

Abstract
Researchers have long noted an excess of patients with schizophrenia were born during the months of January and March. This winter birth effect has been hypothesized to result either from various causes such as vitamin D deficiency (McGrath, 1999; McGrath et al., 2010), or from maternal infection during pregnancy. Infection with a number of viruses during pregnancy including influenza, and rubella are known to increase the risk of schizophrenia in the offspring (Brown, 2006). Animal models using influenza virus or PolyI:C, a viral mimic, have been able to replicate many of the brain morphological, genetic, and behavioral deficits of schizophrenia (Meyer et al., 2006, 2008a, 2009; Bitanihirwe et al. 2010; Meyer and Feldon, 2010; Short et al., 2010). Using a murine model of prenatal viral infection, our laboratory has shown that viral infection on embryonic days 9, 16, and 18 leads to abnormal expression of brain genes and brain structural abnormalities in the exposed offspring (Fatemi et al., 2005, 2008a,b, 2009a,b). The purpose of the current study was to examine gene expression and morphological changes in the placenta, hippocampus, and prefrontal cortex as a result of viral infection on embryonic day 7 of pregnancy. Pregnant mice were either infected with influenza virus [A/WSN/33 strain (H1N1)] or sham-infected with vehicle solution. At E16, placentas were harvested and prepared for either microarray analysis or for light microscopy. We observed significant, upregulation of 77 genes and significant downregulation of 93 genes in placentas. In brains of exposed offspring following E7 infection, there were changes in gene expression in prefrontal cortex (6 upregulated and 24 downregulated at P0; 5 upregulated and 14 downregulated at P56) and hippocampus (4 upregulated and 6 downregulated at P0; 6 upregulated and 13 downregulated at P56). QRT-PCR verified the direction and magnitude of change for a number of genes associated with hypoxia, inflammation, schizophrenia, and autism. Placentas from infected mice showed a number of morphological abnormalities including presence of thrombi and increased presence of immune cells. Additionally, we searched for presence of H1N1 viral-specific genes for M1/M2, NA, and NS1 in placentas of infected mice and brains of exposed offspring and found none. Our results demonstrate that prenatal viral infection disrupts structure and gene expression of the placenta, hippocampus, and prefrontal cortex potentially explaining deleterious effects in the exposed offspring without evidence for presence of viral RNAs in the target tissues.
Keywords: placenta, influenza, brain, microarray, schizophrenia, viral genes
1. Introduction
Increased rates of schizophrenia have been associated with higher latitude and colder climate (Kinney et al., 2009). Different hypotheses have been proposed for this increase including vitamin D deficiency (McGrath, 1999; McGrath et al., 2010) and prenatal viral infection (Lewis et al. 2001; Fatemi, 2005; Brown, 2006). First proposed by McGrath (1999), prenatal vitamin D deficiency as a risk factor for schizophrenia could potentially explain several epidemiological findings including season of birth, latitude, differences in rates of schizophrenia between urban vs. rural populations, and the increased risk in 2nd generation migrants to northern countries (McGrath, 1999; Kinney et al., 2009; McGrath et al., 2010). Animal models of developmental vitamin D (DVD) deficiency have shown changes in brain development (Eyles et al., 2009), synaptic plasticity (Grecksch et al., 2009), and behavior (Grecksch et al., 2009; Fernandes de Abreu et al., 2010; Kesby et al., 2010) consistent with schizophrenia.
Over the past thirty years, numerous reports have established that viral infections during pregnancy increase the risk for the development of schizophrenia in the offspring (Lewis et al. 2001; Fatemi, 2005; Brown, 2006). An excess of schizophrenic patients are born during late winter and spring indicating the potential of influenza infections for these cases. Indeed, studies suggest that 5–15% excess schizophrenic births in the Northern Hemisphere occur during the months of January and March (Hare et al. 1972; Machon et al. 1983; Susser et al. 1999; Boyd et al. 1986; Pallast et al. 1994). Currently, there is debate whether direct infection of the offspring leads to changes ultimately resulting in schizophrenia or whether production of maternal cytokines in response to infection is responsible (Aronson et al. 2001,Aronson et al. 2002; Shi et al. 2009).
The current prevailing opinion is in favor of maternal cytokines as the causative agent of pathology in the developing offspring. A series of studies in different animal models for schizophrenia (Carpenter and Koenig, 2009) shows that maternal infection with human influenza or poly I:C can cause abnormalities in behavioral indices such as PPI and latent inhibition (LI) (Shi et al. 2003; Smith et al. 2007; Zuckerman and Weiner, 2003), both of which are similarly disrupted in subjects with schizophrenia. Recently, it has been demonstrated that injection of pregnant mice with IL-6, but not IFNγ, at E12.5 resulted in deficits in PPI and LI in the adult offspring but not juveniles (Smith et al. 2007), similar to what is observed in schizophrenia. Smith et al. (2007) also found that poly I:C induced behavioral deficits in PPI, LI, and open field tests could be reversed if the poly I:C was co-administered with anti-IL-6 antibodies (Smith et al. 2007). Using IL-6 knockout mice, injection of poly I:C had no effect on PPI, open field test, and social interaction test (Smith et al. 2007). Inhibition of IL-6-mediated JAK2/STAT3 signaling using luteolin and its structural analog diosmin, has been shown to reduce behavioral deficits in social interaction in the offspring of mice born to dams injected with IL-6 on E12.5 of pregnancy (Parker-Athill et al. 2009). Interestingly, overexpression of anti-inflammatory cytokine IL-10 modulates poly I:C-induced behaviors such as PPI and LI (Meyer et al. 2008b). Taken together, these results suggest that pro-inflammatory cytokines, in particular IL-6, are key to mediating the effects of poly I:C infection on behavioral deficits in rodents that mimic similar deficits in schizophrenia and autism (Smith et al. 2007).
