

Abstract
Here, we analyze for the first time the immunological and therapeutic efficacy of a dendritic cell (DC) vaccine based on a cancer-testis antigen, Brother of Regulator of Imprinted Sites (BORIS), an epigenetically acting tumor-promoting transcription factor. Vaccination of mice with DC loaded with truncated form of BORIS (DC/mBORIS) after 4T1 mammary tumor implantation induced strong anti-cancer immunity, inhibited tumor growth (18.75% of mice remained tumor-free), and dramatically lowered the number of spontaneous clonogenic metastases (50% of mice remained metastases-free). Higher numbers of immune effector CD4 and CD8 T cells infiltrated the tumors of vaccinated mice vs. control animals. Vaccination significantly decreased the number of myeloid-derived suppressor cells (MDSCs) infiltrating the tumor sites, but not MDSCs in the spleens of vaccinated animals. These data suggest that DC-based mBORIS vaccination strategies have significant anti-tumor activity in a therapeutic setting and will be more effective when combined with agents to attenuate tumor-associated immune suppression.
Keywords: Immunotherapy of breast cancer, myeloid derived suppressor cells (MDSC), cancer-testis antigen (CTA), Dendritic cell (DC)-based vaccine, Brother of Regulator of Imprinted Sites (BORIS), tumor promoting transcription factor, 4T1 mammary carcinoma
1. Introduction
Breast cancer is currently the second most common type of cancer after lung cancer (10.4% of all cancer incidence) and the fifth most common cause of cancer death [1]. Although radiotherapy, chemotherapy, and surgery are all used in the treatment of breast cancer, there is a low but continuous rate of relapse, with a majority of these patients that relapse succumbing because of their metastatic disease [2]. Therefore, the ability to control, prevent, and/or treat metastases is of great importance for the clinical application of any immunotherapy. Numerous immunotherapeutic strategies have been tested so far for boosting anti-tumor cellular immune responses in breast cancer. However, only a few of them moderately enhanced the frequency of anti-tumor associated antigen-reactive T cells [3,4]. These strategies include adoptive cell transfer (ACT), treatment with cytokines and other immune stimulatory agents, or treatment with antibodies against suppressor cytokines and other suppressive molecules in combination with various vaccines [4,5,6]. A major limitation of vaccines based on tumor-associated antigens (TAA) [7,8] used for treatment of breast cancer is tumor-associated self-tolerance [9]. Even in the cases when tolerance can be broken by immunizations [10], tumors often mutate immunogenic epitopes evading immune attack [11]. Finally, these TAAs may induce potentially harmful autoimmunity through cross-reactivity with self-antigens. Thus, the availability of a sufficiently potent anti-cancer vaccine targeting an optimal TAA is extremely important to eliminate tumors before they can evade the immune attack. Historically, it was believed that the ideal TAA should meet the following criteria: (i) expression should be restricted to neoplastic cells and/or cells residing in immuno-privileged sites; (ii) the ability to evoke a therapeutic anti-cancer immune response following vaccination; and (iii) should be essential for the function of the tumor so that loss of such a TAA would result in loss of tumor activity. Cancer Testis (CT) antigens, in general, meet these criteria and these characteristics along with their immunogenicity make them excellent candidates for cancer vaccines [12,13]. Not surprisingly, immunogenic CT antigens are used as components of several of the most potent therapeutic human vaccines against different cancers [13,14,15], although to our knowledge not for breast cancer immunotherapy in clinical trials.
The CT-gene BORIS was identified as a mammalian paralogue of CTCF [16,17]. This unique epigenetically acting, tumor-promoting, transcription factor expressed in testis, also regulates the expression of other oncogenic molecules including MAGE-A1, NY-ESO-1, and SPANX [18,19,20,21,22]. Based on these observations and an important role for BORIS in oncogenic transformation [23], we tested a zinc finger deleted (modified) BORIS (mBORIS) in preventive studies and showed that delivery of this antigen by adenoviral [24] or DNA plasmid vectors [25,26] generated protective anti-tumor cellular immune responses, inhibited 4T1 mouse mammary tumor growth, and prolonged survival of vaccinated mice subjected to tumor challenge.
Herein, we report for the first time, the application of an mBORIS-directed dendritic cell (DC)-based anti-tumor vaccine, with efficacy in treatment of both preexisting tumor and metastatic mammary carcinoma. Such use of DCs as a system for delivery of antigen to the immune system serves as a cellular adjuvant [27] by the nature of their capacity to elicit robust immune responses. The recent FDA approval of Provenge, a cellular therapy containing human dendritic cells, lends additional credence to immunotherapy strategies incorporating DCs (http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm210174.htm). We hypothesized that loading of DCs, professional antigen-presenting cells, with mBORIS antigen would induce strong cellular immune responses controlling tumor growth and metastatic disease.
2. Materials and Methods
2.1. Animals
Female 8- to 10-wk-old BALB/c mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All animals were housed in a temperature- and light cycle-controlled facility, and their care was under the guidelines of the National Institutes of Health and the approved Institutional Animal Care and Use Committee protocol at the University of California, Irvine.
2.2. Purification of recombinant mBORIS protein, isolation of DC, analysis of surface markers and loading of DC with protein
Purified recombinant mBORIS and gp120 proteins, prepared as described [25], were used for the loading of DC. The level of endotoxin was measured using E-TOXATE Kits as recommended by the manufacturer (Sigma, St Louis, MO), and it was ranged from 1,500 to 2,000 ng bacterial lipopolysaccharide endotoxin (according to FDA RSE) per mg of protein. Thus, DC were loaded with a recombinant protein containing ~80-100 ng of bacterial endotoxin in 1 μM of loaded protein (see below).
DCs were generated from bone marrow isolated from tibia and femurs of mice and cultured in the presence of GM-CSF and IL-4 as described [28]. On day 6, cells were enriched for CD11c+ DC (>92% purity) using positive selection with anti-CD11c-coated magnetic microbeads (MiltenyiBiotec, Auburn, CA). These cells were loaded with 1 μM mBORIS for 24 hrs and analyzed by FACScan (BD Biosciences, CA) or MacsQuant (MiltenyiBiotec, Auburn, CA) cytometers for cell surface markers using fluorophore-conjugated antibodies specific to MHC class II (I-Ad/I-Ed), CD80, CD86, CD40, CD54, DEC205 (BD Pharmingen, San Diego CA). Non-specific “background” was distinguished by staining with isotype control primary Abs (BD Pharmingen, San Diego, CA). The dynamics of mBORIS protein uptake by CD11c+ DC was also analyzed by FACScan using intracellular staining method with the 8112 rabbit-anti mBORIS antisera (this antibody is specific to very N-terminal peptide MAA AEV PVP SGY FTQ IKE KLK PGD LEE EKE EDV C of human BORIS and is cross-reactive with mouse BORIS and was provided by Dr. Loukinov) followed by FITC-labeled secondary antibody (BioLegend, San Diego, CA). DC loaded with mBORIS or irrelevant recombinant antigen (gp120 of HIV-1), each prepared and purified in exactly the same manner, were used for immunization of mice.
