

Abstract
Purpose
VEGFR-2 plays a crucial role in mediating angiogenic endothelial cell responses via the VEGF pathway and angiogenesis inhibitors targeting VEGFR-2 are in clinical use. As angiogenesis is a host-driven process, functional heritable variation in KDR, the gene encoding VEGFR-2, may affect VEGFR-2 function, and ultimately, the extent of tumor angiogenesis.
Experimental Design
We resequenced KDR using 24 DNAs each from healthy Caucasian, African American and Asian groups. Non-synonymous genetic variants were assessed for function using phosphorylation assays. Luciferase reporter gene assays were used to examine effects of variants on gene expression. KDR mRNA and protein expression, and microvessel density (MVD) were measured in non-small cell lung cancer (NSCLC) tumor samples and matching patient DNA samples were genotyped to test for associations with variants of interest.
Results
KDR resequencing led to the discovery of 120 genetic variants, of which 25 had not been previously reported. Q472H had increased VEGFR-2 protein phosphorylation and associated with increased MVD in NSCLC tumor samples. −2854C and −2455A increased luciferase expression and associated with higher KDR mRNA levels in NSCLC samples. −271A reduced luciferase expression and associated with lower VEGFR-2 levels in NSCLC samples. −906C and 23408G, associated with higher KDR mRNA levels in NSCLC samples.
Conclusions
This study has defined KDR genetic variation in three populations and identified common variants that impact on tumoral KDR expression and vascularization. These findings may have important implications for understanding the molecular basis of genetic associations between KDR variation and clinical phenotypes related to VEGFR-2 function.
Keywords: angiogenesis, KDR, resequencing, gene expression, NSCLC, functional validation
Introduction
VEGFR-2 (encoded by kinase insert domain receptor, KDR) is an important factor in tumor development and progression due to its pro-angiogenic effects (reviewed in (1)). VEGFR-2 levels correlate with tumor growth rate, microvessel density (MVD), proliferation, and tumor metastatic potential in several cancers (2, 3). Experimental blockage of the interaction between VEGF-A and VEGFR-2 inhibits tumor growth and metastasis (4). Indeed, angiogenesis inhibitors targeting the tyrosine kinase activity of VEGFR-2 are an effective class of anti-cancer drugs (5, 6). Although small molecule angiogenesis inhibitors interact with several targets, they are all characterized by a strong inhibitory activity of VEGFR-2 (7–9). In addition, the in vivo activity of one of them, sorafenib, is primarily mediated through VEGFR-2-mediated inhibition of angiogenesis (10).
Since gene expression in humans is in part determined by genetic factors (11) and angiogenesis is a host-mediated process (12), functional germline variation in KDR may contribute to variability in tumor endothelial function and, consequently, may affect cancer prognosis and the efficacy of VEGFR-2 inhibitors. Indeed, baseline soluble VEGFR-2 levels have been associated with a reduction in tumor size and clinical benefit in response to sunitinib treatment (13). Although no molecular marker is currently used to guide anti-angiogenesis therapy (14), the efforts towards identifying genetic prognostic biomarkers and predictive markers of response to VEGFR-2 inhibitors could be highly informed by studies characterizing KDR genetic variants. Furthermore, limited data are available on KDR germline genetic variation in the population, its ethnic differences and its effect on gene function or tumoral expression. Hence, in this study, we defined common germline KDR variation in different ethnic groups and assessed the phenotypic impact of putative functional variants. These aims were achieved by resequencing healthy Caucasian, African American and Asian individuals; performing bioinformatic and in vitro functional analyses; and finally, rationally selecting variants to test their association with KDR expression and microvessel density (MVD) in non-small cell lung cancer (NSCLC) tumor specimens.
Materials and Methods
KDR resequencing
Twenty-four germline DNA samples each from healthy Caucasians, Asian-Chinese, African-Americans obtained from the Coriell Institute Human Variation Collection * were chosen for resequencing. All 30 exons were sequenced (Figure 1). In addition, sequenced non-coding regions comprised flanking intronic sequences, promoter and the 5’-upstream region containing, evolutionarily conserved non-coding genomic regions (determined by comparative genomics using the UCSC genome browser – Supplementary Table S1), and regions determined to contain transcription factor binding sites according to computational prediction using Cluster-Buster†- Supplementary Table S2) (15). Primers used for PCR amplification are listed as in Supplementary Table S3. PCR reactions were set up using forward and reverse primers, HotStar DNA polymerase (Qiagen, Hilden, Germany) and 10 ng of DNA. After initial 15 min of activation at 95°C, touch down cycles were performed at: 95°C for 30 s, touch down annealing from 65°C to 54.5°C (−1.5°C per cycle) for 30 s and 72°C for 1.5 min for 7 cycles, following the standard cycle of 95°C for 30 s, 55°C annealing for 30 s, and 72°C for 1.5 min for 30 cycles. PCR products were purified using the MultiScreen PCR Purification Kit (Millipore, Billerica, MA) and eluted in 30 µl elution buffer. DNA sequencing was performed at University of Chicago DNA Sequencing Core Facility by Sanger dye-terminator sequencing. Sequence analysis was performed using Sequencher (Gene Codes, Ann Arbor, MI), Version 4.7.
Figure 1. Genomic structure of KDR and SNPs investigated in this study.

SNPs which were examined in our studies are shown with boxes indicating exons. SNPs are numbered in reference to the first base of the ATG start codon. Non-synonymous SNPs are shown with the appropriate amino acid variation. The figure is not to exact scale.
Linkage disequilibrium analysis and selection of tagging SNPs
Summary statistics of DNA sequence variation were calculated using SLIDER*. Linkage disequilibrium (LD) analysis of SNPs among the three ethnic groups was performed using the VG2 program†. For selection of haplotype tagging SNPs (tSNPs), SNPs were clustered using LD Select (16) integrated in the VG2 program. The parameters for the selection of tSNPs were r2≥0.8 and a minor allele frequency (MAF) ≥5%. Pair-wised LD analysis was done by the LD Plotter and r2 values are calculated using an iterative EM algorithm (17).