There is also evidence that the virus itself crosses the placenta to directly infect the fetus. Using a mouse animal model Aaronson et al (2001,2002) has shown that viral RNA for influenza A/WSN/33 strain (H1N1) persisted in the brains of offspring of virally infected dams. Recently a human postmortem study (Gu et al. 2007) demonstrated that the avian influenza virus (H5N1) can be transmitted across the placenta from mother to fetus. Taken together, these data show that both strains, at the expected doses contracted, can infect and be transmitted transplacentally to the fetus. These studies further provide clear evidence that one does not need viremia to induce placental passage of the virus.
An important aspect neglected in this debate is that the site of pathology may be the placenta. Whether the virus, traveling from the lungs, crosses the placenta; or whether in response to infection, maternal cytokines are produced which cross the placenta remains unknown. In the current study we examined the placenta as a site of pathology following prenatal viral infection on E7, in addition to prefrontal cortex and hippocampus in the exposed offspring. We hypothesized that viral infection would lead to gene expression and anatomical abnormalities in the exposed mice.
2. Materials and Methods
2.1. Viral infection
All experimental protocols used in this study were approved by the Institute for Animal Care and Use and Institutional Biosafety Committees at the University of Minnesota. A sublethal dose of influenza A/NWS/33 (H1N1) (diluted to 10−4.5) was administered intranasally (i.n.) on the seventh day of pregnancy (E7) to pregnant C57BL6J mice (N=41) (Charles River, Wilmington, MA) as previously described (Fatemi et al. 2008a,b, 2009a,b). A second group of control dams (N=41) were sham-infected i.n. with 90 μl of minimum essential medium (MEM). Both sets of mice received drinking water containing 0.006% oxytetracycline (Pfizer, New York, NY) to control for the possibility of bacterial infections.
2.2. Placental collection and dissection
Pregnant mice were deeply anesthetized as previously described (Fatemi et al. 2008a,b, 2009a,b) and placentae were harvested on day 16 of pregnancy (E16). We chose E16 because the placenta was still intact and we were able to isolate it along with the concepti for future experiments. One set of placentae (N=3 infected, N=3 control) was removed following perfusion with phosphate-buffered 4% paraformaldehyde (pH 7.4) with later immersion in the same fixative for 14 days at 4°C for light microscopic studies in a different set of mice. Placental tissue was separated from the developing embryo and was snap-frozen by immersion in liquid nitrogen and stored at −80°C for further assays.
2.3. Brain collection and dissection
Pregnant mice were allowed to deliver pups (N=18 infected; N=16 control). Day of delivery was designated as postnatal day zero (P0). Male littermates at P0, P14, P35, and P56 were obtained for experiments. Groups of infected and sham-infected male neonates were deeply anesthetized as previously described (Fatemi et al. 2008a,b, 2009a,b). Brains were removed from skull cavities following perfusion as previously described (Fatemi et al. 2008a,b, 2009a,b) for all imaging studies (see section 2.6). For gene array studies, unfixed brains were snap-frozen by immersion in liquid nitrogen and stored at −80°C. Prefrontal cortex and hippocampus were dissected as previously described (Fatemi et al. 2009a,b).
2.4. Microarray
Placentae (N=3 control and N=3 infected) were harvested at E16, snap frozen in liquid nitrogen and stored at −80°C for microarray studies. Microarray was performed as previously described (Fatemi et al., 2008a, 2009a,b).
Simultaneously, as described in section 2.3, a separate group of mothers were allowed to deliver their pups. Prefrontal cortex and hippocampus were dissected as previously described (Fatemi et al., 2008a, 2009a,b) from exposed (N=3) and control (N=3) offspring at each of two postnatal time points: P0 (birth) and P56 (adulthood). Following dissection, brain tissue was snap frozen in liquid nitrogen and stored at −80°C for microarray analysis, which was performed as previously described (Fatemi et al. 2008a, 2009a,b).
Mouse Genome 430 2.0 array was obtained from Affymetrix. The array provided comprehensive coverage of the transcribed mouse genome on a single array analyzing 39,000 transcripts and variants for over 14,000 genes. The oligonucleotide sequences, their locations on the array, and other annotations are available in files that can be viewed or downloaded from www.netaffx.com.