2.3. Immunizations and analyses of tumor growth or clonogenic lung metastases
For the in vitro immunologic studies two groups of BALB/c mice (n=8 per group) were injected s.c. 3 times with 5×105 of DC/mBORIS or DC/gp120 into the right flank. A third group of mice were injected 3 times with 100μg/mouse of recombinant mouse mBORIS formulated in QuilA (Brenntag, Denmark). Mice were sacrificed one week after the last immunization for the in vitro analyses. For the therapeutic studies, unmodified 4T1 mammary carcinoma cells were freshly prepared and 7×103 tumor cells were injected at day 0 into the mammary fat pads as described [24,26] followed by weekly immunizations with DC/mBORIS or DC/gp120. Tumor growth was monitored daily starting at day 12 when tumors became palpable as previously described [24,26]. Tumor volumes were determined by two-dimensional measurement and calculation using the formula (a × b2)/2, where a represents the largest diameter and b the smallest diameter of the tumor. The lowest measurable volume of tumor was approximately 0.005 cm3. On day 22 after tumor implantation, control (non-immunized or DC/gp120 immunized) and experimental mice were sacrificed, and the number of clonogenic metastases in the lungs was analyzed as described [29,30]. Blue colonies of clonogenic metastases were calculated by two independent observers.
2.4. Analysis of T cell responses and antibody production
BORIS-specific CD4+ T cell proliferation was evaluated using CFSE dilution flow cytometry-based assay [25,26], (delta Δ=percent of proliferating CD4+ T cells in re-stimulated culture minus that in non-stimulated culture). A standard ELISpot assay was used to detect production of IFN-γ as previously described and cytotoxic T lymphocyte (CTL) activity of splenocytes from immune and control mice was analyzed by FACScan using as a target unmodified 4T1 or B16 tumor cells, as previously described [25,26]. Anti-BORIS antibodies were detected in the sera of experimental and control mice by ELISA, as previously described [25,26].
2.5. Analysis of splenic myeloid-derived suppressor cells (MDSC)
Flow cytometry was used to determine and quantify the percentage of the most common population of suppressor cells, MDSC, in spleens and tumors. For detection of CD11b+, Gr1+ MDSC, we stained splenocytes from experimental or control mice with APC-conjugated anti-CD11b and FITC-conjugated anti-Gr-1 antibodies (Miltenyi Biotec, Auburn, CA). As a control we used freshly isolated splenocytes from naïve (tumor-free) mice. A standard surface staining protocol from Miltenyi Biotec was used.
2.6. Analysis of suppressor activity of splenocytes of tumor-bearing mice
For analyses of suppressor activity, splenocytes from tumor-bearing vaccinated mice were mixed with CFSE-labeled splenic cells isolated from tumor-free (naïve) BALB/c mice at ratios 10:1, 2:1, 1:1, 1:2, 1:10. Cultures of splenocyte mixtures were stimulated with 10 μg/ml immobilized anti-CD3 and 1μg/ml of soluble anti-CD28 antibodies (both from BD Pharmingen, San Diego, CA). After 5 days, T cell proliferation (as measured by dilution of CFSE) was detected in CD4+ T cells by flow cytometry using a MacsQuant cytometer (Miltenyi Biotec, Auburn, CA). The percent of suppression was calculated with the proliferation of CD4+ splenocytes isolated from tumor-free naïve mice considered to be a 100%.
2.7. Analysis of tumor-infiltrating cell populations
On day 22 after tumor implantation, mammary fat pad tumors were surgically removed from tumor-bearing mice as described [29,31] and used for analysis of tumor-infiltrating cell populations. Briefly, tumors were minced and digested in 1mg/ml collagenase type IV (2hr, 4°C) on a rotating platform. Digested tumors were then subjected to filtration and washing, and cells were stained with anti-CD4-PerCP, anti-CD8-FITC (both from BD Pharmingen, CA), anti-CD11b-APC, anti-Gr1-FITC, antibodies (both from Miltenyi Biotec, Auburn, CA). Samples were analyzed by flow cytometry using a MacsQuant cytometer (Miltenyi Biotec, Auburn, CA). Results are presented as number of cells in the designated subset per 106 total cells.
2.8. Statistical Analyses
All statistical parameters were calculated as described [20]. Correlations between tumor volumes and number of tumor-infiltrated MDSCs in mice vaccinated with DC/mBORIS or injected with DC/gp120 were analyzed using GraphPad Prism 3.0 Software to determine the Pearson’s R correlation coefficient between tumor volumes and number of MDSCs.
3. Results
3.1. Characterization of DC/mBORIS vaccine
Before initiating vaccination experiments, we documented the production of recombinant mBORIS protein (Fig 1A), characterized BMDC, and the loading of mBORIS protein into these cells (Fig. 1B,C). Preparations of purified and mBORIS and gp120 (the irrelevant control antigen) loaded CD11c+ BMDC uniformly expressed CD40, CD54, CD80, MHC II, CD86, and DEC205. Of note, the expression level of DEC205 was almost the same in semi-mature BMDC before and after loading with mBORIS (data not shown). Thus, we generated mature BMDC (Fig. 1C) loaded with mBORIS or gp120 recombinant proteins [28] (Fig. 1B) for use in the immunizations [32,33].
Figure 1.
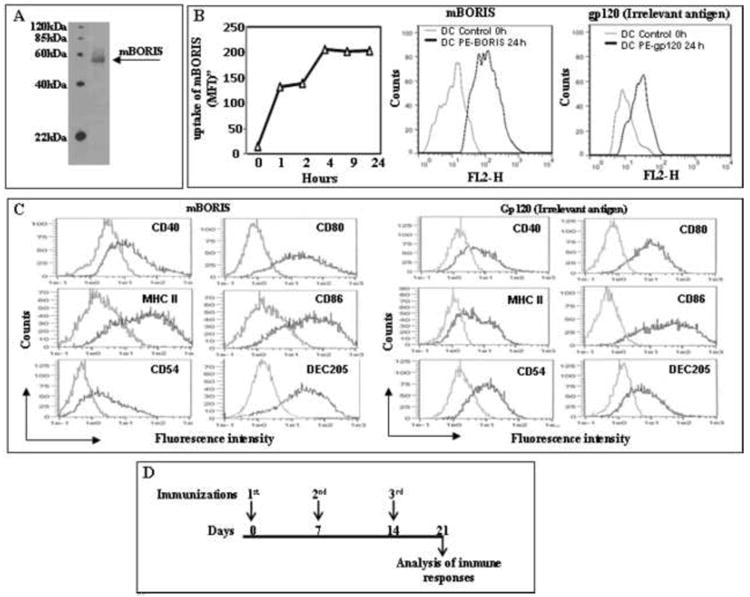
Preparation of DC/mBORIS vaccine and experimental design of immunological studies. (A) Analysis of purified mBORIS recombinant protein in 10% Bis-Tris gel after Coomassie blue staining. (B) The levels of mBORIS proteins (1μM) uptake by CD11c-enriched DC at the indicated time points was analyzed by FACScan using intracellular staining method with polyclonal antibody specific to mBORIS followed by secondary anti-rabbit antibodies. Representative histograms showing uptake of mBORIS and control gp120 proteins after 24 hours are presented. (C) Flow cytometric analyses of DC cell surface phenotype. Cells were incubated in the presence of GM-CSF/IL-4 (see Materials and Methods) and loaded with mBORIS and gp120, respectively, followed by detection of CD40, CD80, CD86, MHC class II, CD54, and DEC205 molecules (black line) or isotype control primary Abs (gray line) in the CD11c-enriched cell population. Representative histograms from 2 experiments are presented. (D) Immunization schedule for administration of DC-based mBORIS and gp120 vaccines.