Bioinformatic analyses
The putative functional effects of SNPs were examined using FastSNP (18). The transcription factor binding site analysis of FastSNP was complemented by using PROMO (19). WWW Promoter Scan* was used to predict functional KDR promoters. NCBI ORF finder† was used to determine open reading frames.
VEGFR-2 phosphorylation assays
The coding region of KDR was PCR amplified from cDNA using the following primers 5’-TTAAACTTAAGCTTGGTACCATGGAGAGCAAGGTGC-3’ and 5’-TAGACTCGAGCGGCCGCTCACAGATCCTCTTC-3’. The pcDNA5 FRT vector (Invitrogen, California, USA) was PCR amplified using primers 5’-GCGGCCGCTCGAGTC-3’ and 5’-GGTACCAAGCTTAAG-3’. Vector and insert were joined by In-Fusion Cloning (Clontech, Mountain View, CA, USA) according to kit instructions. The KDR variants R106W, V297I, Q472H and C482R were generated by using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA) as per the manufacturer’s instructions.
HEK293 (ATCC) cells were grown for 24 h in DMEM with 10% FBS and transfected with Ca3(PO4)2 precipitation. At 30 h after transfection, cells were starved overnight in DMEM supplemented with 1% BSA. Transfected cells were stimulated with 1.5 nM VEGF-A165. Cell lysates were resolved on SDS gels, blotted to PVDF membranes, and immuno-decorated with phospho-specific pY1175 or VEGFR-2 55B11 antibodies (Cell Signaling, Danvers, MA, USA). All experiments were performed in triplicates, and immunoblots were quantified by densitometric scanning using ImageQuant RT ECL (Affymetrix-GE Healthcare, Freiburg, Germany).
Dual-Luciferase assays
A pGL2 plasmid (Promega, Madison, WI, USA) with KDR 5’ upstream promoter region and Firefly luciferase gene was a gift from Dr. Patterson (20). Site-directed mutagenesis was performed by PCR according to the method of the QuikChange Site-Directed Mutagenesis Kit. Two allelic variations were introduced (−367C/T, −271G/A). Plasmids containing all major alleles (−367C, −271G), the −367T allele, the −271A allele and −367T/−271A alleles were generated.
To generate constructs containing variants at the −2854, −2455, −2008 and −1942 KDR loci, two PCRs of overlapping sequence were designed. Primers for the PCR amplifying the upstream region were the forward primer 5’-ACCCCAGTTCCTGGTTCAATGCCT-3’ and the reverse primer 5’-GGGAAGCTTGTCTTTTACCTCCCAGA-3’. These primers were used to amplify human genomic DNA with Phusion High-Fidelity polymerase (NEB, Ipswich, MA, USA). This fragment was cloned into pGL4.20 (Promega) using BglII and HindIII sites. The second PCR was amplified using the forward primer 5’-GCAATTGTGGGAAGAGAAGGGTGAC-3 ’ and the reverse primer 5 ’-GTAAGCTTCCGCAGCGCAGGACAGTT-3’. This PCR fragment was cloned into the construct containing the BglII-HindIII fragment in pGL4.20 using the HindIII site in the reverse primer and an AseI site in the initial construct which was also present in the overlapping PCR fragment. The final construct contained a fragment between −2659 and +235 of KDR. Site-directed mutagenesis was performed as before and allelic variations were introduced: −2854 A/C (rs1551645), −2455 G/A (rs1551641), −2008 A/G (rs28517654) and −1942 A/G (rs28481683).
SVEC4-10 endothelial cells (a gift from Dr. Mark Lingen) were cultured in Dulbecco’s Modified Eagle’s medium with 10% FBS (fetal bovine serum). SVEC4-10 endothelial cells in 24 well plates were transfected by lipofectamine method (Invitrogen, Carlsbad, CA, USA) using the reporter gene plasmid construct of interest and a Renilla TK plasmid (Promega). Cells were lysed 40 h after transfection and the luciferase assays were then performed as per the manufacturer’s instructions. Each construct was transfected three times for each experiment using triplicate wells. The ratio of Firefly to Renilla luciferase served as a measure of the luciferase activity. The luciferase activity in each experiment was normalized to the luciferase activity of the wild-type construct.
Non-small cell lung cancer (NSCLC) patients and tissue-microarray (TMA) preparation
The cohort consisted of 170 sequential Polish Caucasian patients who were systematically diagnosed with resectable NSCLC and from whom tumors were deposited into the tissue bank at the Medical University of Gdansk, Poland. The majority of patients were males, with squamous cell histology, smokers, and older than 60 years. Further details of the cohort are described in Dziadziuszko et al. (21). Primary tumors were fresh frozen at the time of surgery and stored at −80°C.
Representative regions of the blocks of consecutive NSCLC patients were selected from an archive of the Medical University of Gdansk (Poland) and microscopic diagnosis was verified by a board certified pathologist. Hematoxylin-eosin stained tissue sections with greater than 70% tumor cellularity were selected by the study investigator (RD) and verified by two pathologists at the University of Colorado Cancer Center. Three cylindrical tissue cores (1.5 mm in diameter) were punched from neoplastic areas by puncher provided by customized tissue microarray service (MaxArray, Invitrogen, South San Francisco, CA). Total RNA and genomic DNA were prepared from corresponding fresh-frozen tumor samples using AllPrep DNA/RNA kit (Qiagen).