2.5. QRT-PCR
Quantitative RT-PCR for selected genes of interest from our microarray data set, including genes that have previously been associated with autism or schizophrenia was performed as previously described (Fatemi et al. 2008a, 2009a,b) using the TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA) with GAPDH and β-actin as endogenous control assays. Primary analysis of the acquired signal data was performed in SDS 2.3 and RQ Manager 1.2 (Applied Biosystems). Outlier reactions were removed after Grubb’s test identification and differential expression was calculated using the ΔΔCT method (Livak and Schmittgen, 2001). Primers for H1N1 A/NWS/33 viral genes were: matrix protein 1/matrix protein 2 (M1/M2) forward primer – AGTGGCATTTGGCCTGGTAT, reverse primer – GAGACCGATGCTGGGAGTCA; neuraminidase (NA) forward primer – CCTTCCCCTTCTCGATCTTGA, reverse primer – CCGATGGCCCAAGTGATG; and nonstructural protein 1 (NS1) forward primer – TCATGCCCAAGCAGAAAGTG, reverse primer – CATGATCGCCTGGTCCATTC; based on the work of Lo et al (2003).
2.6. DTI and MR scanning
Brains from C57BL/6 male neonates born to infected and sham infected E7 mothers at P14 (which corresponds to childhood) and P35 (which corresponds to adolescence) (N=3 infected, N=3 control, 1 male/litter per group) were perfusion fixed in 4% paraformaldehyde in PBS buffer (pH 7.4) and subjected to DTI and MR scanning in PBS at room temperature as previously described (Fatemi et al. 2008a, 2009a,b; Mori et al. 2001).
2.7. Light microscopy
For light microscopy placentae were embedded in resin and sectioned using an ultratome at 40 μm (N=3 placentae infected; N=3 placentae control). Slides were stained using toluidine blue or epoxy tissue stain and examined for morphology as previously described (Liu et al. 2007).
2.6. Statistical analysis
Two-tailed student’s t-tests were used to test for differences in mRNA expression levels between infected and sham-infected mice. For microarray analysis, significant differences were defined as those with at least a 1.5 fold change and a p value < 0.05. For qRT-PCR, signal values for GAPDH and β-actin were averaged and used for sample normalization. Two-tailed student’s t-tests were then performed to examine differences between infected and sham-infected placentae or brain samples. T-tests were also used to measure differences in brain volume and fractional anisotropy between infected and sham-infected mice. Significance was defined as p<0.05.
3. Results
3.1. Gene expression changes in placenta following infection at E7
Microarray analysis of placental tissue found a significant (fold change of at least 1.5, p<0.05) upregulation of 77 genes and downregulation of 93 genes (Appendix A). Among the important downregulated genes were: Glutamate receptor, ionotropic, AMPA1 (alpha 1) (Gria1), catenin (cadherin associated protein), delta 1 (Ctnn1), forkhead box O3 (fox03), and dysferlin (Dysf) (Table 1). Important upregulated genes included phosphodiesterase 10A (Pde10a), fyn proto oncogene (Fyn), programmed cell death 2 (Pdcd2), and quaking (Qk). QRT-PCR verified the direction and magnitude of change for 11 placental genes: Fyn, GPI-anchored HDL-binding protein 1 (Gpihbp1), guanine nucleotide binding protein (G protein), gamma 12 (Gng12), insulin-like growth factor binding protein 3 (Igfbp3), macrophage migration inhibitory factor (Mif), myocyte enhancer factor 2C (Mef2c), ribonuclease L (2′, 5′-oligoisoadenylate synthetase-dependent) (Rnasel), runt related transcription factor 1 (Runx1), TAF1 RNA polymerase II, TATA box binding protein (TBP)-associated factor (Taf1), T-cell specific GTPase (Tgtp), and inactive X specific transcripts (Xist) (Table 1).
Table 1.
Microarray and qRT-PCR for selected placental and brain genes
| Gene Name | Symbol | Region | Microarray | qRT-PCR | ||
|---|---|---|---|---|---|---|
| FC | p | FC | p | |||
| Fyn proto-oncogene | Fyn | Placenta | 1.69 | 0.0374 | 1.17 | 0.028 |
| GPI-anchored HDL-binding protein 1 | Gpihbp1 | Placenta | 0.45 | 0.0078 | 0.72 | 1.91 × 10−5 |
| guanine nucleotide binding protein (G protein), gamma 12 | Gng12 | Placenta | 1.77 | 0.031 | 2.75 | 1.65 × 10−7 |
| insulin-like growth factor binding protein 3 | Igfbp3 | Placenta | 0.583 | 0.018 | 0.80 | 0.004 |
| macrophage migration inhibitory factor | Mif | Placenta | 0.60 | 0.0176 | 0.88 | 0.020 |