3.2. Evaluation of immunogenicity of DC/mBORIS vaccine
Mice were immunized with DC/mBORIS, DC/gp120, or mBORIS recombinant protein for assessment of humoral and cellular immune responses (Fig. 1D). DC/mBORIS induced robust antigen-specific proliferation of the CD4+ T cells that was significantly higher (P<0.001) in comparison to that detected in splenocytes isolated from mBORIS protein vaccinated mice (Fig. 2). CD4+ T cell proliferation in splenocytes isolated from DC/gp120-injected mice was at background levels (Fig. 2A). Vaccination with DC/mBORIS induced high frequencies of IFN-γ producing cells that were increased in comparison to those in mice vaccinated with mBORIS protein alone (P<0.01) (Fig. 2B). Splenocytes from control DC/gp120 (Fig. 2B) mice matched the background level of IFN-γ producing cells in non-vaccinated animals (data not shown).
Figure 2.
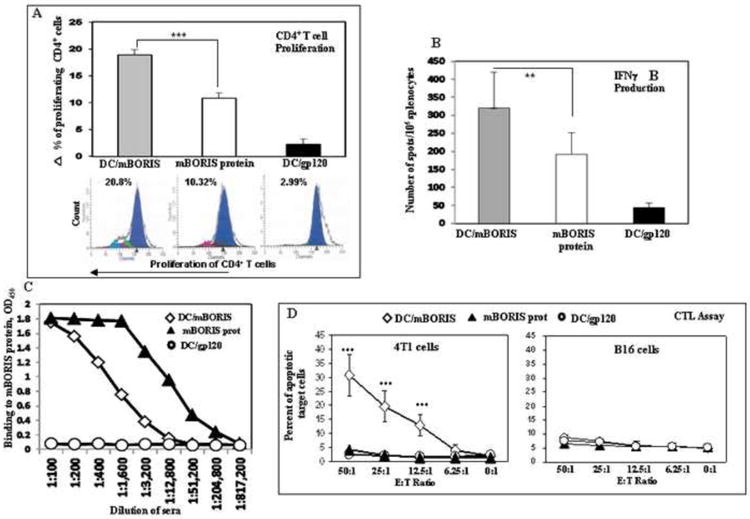
DC/mBORIS vaccine induces strong antigen-specific cellular responses. (A) mBORIS-specific proliferation of CD4+ T cells detected by CFSE assay. Representative histograms for each group are presented. (B) The activation of IFN-γ producing splenocytes detected by ELISpot analysis. (C) mBORIS-specific humoral immune responses detected by ELISA using pooled sera from 8 mice per group, combined results of two separate measurements. (D) Splenocytes isolated from DC/mBORIS mice killed MHC I-matched 4T1 (H2d), but not unmatched B16 (H2b) target tumor cells.
Data in A, B, and D are representative results from two separate experiments with 8 mice/group in each experiment (bars represent SD: **P<0.01 and ***P<0.001 relative to designated controls).
Since both DC/mBORIS and mBORIS vaccines induced significant T helper immune responses, we measured anti-BORIS antibody titers in the sera of experimental and control animals. Analysis of humoral immune responses demonstrated that the DC/mBORIS vaccine, which induced the strongest CD4+ T cell proliferation, generated a modest level of anti-BORIS antibodies (end titer is ~1:10,000) while mBORIS protein formulated in QuilA induced much higher titers of anti-mBORIS antibodies (~1:205,000) (Fig. 2C). Of note, it was not unexpected that mice immunized with mBORIS protein induced CD4+ T cell proliferation, splenocytes producing IFN-γ, and elicited high levels of anti-mBORIS antibodies, since this antigen was formulated in a strong adjuvant [34]. However, the absence of significant antigen-specific cytotoxicity (see below) argues for the use of DC loaded with the recombinant protein over recombinant protein even in the presence of a strong adjuvant.
Previously, we reported the natural expression of BORIS transcript and protein in 4T1 mammary carcinoma [24,25] and B16 melanoma cell lines (data not shown). We used these cell lines as unmodified targets for the detection of cytolytic activity of freshly isolated immune or control splenocytes. Of note, this direct killing of tumor cells was performed without in vitro re-stimulation of these cells. Mice vaccinated with DC/mBORIS killed ~30% of 4T1 target cells at Effector:Target (E:T) ratio 50:1, and significant killing was observed even at E:T ratio 12.5:1. The effector cells isolated from not only DC/gp120, but also mBORIS immunized mice showed only background level of the 4T1 target cells killing (Fig. 2D). This cytotoxicity was associated with CD8+ T cells because it was MHC class-I restricted: the same effector cells from DC/mBORIS vaccinated mice kill only 8.7% of B16 target cells with the H-2b haplotype at an E:T ratio of 50:1 (Fig. 2D). This level of cytotoxicity may be attributable to NK cell activity.
3.3. Evaluation of therapeutic potency of DC/mBORIS vaccine
After demonstrating the immunogenicity of our DC-based vaccine, we proceeded to evaluate its therapeutic potency in a very aggressive and highly metastatic mouse mammary carcinoma model based on 4T1 cells. We previously evaluated various formulations of BORIS-based vaccines in the setting of protection against a tumor challenge using this mouse model of breast cancer [24,26]. Although these studies are important for initial proof of concept and demonstration of efficacy, such protection experiments do not accurately reflect the clinical situation. Therapeutic models with treatment initiated after tumor implantation are more representative of the human clinical situation. Accordingly, we choose to evaluate mBORIS-based immunotherapeutic strategies under therapeutic conditions (Fig. 3A). Because we have already demonstrated that the mBORIS protein with strong adjuvant was ineffective in inhibition of tumor growth in preventative vaccination studies, we did not include this group into the therapeutic setting of experiments [26]. As shown in Fig. 3B, starting at day 19, tumor volumes of mice from the DC/mBORIS group were significantly smaller compared to both control groups. The difference in tumor volumes of mice from DC/mBORIS group remained significant and was appreciably smaller than in control animals persisting up to day 22. Importantly, 3 out of 16 DC/mBORIS-immunized mice (over two independent experiments) remained tumor-free during 22 days, while all control mice developed tumors by days 15-17 after tumor implantation. Of note, one mouse in DC/gp120 group developed a very small tumor on day 17 reaching 0.004 cm3 on the day of termination of the experiment (Fig. 3B).
Figure 3.