VEGFR-2 and microvessel density (MVD) staining by immunohistochemistry
Immunohistochemical staining was performed using horseradish peroxidase (HRP)-labeled dextrose-based polymer complex bound to secondary antibody (DAKO Cytomation, Carpinteria, CA). In brief, four-micron TMA sections paraffin sections were deparaffinized in xylene, rehydrated through graded ethanol solutions to distilled water and then washed in Tris-buffered saline. Antigen retrieval was carried out by heating sections in Citra Plus Buffer (Biogenex, San Ramon, CA, USA), pH=6, for 15 min in a microwave oven. Endogenous peroxidase activity was quenched by incubation in 3% H2O2 in methanol for 5 min. Non-specific binding sites were blocked using Protein Block (DAKO, Glostrup, Denmark) for 20 min. Then tissue sections were incubated for 1 h at room temperature with the rabbit polyclonal antibody against a 1:100 dilution of VEGFR-2 (Cat. No. 676488; Calbiochem, Merck, Darmstadt, Germany) for VEGFR-2 staining and with mouse monoclonal antibodies against CD31 (clone JC70A, 1:40, DAKO) for MVD. This step was followed by 30 min incubation with goat anti-rabbit IgG conjugated to a horseradish peroxidase (HRP)-labeled polymer (EnvisionTM System, DakoCytomation, Carpinteria, CA) for VEGFR-2 staining and with goat anti-mouse IgG conjugated to a horseradish peroxidase-labeled polymer (EnvisionTM+ System, DAKO) for MVD. Slides were then developed for 5 min with 3-3’-diaminobenzidine chromogen, counterstained with hematoxylin, and cover-slipped. Negative controls were performed by substituting primary antibody with non-immune rabbit immunoglobulins. The staining quantification was performed using the automated cellular imaging system at the Human Tissue Research Center at University of Chicago.
For the automated VEGFR-2 scoring, microarrays were scanned at 40X. Each core was analyzed separately by identifying the most representative tumoral area and scoring it. The average score among the three areas chosen was then calculated. Positive staining was calculated by applying two thresholds with one recognizing blue background (hematoxylin stained) cells and another recognizing brown positive cells. The percentage of positivity was the area detected by the brown threshold divided by the sum of the area detected by the brown and blue thresholds. The intensity was calculated by masking out all areas not selected by the brown threshold and calculating the integrated optical density of brown within the remaining area. This value was divided by the area in pixels of the brown mask to calculate an average intensity of a selected area.
For the automated MVD determination, CD31 was analyzed by an ACIS (ChromaVision Medical Systems, San Juan Capistrano, CA, USA). The image analysis system's MVD application was configured for vessel detection using CD31 stained slides. This was based on applying chromogen masks for high-chromogenic staining (brown threshold) and counterstaining (blue threshold), and the minimal and maximal size of the vessels. After selection of appropriate regions for MVD counting, the following measurements were captured digitally for each selected region of tissue: total vessel count, mean vessel area (µm2 occupied by positively stained vessels), and vessel to tissue area ratio (vessel density percentage calculated by area of blood vessels occupying area of counterstained tissue). The vessel count was then averaged for each case. MVD was calculated as positively staining vessel areas in µm2 divided by total counterstained tissue area in µm2.
Genotyping of KDR SNPs
Patients from the NSCLC cohort (n=170) were genotyped for −2766A/T, −906T/C, 18487A/T (Q472H) and 23408T/G using Taqman® SNP genotyping assays (Applied Biosystems, Foster City, CA, USA), as per the manufacturer’s instructions, using a CFX96 Real-Time System (Bio-Rad, Hercules, CA, USA. Genotyping of −271G/A was performed using a single base extension (SBE) genotyping assay using the following primers: 5 ’-CCACCCTGCACTGAGTCCC-3 ’ (forward PCR primer) 5 ’-GCAGCGGAGGACAGTTGAG-3’ (reverse PCR primer with one base edited from C to G) 5’- GAAACGCAGCGACCACACA -3’ (downstream extension primer). Denaturing high performance liquid chromatography was used for separation of SBE products.
cDNA Synthesis and quantitative PCR (qPCR)
RNA extracted from lung tumor samples was reverse transcribed into cDNA using a High Capacity RT Kit as per the manufacturer’s instructions (Applied Biosystems). KDR primers originally designed by An et al. (22) were used. 18S served as a control gene as it exhibits stable expression in NSCLC tumor tissue (23) and was amplified with sense 5’-CGATGCTCTTAGCTGAGTGT-3’ and antisense 5’-GGTCCAAGAATTTCACCTCT-3’ primers. qPCR of cDNA samples was completed in three independent experiments using a Bio-Rad CFX96 Real-Time System, iQ SYBR Green Supermix (Bio-Rad) and 6 µl cDNA. Thermal cycling parameters were followed by a disassociation step. Absolute quantification of mRNA levels was achieved using commercial human lung total RNA (Ambion, Applied Biosytems) converted to cDNA as above and serially diluted as an internal reference standard curve. mRNA levels were expressed as the ratio of KDR to 18S.
Statistical analyses
For the luciferase reporter gene and phosphorylation assays, comparisons were made between the variant and the reference alleles by paired t-tests. The association between each SNP and either log2-transformed KDR mRNA, VEGFR-2 or MVD levels was tested using univariate linear regression by assuming additive (AA=0, AB=1, BB=2), dominant (AA=0, AB/BB=1) and recessive (AA/AB=0, BB=1) models. As these are exploratory studies, a nominal p-value of 0.05 was regarded as significant. For the significant SNP-phenotype associations, we investigated whether age, sex, histology and pathological stage were potential confounding variables by multivariate regression in a model including significant SNPs and patient/tumor characteristics. For mRNA expression, the best combination of two associated SNPs was determined by multiple regression analysis. Since we had available only a smaller subset of samples (n=66) for mRNA analysis, and hence greater chance of detecting false discoveries than in the wider cohort of patients, for these analyses with SNP genotypes, false discovery rate estimates (q-values) were determined using the adaptive two-stage procedure defined by Benjamini et al. (24) and the Excel-based calculator described by Pike (25). All other analyses were performed using the lm library in the R Statistical Package (26) and Prism (GraphPad, La Jolla, CA, USA).