| myocyte enhancer factor 2C | Mef2c | Placenta | 1.66 | 0.023 | 3.60 | 6.47 × 10−10 |
| ribonuclease L (2′, 5′-oligoisoadenylate synthetase-dependent) | Rnasel | Placenta | 2.38 | 0.021 | 3.37 | 8.32 × 10−9 |
| runt related transcription factor 1 | Runx1 | Placenta | 0.283 | 0.001 | 0.36 | 4.02 × 10−6 |
| TAF1 RNA polymerase II, TATA box binding protein (TBP)-associated factor | Taf1 | Placenta | 1.62 | 0.010 | 1.30 | 4.54 × 10−6 |
| T-cell specific GTPase | Tgtp | Placenta | 2.12 | 0.0030 | 2.46 | 0.006 |
| inactive X specific transcripts | Xist | Placenta | 10.92 | 0.0001 | 6.47 | 2.39 × 10−6 |
| contactin 1a | Cntn1 | PFC | 1.69 | 0.0298 | 1.94 | 0.0009 |
| Hermansky-Pudlak syndrome 1 homolog (human) a | Hps1 | PFC | 0.62 | 0.0135 | 0.79 | 0.0401 |
| nuclear receptor subfamily 2, group F, member 1a | Nr2f1 | PFC | 1.69 | 0.0290 | 2.11 | 1.85 × 10−5 |
| neurotrophic tyrosine kinase, receptor, type 3a | Ntrk3 | PFC | 1.55 | 0.0424 | 1.79 | 0.0002 |
| platelet factor 4a | Pf4 | PFC | 0.67 | 0.0004 | 0.10 | 6.74 × 10−5 |
| glutamate receptor interacting protein 1b | Grip1 | PFC | 1.51 | 0.014 | 2.03 | 0.002 |
| paralemmin 2a | Palm2 | Hipp | 0.63 | 0.0128 | 0.59 | 4.48 × 10−7 |
| PTK2 protein tyrosine kinase 2 betaa | Ptk2b | Hipp | 1.61 | 0.0026 | 1.27 | 0.0066 |
P0;
P56; FC, fold change; Hipp, hippocampus
3.2. Gene expression changes in hippocampus and prefrontal cortex in the exposed offspring following infection at E7
Our gene expression studies showed significant (fold change of at least 1.5, p<0.05) changes in gene expression in prefrontal cortex (6 upregulated and 24 downregulated at P0; 5 upregulated and 14 downregulated at P56; Appendix B.1) and hippocampus (4 upregulated and 6 downregulated at P0; 6 upregulated and 13 downregulated at P56; Appendix B.2). qRT-PCR for selected genes identified in the microarray dataset revealed significant changes in magnitude and direction in prefrontal cortex (Table 1) and hippocampus (Table 1). Genes in the prefrontal cortex for which the significant change in mRNA was verified by qRT-PCR included: glutamate receptor interacting protein I (Grip1), platelet factor 4 (Pf4), contactin 1 (Cntn1) and neurotrophic tyrosine kinase receptor, type 3 (Ntrk3) (Table 1). In hippocampus, important genes that displayed altered expression included paralemmin 2 (Palm2) and protein tyrosine kinase 2 beta (Ptk2b) (Table 1).
3.3. No detection of H1N1 viral genes in placenta or brains of offspring following infection at E7
We investigated whether H1N1 A/NWS/33 viral genes were present in placenta of mothers infected at E7 or in the brains of their offspring at various postnatal dates. Three genes were analyzed via qRT-PCR: matrix protein 1/matrix protein 2 (M1/M2), neuraminidase (NA), and nonstructural protein 1 (NS1) using the probes previously used by Lo et al. (2003). We were unable to detect amplification for any of the three genes in placenta, hippocampus or prefrontal cortex, suggesting that the virus either did not cross the placenta, or could not be detected using our technique.
3.4. Morphometric analysis and fractional anisotropy of brains of exposed offspring
Morphometric analysis of brain following infection or sham-infection of C57BL/6 mice at E7 revealed no significant changes in volumes of hippocampus, cerebellum, total brain or ventricles at P14 and P35 (Table 2). However, there was a trend towards reduced hippocampal volume at P35 (p<0.064). Brain imaging also showed significantly increased fractional anisotropy (FA) in white matter of the right middle cerebellar peduncle (MCP-r) at P35 (p<0.046) (Table 3). There was also a trend towards reduced FA of white matter in the hippocampus at P14 (p<0.076).
Table 2.
Hippocampus, cerebellum, brain, and ventricular volume measurements at P14 and P35 following infection at E7
| Hippocampus | Control | Infected | p |
|---|---|---|---|
| P14 | 21.7 ± 0.679 | 21.6 ± 0.152 | 0.81 |
| P35 | 24.5 ± 0.34 | 23.2 ± 0.767 | 0.064 |
| Cerebellum | Control | Infected | P |
| P14 | 40.3 ± 1.68 | 38 ± 1.05 | 0.12 |
| P35 | 51.6 ± 1.57 | 50.1 ± 1.0 | 0.57 |
| Brain | Control | Infected | P |
| P14 | 377 ± 5.70 | 373 ± 3.29 | 0.41 |
| P35 | 442 ± 3.88 | 0.424 ± 14.8 | 0.11 |
| Ventricle | Control | Infected | P |
| P14 | 0.073 ± 0.032 | 0.088 ± 0.020 | 0.52 |
| P35 | 0.158 ± 0.021 | 0.434 ± 0.535 | 0.42 |
Table 3.