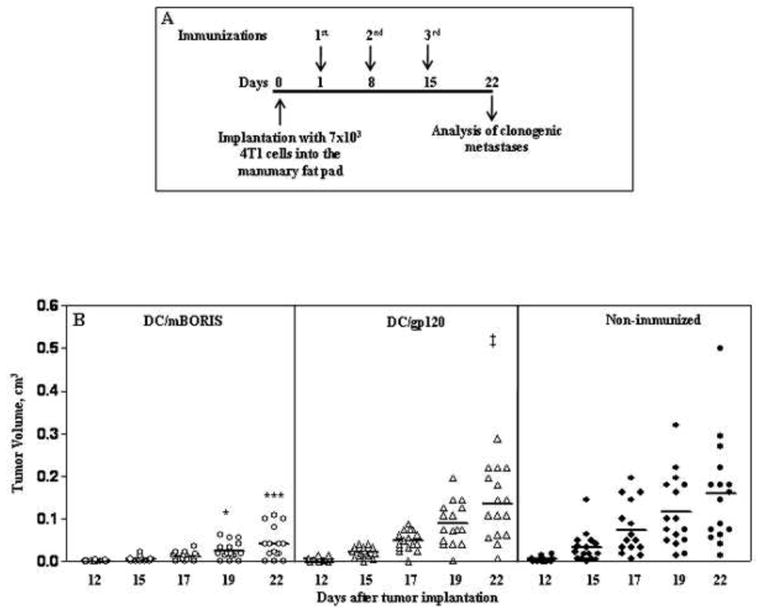
Therapeutic vaccination with DC/mBORIS inhibits the tumor growth. (A) The experimental design of the therapeutic study; mice received the immunotherapy with DC/mBORIS one day after injection of 7×103 4T1 mammary carcinoma cells. (B) Depicts the tumor size of individual animals on the designated days for each treatment along with the mean tumor volume noted by the horizontal bars. Comparison of DC/mBORIS with DC/gp120 immunized animals revealed significant differences for the designated individual days post tumor inoculation, *P<0.05 and ***P<0.001. There was no statistically significant difference between animals receiving DC/gp120 or no therapy whatsoever. ‡ In group DC/gp120 in one mouse small tumor appeared on day 17 (palpable size) reaching a volume of 0.004 cm3 on day 22.
It is well known that people die from metastatic disease and only rarely from a direct primary tumor growth. Therefore, we measured clonogenic metastases in the lungs of vaccinated and control animals on day 22 after tumor implantation. The day of termination was chosen based on data that as early as day 14 and as late as day 18 after primary inoculation, distant spontaneous metastases are observed in the lungs of 100% mice bearing 4T1 tumors. Importantly, when a 4T1 tumor reaches approximately 0.032 cm3 [the tumor diameter (TD) of 4 mm], it exhibits all characteristics of advanced human breast cancer [29]. In our studies (Fig. 4) 50% of mice (8 out of 16) immunized with DC/mBORIS were free of lung metastases. Five mice out of the remaining 8 had less than 5 metastases, while 100% of DC/gp120 immunized and non-immunized animals had greater than 5 clonogenic metastases in the lungs. Of note, average numbers of clonogenic metastases were 6.68, 38.9, and 57.7 in mice vaccinated with DC/mBORIS, DC/gp120, and control animals, respectively.
Figure 4.
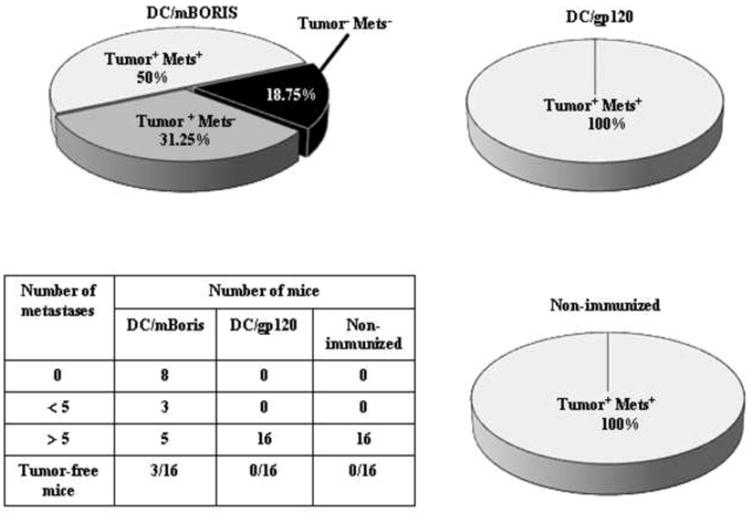
Effect of DC/mBORIS vaccine on metastatic tumor development. Pie charts depict the percentage of animals that had persistent tumor at 22 days with or without the presence of clonogenic lung metastases and the percentage of animals without persistent tumor and without lung metastases. Of note, no animals with eradication of the primary tumor were demonstrated to have lung metastases. The table depicts the range of distribution of number of clonogenic lung metastases present in the animals from the various treatment cohorts.
3.4. Analysis of the influence of the DC/mBORIS vaccine on the suppressive environment in tumor-bearing mice
Tumor development is often associated with progressive accumulation of different subpopulations of suppressor cells in the spleens and at the tumor site of animals. The impact of DC based anti-tumor vaccines on tumor-associated immune suppression is not well characterized. Accordingly, we analyzed the impact of DC/mBORIS vaccination on the micro-environment and cellular elements of tumor-associated immune suppression, in tumor bearing mice. More specifically, we analyzed the impact of vaccination on levels of the most central component of suppressive cell subsets, MDSC, which can impair anti-tumor immune responses at the tumor site and in the spleen [35]. Data presented in Fig. 5A demonstrated that vaccination with DC/mBORIS was associated with significantly decreased levels of tumor-infiltrated MDSC compared to immunization with DC/gp120. In experimental and control groups we found significant correlation between tumor volumes and numbers of tumor-infiltrated MDSC (Fig. 5B). This association of tumor volume with tumor infiltrating MDSC could be secondary to the effect of the DC/mBORIS immunotherapy on tumor growth or a combination of effects on the MDSC population and tumor growth. Of note, this decreased infiltration of MDSC in tumors of DC/mBORIS vaccinated mice was also associated with increased infiltration of CD4+ and CD8+T cells: the significantly higher numbers of these subsets of immune cells have been detected in vaccinated mice compared to that in control animals injected with DC/gp120 (Fig. 5C).
Figure 5.
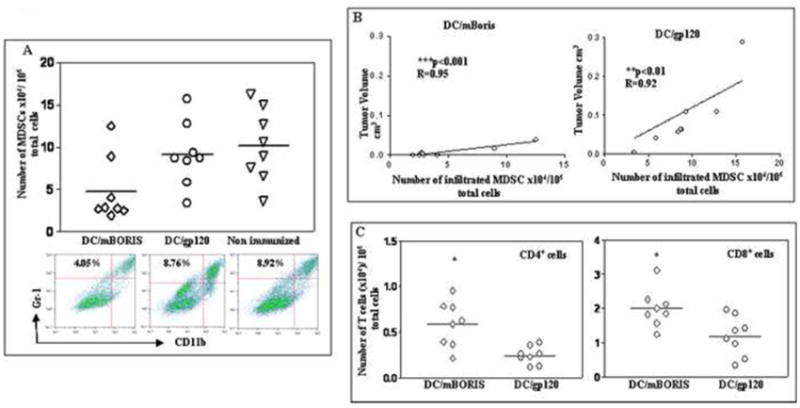
Analysis of tumor-infiltrated suppressors and effectors cells in DC/mBORIS and control (DC/gp120) immunized mice and non-immunized control mice. (A) The numbers of tumor-infiltrated MDSC significantly (*P<0.05) decreased in mice immunized with DC/mBORIS compared with the numbers of these cells infiltrated in tumor sites of control mice. Representative plots for each group are presented. (B) The significant correlations between tumor-infiltrated MDSC and tumor volumes have been observed in both DC/mBORIS vaccinated (***P<0.001, R=0.95, n=8) and DC/gp120 control (**P<0.01, R= 0.92, n=8) animals. (C) Infiltrations of both CD4+ and CD8+ T cells significantly increased (*P<0.05) in mice vaccinated with DC/mBORIS vs of that in control animals.