RESULTS
KDR resequencing
KDR resequencing led to the identification of 113 SNPs and 7 insertion/deletions in the three ethnic groups (data summarized in Supplementary Table S4). Twenty five variants were not found in the dbSNP database* (build 132). In general, the available HapMap data showed that SNP MAFs in HapMap (matched by ethnic group) were similar to the MAFs generated by our resequencing (Supplementary Table S4). Fifteen SNPs were detected in the KDR coding region and eight of these variants were non-synonymous. R106W and P839L had not been previously reported in dbSNP.
Comparison of the variants across the different ethnic groups demonstrated inter-ethnic differences, with African-Americans carrying 94 variants compared with 58 for Asians and 51 for Caucasians. A stronger pattern of LD was found in Caucasians and Asians than African-Americans (Supplementary Figure S1). Thirty eight variants were found in all three groups. Out of the variants with MAF>5%, 45 were found in Caucasians, 49 in Asians, and 53 in African-Americans. Overall, KDR genetic variation was characterized by a low level of LD and this study indicates that 20, 21, and 28 tSNPs should be used to interrogate KDR common variation in Caucasians, Asians, and African-Americans, respectively (Supplementary Figure S2).
Effect of non-synonymous SNPs on VEGFR-2 phosphorylation
Bioinformatic analysis of KDR variants (Supplementary Table S5) guided the prioritization of SNPs for in vitro functional testing. Of the non-synonymous variants, R106W, Q472H and C482R were predicted to affect protein function or be potentially damaging and so we examined their effects on VEGFR-2 phosphorylation. In addition, we also tested V297I, due to its high frequency in the three populations. In HEK293 cells, Q472H showed a 46% increase in VEGFR-2 phosphorylation after VEGF-A165 stimulation (p=0.035) (Figure 2). The other non-synonymous variants did not show any effect on VEGFR-2 phosphorylation.
Figure 2. VEGFR-2 phosphorylation in HEK293 cells and effect of non-synonymous variants.

The y axis corresponds to the ratio of phosphorylated VEGFR-2 to total VEGFR-2. Values are normalized to basal phosphorylation levels of reference sequence KDR. “−” refers to basal phosphorylation levels and “+” refers to VEGF-A165 stimulation. Wt = reference sequence cDNA. The mean ± SEM of 3 experiments in triplicate is shown.
Effect of potential regulatory SNPs on KDR mRNA and protein expression
Bioinformatic analysis of the KDR variants identified 58 potential regulatory variants in non-coding gene regions (Supplementary Table S5). We were particularly interested in common SNPs in the 5’ flanking and promoter regions of the gene, which could be assayed using reporter gene assays. We searched bioinformatically for the KDR promoter and identified a region from −599 to −349 bp upstream of the start codon. The only two common SNPs identified in the promoter and the adjacent 5’UTR were −367T/C and −271G/A. Both SNPs were found to alter transcription factor binding sites after further bioinformatic analysis using PROMO (Supplementary Table S5). In reporter gene assays, we found that constructs containing −271A reduced expression to 49–55% (p<0.0001) of that from the construct with the reference, −271G, allele (Figure 3A). Presence of −367C did not alter expression (p>0.05).
Figure 3. Function of −367T/C and −271G/A (A), and 2854A/C, −2455T/A, −2008A/G and −1942A/G (B) according to luciferase assays in SVEC4-10 endothelial cells.

The mean ± SEM of experiments in triplicate is shown. Panel A shows assays examining the −367T/C and −271G/A variants; and panel B shows assays examining the −2854A/C, −2455G/A, −2008A/G and −1942A/G variants. The significant results of paired t-tests between the wild-type reference and variant alleles are shown.
We also examined the 5’ flanking region of the gene and identified four SNPs which are predicted to alter transcription factor binding sites: −2854A/C, −2455T/A, −2008A/G and −1942A/G. The effect of these SNPs was tested singly but since two pairs of SNPs (−2854A/C and −2455T/A; −2008A/G and −1942A/G) were in perfect LD in all three ethnic groups (Supplementary Figures S1 and S2), reporter gene constructs were also created which contained the variants linked by LD. In SVEC4-10 cells, constructs containing −2455A, −2854C, or both variants, had 20% (p<0.001), 10% (p<0.05) and 10% (p<0.01) greater expression, respectively, than the reference construct (Figure 3B).
Association studies of KDR variants with KDR mRNA and protein expression, and MVD in a NSCLC cohort
Since the −271A, −2455A, −2854C and Q472H (18487T) variants had functional effects in vitro, we prospectively tested their effect on KDR gene expression and MVD in tumors. We genotyped −271G/A, −2766A/T (a tagging SNP for both −2854A/C and −2455T/A; Supplementary Figure S2) and Q472H in a Caucasian NSCLC cohort. In addition, we also genotyped two further variants: −906T/C and 23408T/G. The −906C variant has been associated with serum VEGFR-2 levels and been shown to affect expression in reporter gene assays (27). Bioinformatic analysis predicted that the −906C allele introduces an IKZF2 binding site (Supplementary Table S5) so, as this transcription factor may be involved in mediating KDR expression, another KDR variant which introduces an IKZF2 binding site, the 23408T allele, was also genotyped to determine the impact of this binding site motif on expression. After genotyping in the NSCLC patients, the MAFs were determined to be 0.33 (n=169 patients), 0.46 (n=170), 0.44 (n=166), 0.29 (n=168) and 0.34 (n=169) for −2766A/T, −906T/C, −271G/A, Q472H and 23408T/G, respectively, which were comparable to the MAFs of Caucasians from our resequencing and HapMap (Supplementary Table S4). No significant deviation from Hardy-Weinberg equilibrium was observed for any of the SNPs.
We assayed tumoral VEGFR-2 protein and MVD levels in the same individuals by immunohistochemistry (n=170) and quantified KDR mRNA levels in a subset of these patients (n=66). Analysis of protein and mRNA levels determined that there was a very modest inverse correlation (p=0.03, r2=0.07; Supplementary Figure S4); hence, we used both mRNA and protein levels to infer the molecular effects of KDR SNPs on gene expression. VEGFR-2 protein levels were positively correlated with MVD (p=8e-09; r2=0.18; Supplementary Figure S5).