Fractional anisotropy of hippocampus, corpus callosum, middle cerebellar peduncle, and internal capsule at P14 and P35 following infection at E7
| Hippocampus | Control | Infected | p |
|---|---|---|---|
| P14 | 0.605 ± 0.012 | 0.551 ± 0.037 | 0.076 |
| P35 | 0.67 ± 0.061 | 0.71 ± 0.065 | 0.51 |
| Corpus callosum | Control | Infected | P |
| P14 | 0.562 ± 0.062 | 0.623 ± 0.032 | 0.2 |
| P35 | 0.681 ± 0.045 | 0.652 ± 0.07 | 0.53 |
| Right middle cerebellar peduncle | Control | Infected | P |
| P14 | 0.737 ± 0.096 | 0.666 ± 0.075 | 0.37 |
| P35 | 0.838 ± 0.006 | 0.846 ± 0.001 | 0.046 |
| Left middle cerebellar peduncle | Control | Infected | P |
| P14 | 0.709 ± 0.026 | 0.627 ± 0.071 | 0.13 |
| P35 | 0.803 ± 0.021 | 0.817 ± 0.014 | 0.37 |
| Right internal capsule | Control | Infected | P |
| P14 | 0.661 ± 0.024 | 0.624 ± 0.030 | 0.17 |
| P35 | 0.773 ± 0.057 | 0.760 ± 0.054 | 0.68 |
| Left internal capsule | Control | Infected | P |
| P14 | 0.678 ± 0.011 | 0.591 ± 0.081 | 0.14 |
| P35 | 0.776 ± 0.020 | 0.778 ± 0.023 | 0.89 |
3.5. Prenatal viral infection at E7 results in placental structural abnormalities
When compared with controls, placentae from infected mothers displayed a number of histological abnormalities (Fig. 1). The labyrinth zone was absent from all sections of infected placenta, which could be due to the extensive disorganization of the infected tissues, rendering the labyrinth zone unrecognizable. Thrombi, which varied in size from 40–120 microns in diameter, were present in various areas of the placenta including the junctional zone (Fig. 1B, D). There was also an increased number of inflammatory cells in the junctional zone of infected animals (Fig. 1D).
Figure 1.
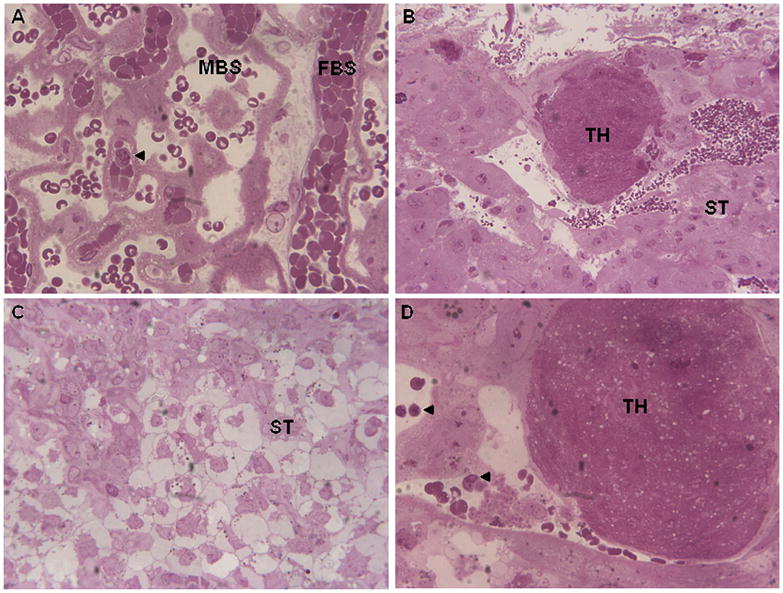
Placental abnormalities following prenatal viral infection. A. Labyrinth zone of placenta (60X) from sham-infected dam showing fetal blood space (FBS) and maternal blood space. Arrow points to an inflammatory cell. B. Junctional zone of placenta (20X) from infected dam showing presence of thrombosis (TH). ST, spongiotrophoblast cells. C. Junctional zone of placenta (40X) from infected dam showing disorganization of ST cells. D. Junctional zone of placenta (60X) from infected dam showing presence of TH and inflammatory cells (arrows).
4. Discussion
In the current study we found that prenatal viral infection at E7 (approximately middle first trimester) resulted in a number of changes to the placentae of infected dams and brains of exposed offspring. We identified gene expression changes in the placentae of infected dams and in the hippocampi and frontal cortices of exposed offspring. Importantly, we did not detect mRNA for three H1N1 influenza viral genes in any of the tissues examined. Placentae of infected dams displayed a number of structural abnormalities that could potentially impact the developing embryos. Morphometric analysis of brains from exposed offspring showed minimal changes when compared with controls in contrast to what our laboratory has observed following infection at later time points.
This is the first study to examine the placenta as a site of pathology following influenza infection during pregnancy. Interestingly, our microarray results for placenta showed that 20% of the genes that showed altered expression are involved in apoptotic or anti-apoptotic pathways (i.e., Atrx, Mef2c, Taf1, Pdcd2, Igfbp3), 10% are associated with immune response (i.e., Atg5, Dysf, Msln, Pias3, Plek2, Pus1, Tac4, Tgtp), approximately 11% deal with hypoxia (i.e., Atg5, Foxo3, Smad4), and approximately 11% are involved with inflammation (i.e., Rnasel, Gng12, Smad4, Tac4) (Table 4). Moreover, 9.4% were associated with major mental disorders including schizophrenia (i.e., Fyn, Gria1; Ohnuma et al., 2003; O’Connor and Hemby, 2007), bipolar disorder (i.e., Fyn, Pura; Szczepankiewicz et al. 2009; Nakatani et al. 2006), major depression (i.e., Pten, Zfp36; Hsiung et al. 2003; Thalmeier et al. 2008) and autism (i.e., Pten, Robo1; Anitha et al. 2008; McBride et al. 2010) (Table 4). In the categories of inflammation, immune response, and hypoxia, multiple genes fit into two categories (i.e. Atg5, Igfb3, Mif, Prnp, Tac4, F2r, Pten, Pdcd2, Tgtp, and Dysf) or all three categories (Foxo3, Smad4, Hes1, Mnt, Zfp36, and Fyn). This overlap is not unexpected as these three processes are often interconnected. The altered expression of these genes suggests that following viral infection, the placental environment is challenged, possibly due to the maternal or fetal immune response.