Tumor-infiltrated cell populations were analyzed in tumors isolated from individual animals (n=8/group) and numbers of CD4+, CD8+ and MDSC, per 106 total cells are presented in A and C.
We also measured the percentage of MDSC in spleens of experimental and control animals. In immunized animals, regardless of immunogen, the levels of MDSC in spleens of tumor-bearing mice were significantly elevated compared with tumor free naïve mice (Fig. 6A). Of note, the levels of MDSC detected in spleens isolated from DC/mBORIS and DC/gp120 immunized mice were similar, suggesting that this anatomic compartment did not experience any changes due to the administration of antigen-specific DC based immunotherapy. Not only was there no change in the number of immune suppressive cells in the spleen, but vaccination also did not alter the suppressive activity of these cells. Splenocytes isolated from DC/mBORIS and control DC/gp120 vaccinated tumor-bearing mice mixed with splenocytes isolated from naïve tumor-free mice dramatically inhibited proliferation of T-cells activated with anti-CD3 and anti-CD28 antibodies (Fig. 6B).
Figure 6.
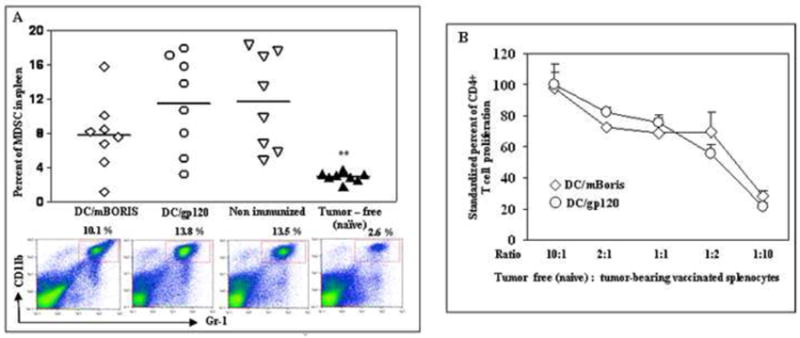
Flow cytometric analysis of splenic MDSC from individual tumor-bearing and tumor-free mice. (A) depicts the levels of MDSC within all nucleated spleen cells. Representative plots for each group are presented. (B) Splenocytes from DC/mBORIS vaccinated and DC/gp120 injected control tumor-bearing mice inhibit polyclonal activation of CFSE-labeled naïve splenic CD4+ T lymphocytes from tumor-free mice (see details in Materials and Methods). Graphs of results from flow cytometric analysis (A) of individual animals and the results from each individual animal are depicted as a symbol and the mean of all animals in each group is noted by the horizontal bar. MDSCs were significantly elevated compared to naïve tumor-free animals (**P<0.01). Average data and SD in panel B are generated from two independent experiments with splenocytes isolated from 4 naïve and 4 tumor-bearing mice.
4. Discussion
In general, metastatic disease but not primary tumors lead to significant morbidity and mortality in cancer patients [36]. Thus the promise of anti-tumor immunotherapy is the elimination of occult micro-metastases by the immune system. Recent studies have provided proof of concept for the utility of anti-cancer immunotherapy strategies in the clinical setting [3,4,37,38]. Even though numerous anti-cancer vaccines have been developed utilizing a wide range of TAAs, relatively little clinical progress in treating a wide variety of tumors has been achieved. In fact, many TAA antigens that were effective in different animal models did not induce strong anti-cancer immunity in humans, hence limiting their potential use for therapy [38]. Recent success in understanding the molecular biology and immunology of cancer provides new opportunities for generation of anti-cancer vaccines and therapeutics targeting various antigens that are not expressed in normal cells (e.g., CT-antigens) that should be recognized by the immune system as foreign molecules [8,12,13].
Previously, we demonstrated the protective efficacy of one such CT-antigen, mBORIS delivered by viral or plasmid vector [25]. Although protection against tumor challenge is an obvious first step in developing a viable immunotherapeutic strategy, treatment of patients requires that an immunotherapy have activity against pre-existing tumors. Such activity against pre-existing tumor typically requires even more potent strategies than protection against a tumor challenge. The recognition of DCs as extremely potent antigen-presenting and immune stimulating cells suggests that these cells may be effective in breaching the threshold of activity against pre-existing tumor. Thus, we examined the feasibility of a novel CT-antigen, BORIS as a target for immunotherapy of breast cancer through improved delivery of this CT-antigen by very potent antigen-presenting and immunostimulatory cells, DCs [39,40]. Accordingly, we decided to study for the first time, the immunotherapeutic potency of a mBORIS based vaccine delivered by arguably the most effective cellular adjuvant, DC. Importantly, we have demonstrated that vaccination with DCs loaded with mBORIS enhanced the numbers of anti-BORIS specific IFN-γ producing splenocytes and proliferation of anti-mBORIS specific CD4+ helper T cells. The DC/mBORIS vaccine also induced CD8+ T cells that killed 4T1 target cells of H2d haplotype without in vitro restimulation(direct CTL activity), but not B16 melanoma cells of H-2b haplotype. This suggests that this strategy elicits MHC class I restricted CTL activity with its associated T helper class I response. While anti-tumor cellular immunity was dramatically enhanced by delivering mBORIS in vivo through the cellular adjuvant, DCs, under these conditions we did not detect improvement in the elicited humoral immunity. In contrast, a recombinant protein vaccine formulated in Th1-type adjuvant did not induce cytolytic responses despite eliciting robust antigen-specific humoral and CD4+ proliferative responses (Fig. 2). Thus, while DC/mBORIS vaccine elicited only modest levels of anti-BORIS antibodies, it generated strong antigen-specific cellular immunity with a more pronounced Th1 bias in Th2 prone BALB/c mice.