In univariate analyses of mRNA expression, the minor alleles of −2766A/T −906T/C, −271G/A, and 23408T/G correlated with significantly greater expression (Figure 4A–G). The association of −2766A/T and mRNA expression was consistent with the luciferase assay results of the −2854A/C and −2455T/A SNPs, which are tagged by −2766A/T. The minor allele of Q472H associated with significantly lower mRNA levels (Figure 4H–J), probably by altering the splicing machinery due to its vicinity (3 bp) to the intron/exon boundary. After false discovery rate analysis to control for the multiple comparisons, the only association not to maintain significance was −271G/A in the dominant genetic model (q=0.05; Figure 4F). However, the association of this SNP in the additive model remained significant as did all the other nominally significant associations with mRNA levels. Multivariate analyses were performed which incorporated combinations of SNP genotypes. However, possibly due to sample size limitations, the most significant model included only two SNPs: 0.98*genotype (−906 TC/CC; p=0.037) + 1.74*genotype (Q472H AA/AT, p=0.007), overall r2=0.219 and p=0.0004. The effect of these SNPs seems to be independent, as shown by the lack of LD between them (Supplementary Figure S3).
Figure 4. Association between KDR genotypes and KDR mRNA expression in NSCLC tumor specimens.

KDR mRNA expression was measured in 66 tumor specimens. The relationship of −2766A/T and mRNA expression is shown in panels A and B in additive and dominant models, respectively; −906T/C genotype and mRNA expression is shown in panels C and D in additive and dominant models, respectively; −271G/A genotype and mRNA expression is shown in panels E and F in additive and dominant models, respectively; 23408T/G genotype and mRNA expression is shown in a dominant model in panel G; and Q472H genotype and mRNA expression is shown in additive, dominant and recessive models in panels H, I and J, respectively. Nominal statistical significance is denoted by p-values and the results of false discovery rate testing by q-values.
In univariate analyses of protein expression, no significant associations were found between the genotyped variants and VEGFR-2 protein levels. However, when testing the effect of patient/tumor characteristics, protein expression was lower in later stages of disease (Supplementary Figure S6). Controlling for early (I–II) versus late (III–IV) stage of disease, we found that the tumors of patients with −271GG/GA genotypes had greater VEGFR-2 levels compared with tumors from patients with −271AA genotype: −0.32*stage (III&IV; p=0.02) + 0.36*genotype (−271GG/GA; p=0.03); overall p=0.01, r2=0.054.
In univariate analysis of MVD, the major allele, A, of Q472H, coding for wild-type VEGFR-2, associated with lower MVD (additive and dominant: p=0.036 and p=0.040; Figure 5). After controlling for differences in tumor histology (Supplementary Figure S7), patients with the Q472H AA genotype (i.e. expressing only wild-type VEGFR-2) were shown to have tumors with lower MVD than patients with AT/TT genotypes: 0.57*histology (adenocarcinoma and large cell carcinoma; p=0.01) −0.67*genotype (Q472H AA; p=0.05); overall p=0.005, r2=0.050.
Figure 5. Association between Q472H genotypes and MVD in NSCLC tumor specimens.

MVD was measured in 170 tumor specimens. The relationship between Q472H genotype and MVD is shown in panels A and B in additive and recessive models, respectively.
DISCUSSION
This report represents the most extensive molecular genetic study of KDR. Our resequencing study identified 120 KDR variants which included 25 previously unreported. Furthermore, there were considerable differences between the presence of specific variants and their corresponding frequencies among the three ethnic populations. This comprehensive assessment allowed the generation of haplotype-tagging SNPs for genotyping in subjects from Caucasian, African, or Asian backgrounds.
Statistical associations in genetic cancer risk or outcome studies should be supported with molecular mechanistic evidence of SNP function and the lack of mechanistically-based effects of clinical phenotypes is regarded as a major limitation for identifying biomarkers. Very few studies have focused on the identification of common functional KDR germline variants (27, 28). We have tested associations of twelve KDR SNPs and function in two different assays and with molecular phenotypes from tumor samples, and found five SNPs which have a significant association with at least one of these measures (Figure 6). We analyzed six SNPs in reporter gene assays and found three of these with significant effects. The −2455A and −2854C variants modestly increased reporter gene expression and these alleles were shown to be associated with increased KDR mRNA expression in NSCLC tumors, without an effect on VEGFR-2 protein levels. The −271A variant had a strong negative effect on expression in the reporter assay, and, supportive of this result, patients with the −271AA genotype had less tumor protein expression than subjects with −271GA/GG genotypes, after controlling for disease stage. Furthermore, the observation that VEGFR-2 levels in stages III–IV were significantly lower than in earlier stages appears to be a novel finding in NSCLC.
Figure 6. Summary of experimental and analytical approaches used, and significant findings.