Table 4.
Altered expression of placental genes associated with major mental disorders, hypoxia, inflammation, immune response, and apoptosis following infection at E7
| Category | Downregulated Genes | Upregulated Genes |
|---|---|---|
| Schizophrenia | Egfr, Gria1, Pvr | Fyn, Pde10a, Qk, Arhgap18, Robo1, Taf1 |
| Autism | Mif, Pik3cg | Kdm5a, Pten, Robo1 |
| Bipolar disorder | Gria1, Pura | Bag1, Fyn |
| Major depression/Suicide | Zfp36 | Pten |
| Hypoxia | Atg5, Endog, Edn2, Ero1l, Foxo3, Hes1, Igfbp3, Mif, Smad4, Pigf, Prnp, Tbc1d1, Zfp36 | F2r, Fyn, Mnt, Pten, Pdcd2 |
| Inflammation | Foxo3, Gpihbp1, Hes1, Mif, Smad4, Prnp, Runx1, Tac4, Zfp36 | F2r, Dysf, Fyn, Gng12, Kif5b, Mnt, Pdcd2, Rnasel, Tgtp, Tmem9b |
| Immune Response | Atg5, Ddx3y, Foxo3, Hes1, Igfbp3, Smad4, Msln, Plek2, Pias3, Pus1, Tac4, Zfp36 | Dysf, Ebf1, Fyn, Mnt, Pten, Tgtp |
| Apoptosis | Atg5, Csde1, Endog, Foxo3, Git1, Hes1, Igfbp3, Lman1, Mif, Nub1, Ncstn, Prnp, Pias3, Taok1, Zfp36 | Acyp1, Atrx, Bcap31, Bag1, Fyn, Lrig2, Magoh, Mnt, Mef2c, Ogt, Pten, Pdcd2, Pa2g4, Qk, Rb1cc1, Stradb, Supv3l1, Suz12, Taf1 |
The direction and magnitude of change for eleven of the placental genes, Mef2c, Mif, Fyn, Gpihbp1, Gng12, Igfbp3, Rnasel, Runx1, Taf1, Tgtp, and Xist, were verified by qRT-PCR. Of particular interest are Mif and Fyn. Macrophage migration inhibitory factor (Mif) is a pro-inflammatory cytokine that is released by macrophages and activated T-lymphocytes. Mif, in turn, stimulates these cells to release additional pro-inflammatory cytokines and interleukins including tumor necrosis factor alpha (TNFα), IL2, IL6, and IL8 (Chalandra et al. 1994; Bancher et al. 1998; Benigni et al. 2000). Both microarray, and qRT-PCR found downregulation of Mif mRNA, which may argue against increased presence of cytokines at the placenta. Fyn proto-oncogene is a member of the src family of kinases and is involved in glutamatergic signaling through its phosphorylation of N-methyl-d-aspartate (NMDA) receptor subunits. Polymorphisms of Fyn have been associated with bipolar disorder (Szczepankiewicz et al. 2009) and with performance on the Wisconsin Card Sorting Test, which measures prefrontal cortex function, for subjects with schizophrenia (Rybakowski et al. 2007). An earlier study, however, found no association between Fyn polymorphisms and schizophrenia (Ishiguro et al. 2000). Fyn mRNA and protein have been observed to be elevated in prefrontal cortex of subjects with schizophrenia (Ohnuma et al. 2003). Fyn kinase is a binding partner of the herpes simplex virus (Carter, 2009) demonstrating a role in viral infection that may ultimately lead to brain pathology associated with neurodevelopmental disorders such as schizophrenia. The observed upregulation of Fyn mRNA in the placenta of infected dams further implicates Fyn kinase in viral infection.