The development of cancer immunotherapy has traditionally been dependent on the use of murine tumor models. In our prior and current studies [24,26], we have used the spontaneously metastasizing and poorly immunogenic 4T1 mammary carcinoma derived from a spontaneously arising BALB/c mammary tumor as an animal model [41]. This mouse tumor model shares many characteristics with human breast cancer, both in their immunogenicity and in their growth and metastatic properties [29,41]. Among the most significant properties of this model is that the 4T1 tumor spontaneously metastasizes while the primary tumor is in place, in a pattern comparable to human breast carcinoma [42,43]. More specifically, in this model, metastases are predominately found in the lung and liver and less frequently found in the brain, lymph nodes, and bones [29]. These characteristics of 4T1 cells make it an excellent model to evaluate the effects of drugs and vaccines on breast tumor metastases as well as primary tumor growth. Using this stringent mouse mammary carcinoma model, we demonstrated that initiation of DC/mBORIS vaccination after 4T1 tumor implantation significantly inhibited tumor growth. Importantly, 3 out of 16 DC/mBORIS-immunized mice remained tumor-free during the entire experiment, while all control mice developed tumors (Fig. 3, 4). It is likely that tumor growth was inhibited not only by systemic cellular immune responses described above and manifest by the increased numbers of tumor infiltrated immune-effector CD4 + and CD8 + T cells detected in vaccinated vs. control mice (Fig. 5C). Considering our previous data demonstrating that protein based vaccine that activates CD4+ T cells and induces humoral immune responses did not protect mice in preventative setting, we speculate that this inhibition is CD8+ T cell-mediated. However, precise investigation of cells responsible for tumor rejection by in vivo depletion of CD4+, CD8+ and NK cells using i.v. antibody injection will be performed to address this concept. More importantly, in addition to the substantial effect on primary tumor growth, the DC/mBORIS vaccine had a dramatic effect on the development of metastatic disease (Fig. 4). This therapeutic effect of DC/mBORIS vaccine is quite remarkable, as similar results in the 4T1 model have only been obtained when STAT6-/- mice with enhanced type 1 responses were used, after surgical removal of the primary tumor or when 4T1 was implanted s.c. but not in mammary pad [44,45]. Notably, preventive (not therapeutic) vaccination with another CT-antigen, MAGE-b delivered by L. monocytogenes did not have an effect on primary 4T1 tumors, although it did reduce the numbers of metastases in the lymph nodes, diaphragm, liver, kidney, and spleen, but not in the lungs [46]. Here, we demonstrated that even in a therapeutic setting, a novel CT antigen, mBORIS, delivered by DC dramatically inhibited both the tumor growth (18.75% tumor free) and the lung metastatases (50% of animals being free of detectable metastatic disease).
The development of potent immunotherapeutic strategies for common tumors, such as breast cancer, would be expected to improve disease free and overall survival, leading to improved patient quality of life and obvious societal benefit. Recent reviews have described barriers to successful immunotherapy, including immune suppressor cells and cytokines, all of which have implications for the design of anti-tumor immunotherapeutics [47,48]. It has become increasingly clear that immunotherapy is more likely to be successful if it strongly activates anti-tumor immunity while concurrently inhibiting the suppressive environment not only in lymphoid organs, but also at the tumor site. Accordingly, we evaluated the suppressive tumor micro-environment in vaccinated tumor bearing mice while testing the therapeutic potency of our novel mBORIS antigen delivered by DCs. MDSC are one of the major functional cellular elements of this phenomenon. Significant increases in numbers of MDSC in spleens and tumor sites have also been shown in 4T1 mouse model of breast cancer [44,49,50,51]. Our data confirms that the levels of MDSC were significantly elevated in the spleens of 4T1 tumor bearing mice compared to tumor-free naïve animals.
Importantly, the percentages of these cells were unchanged after immunizations with either DC/mBORIS or DC/gp120, and also this did not change the functional activity of the suppressor cells that were present (Fig 6). Importantly, infiltrations of both CD4+ and CD8+ T cells were significantly increased, while the numbers of MDSC were significantly decreased in tumors of mice immunized with DC/mBORIS compared to the numbers of these cells infiltrated in tumor sites of control mice. Previously it was shown that the levels of suppressor cells correlates with tumor size [50,52]. In our experiments the levels of tumor-infiltrated MDSC correlate with tumor size not only in control mice, but also in mice vaccinated with DC/mBORIS. The underlying cause of the discordance between the effects of the immunotherapy on tumor infiltrating immune suppressive cellular elements and splenic populations of these same elements, is unknown. Also, it is unclear whether the smaller tumor size in DC/mBORIS vaccinated mice is the basis of lower number of MDSC, or whether the lower level of these suppressor cells induced by vaccination is allowing immune-effector CD4+ and CD8+ T cells to fight and inhibit tumor growth. We favor the hypothesis that the DC/mBORIS immunotherapy resulted in increased TAA-specific DC activated T effector cells that would be expected to traffic to sites of TAA expression, i.e., the tumor site. The complexity of interactions between activated effector cells and local suppressive cellular elements may explain the local effects that were associated with tumor size. Any effects on these cellular elements in the spleen would be expected to lag behind the local effects. It is obvious that more functional tests and pre-clinical testing need to be done to clarify this important question. For example, vaccination with mBORIS could be performed after surgical removal of tumors while metastatic diseases as well as survival of animals should be tested later. Regardless of the mechanism, it is important that even in the absence of changes in the splenic MDSC populations, the modest, but significant reduction of tumor-infiltrated MDSC in animals vaccinated with DC/mBORIS was associated with strong anti-tumor cellular immunity, significantly inhibited tumor growth, and decreased metastatic disease in mice with pre-existing 4T1 mammary carcinoma.
Current data suggest that tumor-associated immune suppression contributes to the tumor growth and failure of immunotherapeutic strategies in humans [35,48,53]. MDSC are one of the major component of suppressive cell subsets that can impair anti-tumor immune responses in mouse tumor models [35] and in cancer patients [54], this occurs not only through direct inhibition of T cell responses, but also through the activation of Tregs [55] and angiogenesis [56]. A significant increase in MDSC and Treg numbers have been detected in the blood of patients with different types of cancers [35,57], and these increases in circulating MDSC and Treg correlate with cancer stage [57,58] and possibly tumor burden. Therefore, higher numbers of suppressor cells in cancer patients have been associated with decreased efficacy of strategies to elicit anti-tumor immune responses. Data reported herein and that of others argue for strategies that not only elicit strong anti-cancer immunity, but also robust inhibition of a suppressive environment. In fact, several approaches have been tested in the clinic to deplete or inactivate this population of suppressor cells before administration of immune therapies. These include administrating anti-CD25 [57], anti-CTLA-4 antibodies [59], and cyclophosphamide [60] for depletion of Treg cells and tyrosine kinase inhibitor sunitinib [61,62], all-trans retinoic acid (ATRA) [63] etc for depletion of MDSC.
5. Conclusion
In conclusion, by two distinct measures, we have demonstrated that this DC/mBORIS vaccine has therapeutic efficacy in a treatment setting. This vaccine strategy inhibited the growth of pre-existing, very aggressive and poorly immunogenic 4T1 mammary carcinoma. Importantly, it also decreased the metastatic disease in the lungs of all experimental animals. Taken together, the effect of this strategy and target antigen on the growth of primary 4T1 tumors and lung metastases support the use of a potent immunotherapy that can rapidly destroy tumor cells before selection pressure leads to escape variants when the tumor burden is limited, and thus when there is limited tumor-associated immune suppression. These data also suggest that this strategy is an excellent platform on which to build multi-pronged anti-tumor attacks that incorporate both strategies to limit the degree of tumor-associated immune suppression and potent antigen-specific vaccination, particularly when there is substantial tumor burden. Together this combination may improve the anti-tumor efficacy of this vaccine platform and allow us to better understand the mechanisms of effectors/suppressors relationship for the ultimate goal of translation into the clinical arena.
Acknowledgments
We would like to thank students Mr. M. Shugay, Mr. J. Khlghatyan, Mr. Tiraturyan, Ms. A. Davtyan and Ms. A. Hovakimyan for technical help and Dr. I. Petrushina for valuable comments. This work was supported by Susan Komen Foundation BCTR0707720 for ELN and NIH AG-20241 for DHC and MGA; NS-50895 for DHC and MGA; NS57395 for MGA grants and in part by Intramural Research Program of the National Institute of Allergy and Infectious Diseases, NIH.
Footnotes
Declaration of competing interests Author(s) declare that they have no competing interests.