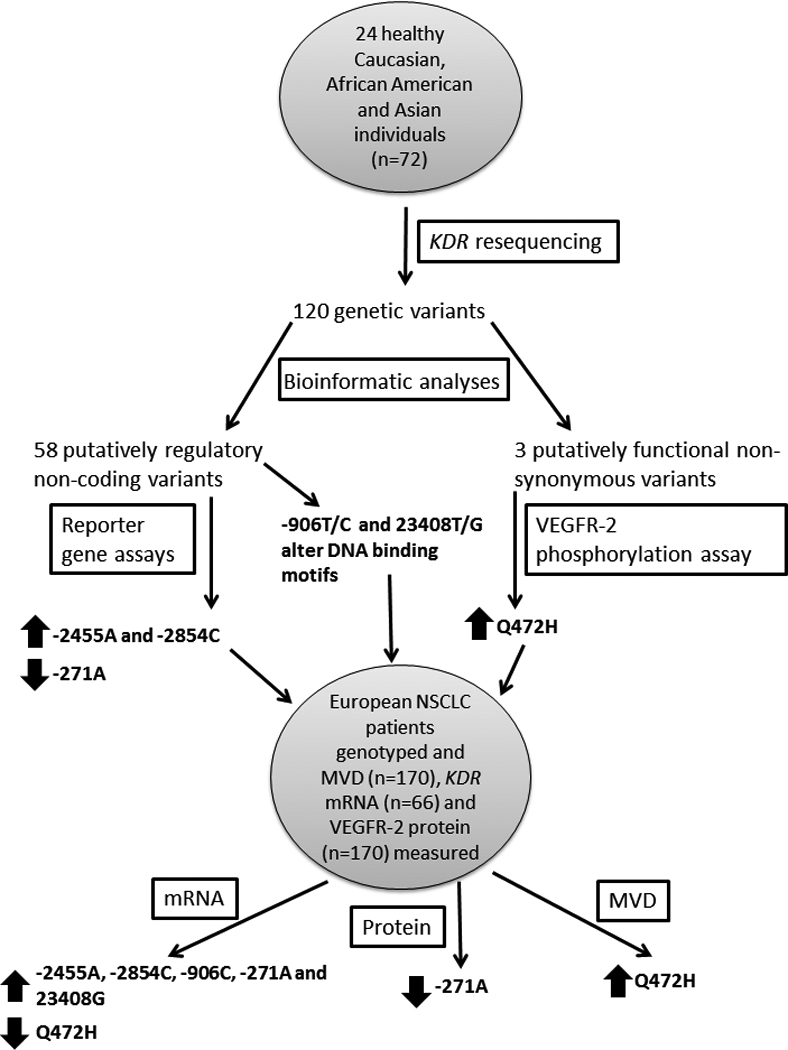
KDR was resequenced in three ethnic groups, each containing 24 healthy individuals. The genetic variants identified from the resequencing were analyzed for function using bioinformatic tools. Putatively functional SNPs were examined using reporter gene or VEGFR-2 phosphorylation assays. Functional SNPs, including two further putatively functional SNPs, were genotyped in a cohort of European NSCLC patients. These genotypes were tested for associations with KDR mRNA, VEGFR-2 protein and MVD levels from matching tumor samples. SNPs significantly (p<0.05) associated with a phenotype of interest are designated by a block arrow. An up arrow indicates that the variant is associated with increased levels of the phenotype; conversely, a down arrow indicates that the variant is associated with decreased levels of the phenotype.
The decreased protein expression associated with −271A appears to be due to a posttranscriptional mechanism. The −271A allele creates a start codon which introduces an upstream open reading frame of 207 bp, in conjunction with an in frame stop codon in the 5’UTR, and thereby inhibits translation of KDR mRNA into protein (29). At the mRNA level in our study, presence of an A allele was associated with greater tumor KDR expression, consistent with the previous association of the −271AA genotype with higher levels of KDR mRNA in NSCLC (30). We have also observed a modest negative correlation between mRNA and protein levels. Indeed, discordance between mRNA and protein levels of KDR has been found under low glucose conditions (31), which are common in the tumor microenvironment (32). The mechanistic explanation of this finding is that glucose depletion may up-regulate KDR mRNA expression through the unfolded protein response but down-regulate VEGFR-2 levels via a protein degradation mechanism (31).
We examined two other putatively regulatory SNPs, −906T/C and 23408T/G, for associations with KDR expression. The −906C variant introduces an IKZF2 binding site and increases reporter gene expression in human umbilical vein endothelial cells (27); in agreement with this observation, we found this variant associated with higher expression of mRNA in NSCLC tumors. Bioinformatic analyses predicted that 23408G disrupts an IKZF2 binding site but this allele was associated with higher mRNA levels. Therefore, it appears that the presence/absence of an IKZF2 binding site motif is not the cause of the differential expression. Instead, this effect may be due to the interaction of another transcription factor, such as GATA2, which putatively binds at this locus (Table S4). Alternatively, there may be a functional SNP in LD with 23408T/G which mediates this effect.
Q472H was predicted to have effects on protein function and this variant had increased phosphorylation after VEGF-A165 stimulation. Wang et al., examined VEGF-A165 binding of VEGFR-2 and found that Q472H increased the binding efficiency (27). In univariate analysis, we observed greater MVD in patients with the Q472H TT genotype. Adenocarcinomas have been shown to have greater MVD (33) and we observed higher MVD in adenocarcinomas and large cell carcinomas (Figure S7). We controlled for tumor histology, and again found a correlation between patients expressing the Q472H variant and greater MVD. Although we observed an inverse relationship between Q472H and mRNA levels, Q472H did not associate with protein levels, measured by IHC of total VEGFR-2 (both phosphorylated and unphosphorylated). Therefore, the effect of Q472H on MVD is likely due to increased activation of the VEGFR-2 receptor through increased phosphorylation, as demonstrated by our finding from the phosphorylation assay. Moreover, this study has identified a germline variant as a potential determinant of increased tumor vasculature in NSCLC.
A potential drawback of this study is the lack of laser-capture microdissection, and the possibility that the molecular phenotypes of KDR in this study are not entirely representative of tumor vasculature. To minimize this effect, we used tumor blocks displaying greater than 70% tumor cellularity. Although it has been clearly shown that VEGFR-2 positive vessels are mainly absent or rare in normal lung vessels, and that VEGFR-2 expression is not detected in malignant lung cells (34), we cannot rule out that VEGFR-2 expression in the stromal cells may be a confounder. However, non-microdissected lung cancer specimens are well accepted in the field as the material for molecular studies (35).