Additional genes, with a variety of important functions, showed changes in mRNA in the same direction and magnitude for both microarray and qRT-PCR. GPI-anchored HDL-binding protein 1 (Gpihbp1) mRNA was significantly downregulated in placenta of infected dams (Table 1). Gpihbp1 is synthesized by endothelial cells, is highly expressed in heart and adipose tissue, and is required for the breakdown of lipids (Beigneux et al. 2007). T-cell-specific GTPase (Tgtp) was found to be elevated in placenta of infected dams. A recent study in mouse found elevated expression in Tgtp protein in mononuclear cells following stimulation with spores from Ganoderma lucidum, a mushroom which is known to modulate the immune system indirectly implicating immune activation in exposed placentas (Ma et al. 2008). Guanine nucleotide binding protein (G protein), gamma 12 (Gng12) has been hypothesized to negatively regulate the lipopolysaccharide (LPS)-induced inflammatory response in a microglial cell line (Larson et al., 2010). Similar to Larson et al (2010), we observed increased mRNA for Gng12 (Table 1) suggesting that Gng12 may play a similar role in placenta following viral infection. Insulin-like growth factor binding protein 3 (Igfbp3) is abundant in the placenta, particularly at the maternal-fetal interface (Han et al. 1996) and behaves as an insulin antagonist (Muzumdar et al. 2006). High concentrations of IGF-I, IGFBP1, and IGFBP3 have been observed in the cerebral spinal fluid of children less than 6 months of age and may play a role in myelination and synapse formation during the development of the nervous system (Bunn et al. 2005). Our observed reduction of Igfbp3 in placenta (Table 1) may impair brain development in the developing embryos. Runt related transcription factor 1 (Runx1) is necessary for formation of haematopoietic progenitor and stem cells from mouse vasculature including vitelline and umbilical arteries, and placenta (Chen et al. 2009). Runx1 is also important for regulating adenosine deaminase (Ada) gene expression in the trophoblast cell lineage of the placenta (Schaubach et al. 2006). Moreover, inactivation of Runx1 in mice has resulted in a decrease of sensory neurons in the trigeminal and vestibulochochlear ganglia (Theriault et al. 2004). The observed downregulation of mRNA for Runx1 (Table 1), may help explain the observed disorganization of the placenta, and like Igfbp3, have an effect on fetal brain development. Ribonuclease L (Rnasel) has been shown to have anti-viral effects (Leib, 2002) and to play a role in inflammation (Nelson et al. 2004; Hsing et al. 2008; Rebbeck et al. 2008). The observed increased expression of Rnasel mRNA (Table 1), reflects these roles. Finally, inactive X specific transcripts (Xist) mRNA was increased in placentae of infected dams (Table 1). Xist is required for inactivation of one of the two X chromosomes in females. It is unclear what the upregulation of mRNA for Xist, may mean for the placenta or for the developing embryos.
Compared with other time points of infection, infection at E7 resulted in a relatively minor number of gene expression changes in hippocampus and PFC of the exposed offspring (Figure 2). At P0, infection at E7 resulted in a total number of 40 genes with altered expression in these two areas compared with 39 at E9 (whole brain), 676 at E16, and 247 at E18 (Fatemi et al. 2005, 2008a,b, 2009a,b). At P56, infection at E7 resulted in changed expression of 38 genes compared with 24 following infection at E9 (PFC only), 349 at E16, and 182 at E18 (Fatemi et al. 2005, 2008a,b, 2009a,b). These data show that infection at later timepoints resulted in greater changes in gene expression in the exposed offspring than infection during the first trimester. These results are in agreement with a recent article by Bitanihirwe et al. (2010) which found that late prenatal immune activation led to marked behavioral deficits and changes in neurotransmitter levels.
Figure 2.
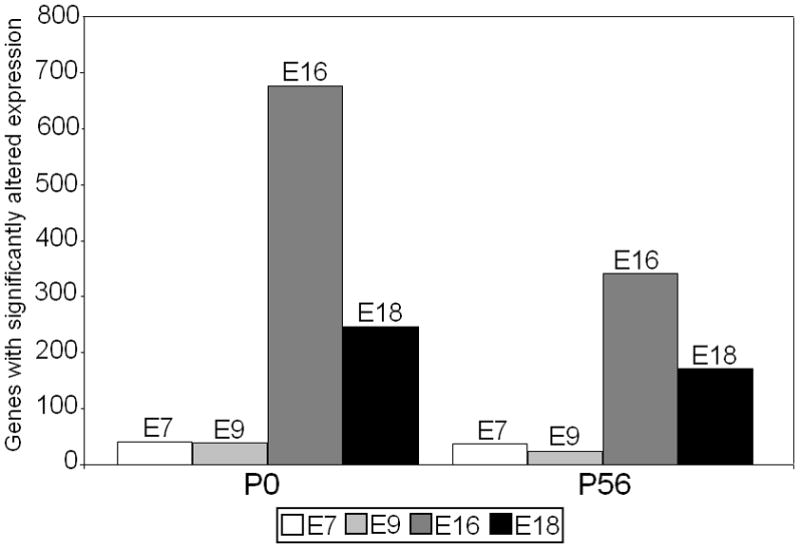
A comparison of the total number of brain genes with significantly (p<0.05, fold change of at least 1.5) altered expression in hippocampus and PFC of infected offspring at P0 and P56 following prenatal viral infection at E7, E9, E16, E18 (Fatemi et al., 2005, 2008a,b, 2009a, unpublished observations). (Note, value for E9 P0 is for whole brain, and value for E9 P56 is for PFC only).