> DC/mBORIS vaccine has therapeutic efficacy in a treatment setting>Vaccination increased the number of tumor-infiltrated effector T cells> Inhibited the growth of pre-existing, aggressive, nonimmunogenic 4T1 mammary carcinoma> Decreased the metastatic disease in the lungs>
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Mikayel Mkrtichyan, Email: mikayel.mkrtichyan@nih.gov.
Anahit Ghochikyan, Email: aghochikyan@immed.org.
Hayk Davtyan, Email: hayk.davtyan@immed.org.
Nina Movsesyan, Email: nmovsesy@uci.edu.
Dmitry Loukinov, Email: DLOUKINOV@niaid.nih.gov.
Victor Lobanenkov, Email: VLOBANENKOV@niaid.nih.gov.
David H. Cribbs, Email: cribbs@uci.edu.
Amanda K. Laust, Email: alaust@uci.edu.
Edward L. Nelson, Email: enelson@uci.edu.
Michael G. Agadjanyan, Email: magadjanyan@immed.org.
References
- 1.WHO. Cancer. 2006 Fact sheet No 297. [Google Scholar]
- 2.Bertucci F, Birnbaum D. Distant metastasis: not out of reach any more. J Biol. 2009;8:28. doi: 10.1186/jbiol128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson KS. Tumor vaccines for breast cancer. Cancer Invest. 2009;27:361–368. doi: 10.1080/07357900802574421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schadendorf D, Algarra SM, Bastholt L, Cinat G, Dreno B, et al. Immunotherapy of distant metastatic disease. Ann Oncol. 2009;20(Suppl 6):vi41–50. doi: 10.1093/annonc/mdp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Overwijk WW. Breaking tolerance in cancer immunotherapy: time to ACT. Curr Opin Immunol. 2005;17:187–194. doi: 10.1016/j.coi.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 6.Waldmann TA. Effective cancer therapy through immunomodulation. Annu Rev Med. 2006;57:65–81. doi: 10.1146/annurev.med.56.082103.104549. [DOI] [PubMed] [Google Scholar]
- 7.Cunto-Amesty G, Monzavi-Karbassi B, Luo P, Jousheghany F, Kieber-Emmons T. Strategies in cancer vaccines development. Int J Parasitol. 2003;33:597–613. doi: 10.1016/s0020-7519(03)00054-7. [DOI] [PubMed] [Google Scholar]
- 8.Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng F, Gabrilovich D, Sotomayor EM. Immune tolerance in breast cancer. Breast Dis. 2004;20:93–103. doi: 10.3233/bd-2004-20111. [DOI] [PubMed] [Google Scholar]
- 10.Smyth MJ, Godfrey DI, Trapani JA. A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol. 2001;2:293–299. doi: 10.1038/86297. [DOI] [PubMed] [Google Scholar]
- 11.Makki A, Weidt G, Blachere NE, Lefrancois L, Srivastava PK. Immunization against a dominant tumor antigen abrogates immunogenicity of the tumor. Cancer Immun. 2002;2:4. [PubMed] [Google Scholar]
- 12.Scanlan MJ, Simpson AJ, Old LJ. The cancer/testis genes: review, standardization, and commentary. Cancer Immun. 2004;4:1. [PubMed] [Google Scholar]
- 13.Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5:615–625. doi: 10.1038/nrc1669. [DOI] [PubMed] [Google Scholar]
- 14.Davis ID, Chen W, Jackson H, Parente P, Shackleton M, et al. Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvant induces broad integrated antibody and CD4(+)and CD8(+) T cell responses in humans. Proc Natl Acad Sci U S A. 2004;101:10697–10702. doi: 10.1073/pnas.0403572101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marchand M, van Baren N, Weynants P, Brichard V, Dreno B, et al. Tumor regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by gene MAGE-3 and presented by HLA-A1. Int J Cancer. 1999;80:219–230. doi: 10.1002/(sici)1097-0215(19990118)80:2<219::aid-ijc10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 16.Klenova EM, Morse HC, 3rd, Ohlsson R, Lobanenkov VV. The novel BORIS + CTCF gene family is uniquely involved in the epigenetics of normal biology and cancer. Semin Cancer Biol. 2002;12:399–414. doi: 10.1016/s1044-579x(02)00060-3. [DOI] [PubMed] [Google Scholar]
- 17.Loukinov DI, Pugacheva E, Vatolin S, Pack SD, Moon H, et al. BORIS, a novel male germ-line-specific protein associated with epigenetic reprogramming events, shares the same 11-zinc-finger domain with CTCF, the insulator protein involved in reading imprinting marks in the soma. Proc Natl Acad Sci U S A. 2002;99:6806–6811. doi: 10.1073/pnas.092123699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Arcy V, Pore N, Docquier F, Abdullaev ZK, Chernukhin I, et al. BORIS, a paralogue of the transcription factor, CTCF, is aberrantly expressed in breast tumours. Br J Cancer. 2008;98:571–579. doi: 10.1038/sj.bjc.6604181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong JA, Kang Y, Abdullaev Z, Flanagan PT, Pack SD, et al. Reciprocal Binding of CTCF and BORIS to the NY-ESO-1 Promoter Coincides with Derepression of this Cancer-Testis Gene in Lung Cancer Cells. Cancer Res. 2005;65:7763–7774. doi: 10.1158/0008-5472.CAN-05-0823. [DOI] [PubMed] [Google Scholar]
- 20.Kouprina N, Noskov VN, Pavlicek A, Collins NK, Schoppee Bortz PD, et al. Evolutionary diversification of SPANX-N sperm protein gene structure and expression. PLoS One. 2007;2:e359. doi: 10.1371/journal.pone.0000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vatolin S, Abdullaev Z, Pack SD, Flanagan PT, Custer M, et al. Conditional Expression of the CTCF-Paralogous Transcriptional Factor BORIS in Normal Cells Results in Demethylation and Derepression of MAGE-A1 and Reactivation of Other Cancer-Testis Genes. Cancer Res. 2005;65:7751–7762. doi: 10.1158/0008-5472.CAN-05-0858. [DOI] [PubMed] [Google Scholar]
- 22.Woloszynska-Read A, James SR, Link PA, Yu J, Odunsi K, et al. DNA methylation-dependent regulation of BORIS/CTCFL expression in ovarian cancer. Cancer Immun. 2007;7:21. [PMC free article] [PubMed] [Google Scholar]
- 23.Dougherty CJ, Ichim TE, Liu L, Reznik G, Min WP, et al. Selective apoptosis of breast cancer cells by siRNA targeting of BORIS. Biochem Biophys Res Commun. 2008;370:109–112. doi: 10.1016/j.bbrc.2008.03.040. [DOI] [PubMed] [Google Scholar]
- 24.Loukinov D, Ghochikyan A, Mkrtichyan M, Ichim TE, Lobanenkov VV, et al. Antitumor efficacy of DNA vaccination to the epigenetically acting tumor promoting transcription factor BORIS and CD80 molecular adjuvant. J Cell Biochem. 2006 doi: 10.1002/jcb.20953. [DOI] [PubMed] [Google Scholar]
- 25.Ghochikyan A, Mkrtichyan M, Loukinov D, Mamikonyan G, Pack SD, et al. Epigenetically acting tumor promoting transcription factor BORIS is widely expressed TAA inducing anti-tumor specific T cell responses. J Immunol. 2007;178:556–573. [Google Scholar]