Our findings highlight the difficulty of identifying common germline variants with large effect sizes in genotype-phenotype association studies. Nevertheless, even small changes in gene expression may increase cancer susceptibility. In mice, a 20% reduction in expression of Pten, a gene involved in the regulation of angiogenesis (36), led to a dramatic rise in tumor incidence (37). Furthermore, we found that a combination of two SNPs can account for 22% of the variation in tumoral levels of KDR mRNA. The contribution of untested functional germline SNPs may partly explain the relatively small effect sizes. Also, we did not examine somatic DNA alterations such as gene amplification which could have significant effects on gene expression. Indeed, increased KDR copy number has been reported in some cancers (38, 39). However, the goal of this study was to characterize regions informative of function by focusing mainly on known regulatory regions.
We acknowledge that the results of this exploratory study are preliminary. The significant associations of SNP genotypes and ex vivo molecular phenotypes observed should be validated in an independent cohort with well-defined microdissected tissue. However, we believe this lack of validation may be somewhat mitigated by supportive findings from - 2854A/C, −2455T/A, −271G/A and Q472H in our in vitro reporter gene and phosphorylation assays, and from the corroborative reports, discussed previously, of the functionality of −906T/C, −271G/A and Q472H.
We have generated the knowledge basis to guide the prospective evaluation of KDR variants for identification of clinical biomarkers and for validating their clinical utility. Our study will inform the selection of SNPs, in accordance with their functional effects and expected frequencies in the corresponding ethnic group. For example, Q472H, which increases VEGFR-2 phosphorylation, associates with MVD levels, and shows a large variation in allele frequencies, ranging from 0.10 in African Americans to 0.52 in Asians (Table S4). Our findings may have implications not only for the molecular genetics of angiogenesis in lung cancer but also in other tumors. Their effect on the outcome of treatment with VEGF-pathway inhibitors should be evaluated prospectively.
Translational relevance.
Angiogenesis is a host-driven process which represents a crucial step in tumor development. The VEGFR-2 receptor mediates angiogenic endothelial cell responses via a downstream signaling pathway and its activity is targeted by several small molecule angiogenesis inhibitors. However, no biomarkers have yet been identified to guide the use of these drugs. We have characterized germline genetic variation of KDR, the gene encoding VEGFR-2, in three ethnic groups and identified functional variants which associate with KDR mRNA and protein levels, and microvessel density in non-small cell lung cancer tumor patient samples. These genetic variants provide strong candidates for the prospective testing of associations with patient outcome, the clinical effect of angiogenesis inhibitors, and other clinical cancer phenotypes related to angiogenesis.
Supplementary Material
Acknowledgments
Financial support: NIH/NCI K07CA140390-01 (FI), Cancer Research Foundation Young Investigator Award (FI), American Cancer Society (FI), Grant ST-23 from Medical University of Gdansk (GD), NIH/NIGMS grant U01GM061393 (FI co-investigator, Ratain/Cox/Dolan PIs), NIH grant 5R01CA125541-04 (RS), Swiss National Science Foundation grant 3100A-116507 (KB-H).
Footnotes
References
- 1.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi Y, Kitadai Y, Bucana CD, Cleary KR, Ellis LM. Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res. 1995;55:3964–3968. [PubMed] [Google Scholar]
- 3.Hara H, Akisue T, Fujimoto T, Imabori M, Kawamoto T, Kuroda R, et al. Expression of VEGF and its receptors and angiogenesis in bone and soft tissue tumors. Anticancer Res. 2006;26:4307–4311. [PubMed] [Google Scholar]
- 4.Hetian L, Ping A, Shumei S, Xiaoying L, Luowen H, Jian W, et al. A novel peptide isolated from a phage display library inhibits tumor growth and metastasis by blocking the binding of vascular endothelial growth factor to its kinase domain receptor. J Biol Chem. 2002;277:43137–43142. doi: 10.1074/jbc.M203103200. [DOI] [PubMed] [Google Scholar]
- 5.Rini BI, Flaherty K. Clinical effect and future considerations for molecularly-targeted therapy in renal cell carcinoma. Urol Oncol. 2008;26:543–549. doi: 10.1016/j.urolonc.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 6.Minguez B, Tovar V, Chiang D, Villanueva A, Llovet JM. Pathogenesis of hepatocellular carcinoma and molecular therapies. Curr Opin Gastroenterol. 2009;25:186–194. doi: 10.1097/MOG.0b013e32832962a1. [DOI] [PubMed] [Google Scholar]
- 7.Harris PA, Boloor A, Cheung M, Kumar R, Crosby RM, Davis-Ward RG, et al. Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-b enzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem. 2008;51:4632–4640. doi: 10.1021/jm800566m. [DOI] [PubMed] [Google Scholar]
- 8.Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–337. [PubMed] [Google Scholar]
- 9.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 10.Chang YS, Adnane J, Trail PA, Levy J, Henderson A, Xue D, et al. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother Pharmacol. 2007;59:561–574. doi: 10.1007/s00280-006-0393-4. [DOI] [PubMed] [Google Scholar]
- 11.Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 13.DePrimo SE, Bello C. Surrogate biomarkers in evaluating response to anti-angiogenic agents: focus on sunitinib. Ann Oncol. 2007;18 Suppl 10:x11–x19. doi: 10.1093/annonc/mdm409. [DOI] [PubMed] [Google Scholar]
- 14.Facchini G, Perri F, Caraglia M, Pisano C, Striano S, Marra L, et al. New treatment approaches in renal cell carcinoma. Anticancer Drugs. 2009;20:893–900. doi: 10.1097/CAD.0b013e32833123d4. [DOI] [PubMed] [Google Scholar]
