [DOI] [PubMed] [Google Scholar]
- Amthor H, Macharia R, Navarrete R, Schuelke M, Brown SC, Otto A, Voit T, Muntoni F, Vrbova G, Partridge T. et al. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc Natl Acad Sci USA. 2007;104:1835–1840. doi: 10.1073/pnas.0604893104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry BA, Ferreira JA, Phillips CL, Brown M. Hindlimb skeletal muscle function in myostatin-deficient mice. Muscle Nerve. 2011;43:49–57. doi: 10.1002/mus.21796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendias CL, Bakhurin KI, Faulkner JA. Tendons of myostatin-deficient mice are small, brittle, and hypocellular. Proc Natl Acad Sci USA. 2008;105:388–393. doi: 10.1073/pnas.0707069105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna F, Costamagna D, Fanzani A, Bonelli G, Baccino FM, Costelli P. Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS One. 2010;5:e13604. doi: 10.1371/journal.pone.0013604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Shan J, Bonne G, Worman HJ, Muchir A. Pharmacological inhibition of c-Jun N-terminal kinase signaling prevents cardiomyopathy caused by mutation in LMNA gene. Biochim Biophys Acta. 2010;1802:632–638. doi: 10.1016/j.bbadis.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchir A, Shan J, Bonne G, Lehnart SE, Worman HJ. Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet. 2009;18:241–247. doi: 10.1093/hmg/ddn343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Muchir A, Shan J, Bonne G, Worman HJ. Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation. 2011;123:53–61. doi: 10.1161/CIRCULATIONAHA.110.970673. [DOI] [PMC free article] [PubMed] [Google Scholar]