- 26.Mkrtichyan M, Ghochikyan A, Loukinov D, Davtyan H, Ichim TE, et al. DNA, but not protein vaccine based on mutated BORIS antigen significantly inhibits tumor growth and prolongs the survival of mice. Gene Ther. 2008;15:61–64. doi: 10.1038/sj.gt.3303044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 28.Kaplan JM, Yu Q, Piraino ST, Pennington SE, Shankara S, et al. Induction of antitumor immunity with dendritic cells transduced with adenovirus vector-encoding endogenous tumor-associated antigens. J Immunol. 1999;163:699–707. [PubMed] [Google Scholar]
- 29.Pulaski BA, Ostrand-Rosenberg S. Reduction of established spontaneous mammary carcinoma metastases following immunotherapy with major histocompatibility complex class II and B7.1 cell-based tumor vaccines. Cancer Res. 1998;58:1486–1493. [PubMed] [Google Scholar]
- 30.Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W. Current Protocols in Immunology. New York: John Willey & Sons; 2004. Mouse 4T1 Breast Tumor Model; p. 20.22. unit. [Google Scholar]
- 31.Pulaski BA, Terman DS, Khan S, Muller E, Ostrand-Rosenberg S. Cooperativity of Staphylococcal aureus enterotoxin B superantigen, major histocompatibility complex class II, and CD80 for immunotherapy of advanced spontaneous metastases in a clinically relevant postoperative mouse breast cancer model. Cancer Res. 2000;60:2710–2715. [PubMed] [Google Scholar]
- 32.Schreurs MW, Eggert AA, de Boer AJ, Vissers JL, van Hall T, et al. Dendritic cells break tolerance and induce protective immunity against a melanocyte differentiation antigen in an autologous melanoma model. Cancer Res. 2000;60:6995–7001. [PubMed] [Google Scholar]
- 33.Wang HY, Fu T, Wang G, Zeng G, Perry-Lalley DM, et al. Induction of CD4(+)T cell-dependent antitumor immunity by TAT-mediated tumor antigen delivery into dendritic cells. J Clin Invest. 2002;109:1463–1470. doi: 10.1172/JCI15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun HX, Xie Y, Ye YP. Advances in saponin-based adjuvants. Vaccine. 2009;27:1787–1796. doi: 10.1016/j.vaccine.2009.01.091. [DOI] [PubMed] [Google Scholar]
- 35.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hortobagyi GN. Treatment of breast cancer. N Engl J Med. 1998;339:974–984. doi: 10.1056/NEJM199810013391407. [DOI] [PubMed] [Google Scholar]
- 37.Waeckerle-Men Y, Uetz-von Allmen E, Fopp M, von Moos R, Bohme C, et al. Dendritic cell-based multi-epitope immunotherapy of hormone-refractory prostate carcinoma. Cancer Immunol Immunother. 2006;55:1524–1533. doi: 10.1007/s00262-006-0157-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palucka K, Ueno H, Fay J, Banchereau J. Harnessing dendritic cells to generate cancer vaccines. Ann N Y Acad Sci. 2009;1174:88–98. doi: 10.1111/j.1749-6632.2009.05000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008;29:372–383. doi: 10.1016/j.immuni.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 41.Miller BE, Roi LD, Howard LM, Miller FR. Quantitative selectivity of contact-mediated intercellular communication in a metastatic mouse mammary tumor line. Cancer Res. 1983;43:4102–4107. [PubMed] [Google Scholar]
- 42.Tomin R, Donegan WL. Screening for recurrent breast cancer--its effectiveness and prognostic value. J Clin Oncol. 1987;5:62–67. doi: 10.1200/JCO.1987.5.1.62. [DOI] [PubMed] [Google Scholar]
- 43.Rutgers EJ, van Slooten EA, Kluck HM. Follow-up after treatment of primary breast cancer. Br J Surg. 1989;76:187–190. doi: 10.1002/bjs.1800760227. [DOI] [PubMed] [Google Scholar]
- 44.Chen L, Huang TG, Meseck M, Mandeli J, Fallon J, et al. Rejection of metastatic 4T1 breast cancer by attenuation of Treg cells in combination with immune stimulation. Mol Ther. 2007;15:2194–2202. doi: 10.1038/sj.mt.6300310. [DOI] [PubMed] [Google Scholar]
- 45.Ostrand-Rosenberg S, Clements VK, Terabe M, Park JM, Berzofsky JA, et al. Resistance to metastatic disease in STAT6-deficient mice requires hemopoietic and nonhemopoietic cells and is IFN-gamma dependent. J Immunol. 2002;169:5796–5804. doi: 10.4049/jimmunol.169.10.5796. [DOI] [PubMed] [Google Scholar]
- 46.Kim SH, Castro F, Gonzalez D, Maciag PC, Paterson Y, et al. Mage-b vaccine delivered by recombinant Listeria monocytogenes is highly effective against breast cancer metastases. Br J Cancer. 2008;99:741–749. doi: 10.1038/sj.bjc.6604526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith FO, Downey SG, Klapper JA, Yang JC, Sherry RM, et al. Treatment of metastatic melanoma using interleukin-2 alone or in conjunction with vaccines. Clin Cancer Res. 2008;14:5610–5618. doi: 10.1158/1078-0432.CCR-08-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–179. doi: 10.1111/j.1600-065X.2008.00602.x. [DOI] [PubMed] [Google Scholar]
- 49.Darrasse-Jeze G, Bergot AS, Durgeau A, Billiard F, Salomon BL, et al. Tumor emergence is sensed by self-specific CD44hi memory Tregs that create a dominant tolerogenic environment for tumors in mice. J Clin Invest. 2009;119:2648–2662. doi: 10.1172/JCI36628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006;176:284–290. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- 51.Stewart TJ, Greeneltch KM, Reid JE, Liewehr DJ, Steinberg SM, et al. Interferon Regulatory Factor-8 Modulates the Development of Tumor-Induced CD11bGr-1 Myeloid Cells. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2009.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer. 2009;124:2621–2633. doi: 10.1002/ijc.24249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 55.Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135:234–243. doi: 10.1053/j.gastro.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 56.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 57.Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115:3623–3633. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, et al. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ribas A. Anti-CTLA4 Antibody Clinical Trials in Melanoma. Update Cancer Ther. 2007;2:133–139. doi: 10.1016/j.uct.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Audia S, Nicolas A, Cathelin D, Larmonier N, Ferrand C, et al. Increase of CD4+ CD25+ regulatory T cells in the peripheral blood of patients with metastatic carcinoma: a Phase I clinical trial using cyclophosphamide and immunotherapy to eliminate CD4+ CD25+ T lymphocytes. Clin Exp Immunol. 2007;150:523–530. doi: 10.1111/j.1365-2249.2007.03521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–2522. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148–2157. doi: 10.1158/1078-0432.CCR-08-1332. [DOI] [PubMed] [Google Scholar]
- 63.Ghosh I, Kumar L, Seth R, Thavraj V. Sustained remission achieved with ATRA and chemotherapy in second relapse of acute promyelocytic leukemia in Down syndrome. J Pediatr Hematol Oncol. 2009;31:539–540. doi: 10.1097/MPH.0b013e3181979494. [DOI] [PubMed] [Google Scholar]