- 15.Frith MC, Li MC, Weng Z. Cluster-Buster: Finding dense clusters of motifs in DNA sequences. Nucleic Acids Res. 2003;31:3666–3668. doi: 10.1093/nar/gkg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74:106–120. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hill WG. Estimation of linkage disequilibrium in randomly mating populations. Heredity. 1974;33:229–239. doi: 10.1038/hdy.1974.89. [DOI] [PubMed] [Google Scholar]
- 18.Yuan HY, Chiou JJ, Tseng WH, Liu CH, Liu CK, Lin YJ, et al. FASTSNP: an always up-to-date and extendable service for SNP function analysis and prioritization. Nucleic Acids Res. 2006;34:W635–W641. doi: 10.1093/nar/gkl236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farre D, Roset R, Huerta M, Adsuara JE, Rosello L, Alba MM, et al. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003;31:3651–3653. doi: 10.1093/nar/gkg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patterson C, Perrella MA, Hsieh CM, Yoshizumi M, Lee ME, Haber E. Cloning and functional analysis of the promoter for KDR/flk-1, a receptor for vascular endothelial growth factor. J Biol Chem. 1995;270:23111–23118. doi: 10.1074/jbc.270.39.23111. [DOI] [PubMed] [Google Scholar]
- 21.Dziadziuszko R, Merrick DT, Witta SE, Mendoza AD, Szostakiewicz B, Szymanowska A, et al. Insulin-like growth factor receptor 1 (IGF1R) gene copy number is associated with survival in operable non-small-cell lung cancer: a comparison between IGF1R fluorescent in situ hybridization, protein expression, and mRNA expression. J Clin Oncol. 2010;28:2174–2180. doi: 10.1200/JCO.2009.24.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.An SJ, Nie Q, Chen ZH, Lin QX, Wang Z, Xie Z, et al. KDR expression is associated with the stage and cigarette smoking of the patients with lung cancer. J Cancer Res Clin Oncol. 2007;133:635–642. doi: 10.1007/s00432-007-0214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguewa PA, Agorreta J, Blanco D, Lozano MD, Gomez-Roman J, Sanchez BA, et al. Identification of importin 8 (IPO8) as the most accurate reference gene for the clinicopathological analysis of lung specimens. BMC Mol Biol. 2008;9:103. doi: 10.1186/1471-2199-9-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benjamini Y, Krieger AM, Yekutieli D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika. 2006;93:491–507. [Google Scholar]
- 25.Pike N. Using false discovery rates for multiple comparisons in ecology and evolution. Methods in Ecology and Evolution. 2011;2:278–282. [Google Scholar]
- 26.Team RDC. Computing RFfS. Austria: Vienna; 2010. R: A language and environment for statistical computing. [Google Scholar]
- 27.Wang Y, Zheng Y, Zhang W, Yu H, Lou K, Zhang Y, et al. Polymorphisms of KDR gene are associated with coronary heart disease. J Am Coll Cardiol. 2007;50:760–767. doi: 10.1016/j.jacc.2007.04.074. [DOI] [PubMed] [Google Scholar]
- 28.Kariyazono H, Ohno T, Khajoee V, Ihara K, Kusuhara K, Kinukawa N, et al. Association of vascular endothelial growth factor (VEGF) and VEGF receptor gene polymorphisms with coronary artery lesions of Kawasaki disease. Pediatr Res. 2004;56:953–959. doi: 10.1203/01.PDR.0000145280.26284.B9. [DOI] [PubMed] [Google Scholar]
- 29.Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci U S A. 2009;106:7507–7512. doi: 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.An SJ, Chen ZH, Lin QX, Su J, Chen HJ, Lin JY, et al. The −271 G>A polymorphism of kinase insert domain-containing receptor gene regulates its transcription level in patients with non-small cell lung cancer. BMC Cancer. 2009;9:144. doi: 10.1186/1471-2407-9-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adham SA, Coomber BL. Glucose is a key regulator of VEGFR2/KDR in human epithelial ovarian carcinoma cells. Biochem Biophys Res Commun. 2009;390:130–135. doi: 10.1016/j.bbrc.2009.09.081. [DOI] [PubMed] [Google Scholar]
- 32.Henning T, Kraus M, Brischwein M, Otto AM, Wolf B. Relevance of tumor microenvironment for progression, therapy and drug development. Anticancer Drugs. 2004;15:7–14. doi: 10.1097/00001813-200401000-00002. [DOI] [PubMed] [Google Scholar]
- 33.Ozbudak IH, Ozbilim G, Kucukosmanoglu I, Dertsiz L, Demircan A. Vascular endothelial growth factor expression and neovascularization in non--small cell lung carcinoma. Int J Surg Pathol. 2009;17:390–395. doi: 10.1177/1066896908323506. [DOI] [PubMed] [Google Scholar]
- 34.Smith NR, Baker D, James NH, Ratcliffe K, Jenkins M, Ashton SE, et al. Vascular endothelial growth factor receptors VEGFR-2 and VEGFR-3 are localized primarily to the vasculature in human primary solid cancers. Clin Cancer Res. 2010;16:3548–3561. doi: 10.1158/1078-0432.CCR-09-2797. [DOI] [PubMed] [Google Scholar]
- 35.Eberhard DA, Giaccone G, Johnson BE. Biomarkers of response to epidermal growth factor receptor inhibitors in Non-Small-Cell Lung Cancer Working Group: standardization for use in the clinical trial setting. J Clin Oncol. 2008;26:983–994. doi: 10.1200/JCO.2007.12.9858. [DOI] [PubMed] [Google Scholar]
- 36.Castellino RC, Muh CR, Durden DL. PI-3 kinase-PTEN signaling node: an intercept point for the control of angiogenesis. Curr Pharm Des. 2009;15:380–388. doi: 10.2174/138161209787315873. [DOI] [PubMed] [Google Scholar]
- 37.Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, et al. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010;42:454–458. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holtkamp N, Ziegenhagen N, Malzer E, Hartmann C, Giese A, von Deimling A. Characterization of the amplicon on chromosomal segment 4q12 in glioblastoma multiforme. Neuro Oncol. 2007;9:291–297. doi: 10.1215/15228517-2007-009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hahtola S, Burghart E, Puputti M, Karenko L, Abdel-Rahman WM, Vakeva L, et al. Cutaneous T-cell lymphoma-associated lung cancers show chromosomal aberrations differing from primary lung cancer. Genes Chromosomes Cancer. 2008;47:107–117. doi: 10.1002/gcc.20513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.