We were unable to detect presence of mRNA for three of the H1N1 virally specific genes - matrix protein 1/matrix protein 2 (M1/M2), neuraminidase (NA) and nonstructural protein 1 (NS1) in the placenta of infected dams or in the hippocampus or frontal cortex of infected offspring. NS1 has roles in promoting viral replication and inhibition of the host immune response (Hale et al. 2008). Specifically, NS1 is an antagonist of interferon production (Kochs et al. 2007). One RNA segment of the viral genome codes for both the matrix proteins M1 and M2 (Okuda et al. 2001; Gaanagé et al. 2009). M1 plays roles in viral replication, assembly, and budding (Barman et al. 2001) while M2 has proton channel activity (Ilyinskii et al. 2007). NA facilitates the release of virus from infected cells by removing the sialic acids that binds hemagglutinin (HA) to the infected cell (Zambon, 2001; Huang et al. 2008). NA is a target for drugs such as peramivir and oseltamivir which have been developed to prevent influenza infection. Our results are similar to those of Shi et al. (2005) who detected viral mRNA in the lungs of infected dams but only rarely in the placentae and not at all in the brains of exposed offspring. These data suggest that the virus did not cross the placenta to directly infect the offspring or alternately, using our technology, we could not identify evidence for presence of this virus. Thus, our results provide further indirect evidence that the observed deleterious changes observed in animal models of prenatal viral infection are perhaps the result of action of fetal or maternal cytokines.
Our laboratory has shown a number of deleterious effects of prenatal viral infection at E9, E16, and E19 on brain structure (Fatemi et al. 1999, 2002, 2008a, 2009a,b). Viral infection at E9 resulted in abnormal corticogenesis of the hippocampus and prefrontal cortex of exposed offspring at P0, which was accompanied by a reduction in Reelin, a molecule that is crucial for proper laminar organization of the brain (Fatemi et al. 1999). Infection at E9 also resulted in long-term effects on brain development resulting in macrocephaly and pyramidal cell dysregulation in adulthood (Fatemi et al. 2002). Infection at E18 and especially at E16 resulted in more deleterious effects including reductions in volume of the hippocampus, cerebellum, and total brain and changes in white matter FA in multiple regions (Fatemi et al. 2008a, 2009a,b). In contrast, infection at E7 had a relatively minor effect on brain morphology in the exposed offspring, with the exception of increased FA in the right middle cerebellar peduncle at P35, when compared with infection during late first trimester (E9) or middle or late second trimester (E16 and E18, respectively). In mouse 1st trimester roughly corresponds with E0-E10 while 2nd trimester corresponds to E10 – E19/20, respectively (Rodier, 1980) with organogenesis occurring during E10 – E14; and fetal growth and development occurring during E14 – E19 or 20 (Hogan et al. 1994). Individual brain areas follow specific timetables for growth (Morgane et al. 1992; Avishai-Eliner et al. 2002; Boksa and Luheshi, 2003), such as the cerebral cortex (E11.5–E16) and hippocampus (E11–E15.5) (Rodier 1980). As infection at E7 occurs prior to these critical periods, it may explain the relatively low impact on brain morphology.
The various structural abnormalities we observed in placentae of infected mothers demonstrate that sublethal doses of H1N1 can result in loss of the LZ, thrombus formation, apoptosis and necrosis of placenta, and inflammatory cell recruitment. Similar to our observed reduction of the LZ, infection with Campylobacter rectus has also been shown to reduce the LZ of the murine placenta (Offenbacher et al. 2005). The authors suggest that this change might result in impairment of respiratory gas exchange, and flow of nutrients and wastes between the mother and fetus (Offenbacher et al. 2005). Interestingly, a study of a mouse model of diabetes likewise found reduced LZ which was accompanied by an over-representation of spongiotrophoblast cells, suggesting a similar compromise of placental transport (Yu et al., 2008). As we did not detect viral RNA in the placenta of infected dams, these changes may be due to production of either maternal or fetal cytokines probably due to increased abundance of inflammatory cells in the infected placentae. Bacterial infection by C. rectus resulted in dramatic increase of interferon gamma (INF-γ) (Offenbacher et al. 2005) which acts as a mediator of neuronal oxidative stress and neuronal apoptosis (Vartanian et al. 1995). A guinea pig model of placental insufficiency, in which uteroplacental blood flow was restricted, resulted in offspring that displayed reduced PPI and brain structural abnormalities similar to those observed in subjects with schizophrenia (Rehn et al. 2004). Similar mechanisms may be at work following prenatal viral infection, impacting gene expression in the brains of exposed offspring and behavioral changes previously observed with this our model (Shi et al., 2003).
A limitation to the current study is the limited sample size/statistical power. Because of the low N, variability among samples may have masked changes in gene expression and brain morphology. Further experiments are necessary to confirm the present findings. Future studies include western blotting of placental genes that demonstrated significantly altered expression. Measurement of cytokines in placental tissue, especially, IL-6 would provide important information on potential roles of inflammation on the observed results.
5. Conclusions
Prenatal viral infection at E7 led to gene expression and morphological changes in placenta at E16. Many of the altered placental genes were associated with hypoxia, inflammation, immune system response, and apoptosis, suggesting that immune challenge had deleterious effects on the placental environment. Moreover, infection at E7 led to gene expression changes in the hippocampus and PFC of exposed offspring at birth and these changes persisted into young adulthood. Infection at E7 had minimal effect on brain morphology and white matter fractional anisotropy of the exposed offspring. Finally, a qRT-PCR experiment found no evidence for mRNA for three H1N1 viral genes in the placentae or in the brains of exposed offspring. These data suggest that the multiple effects of viral infection are not due to direct infection of the offspring.
Supplementary Material
Acknowledgments
Grant support by the National Institute of Child Health and Human Development (#5R01HD046589-04 and #5R01HD046589-04S1) to SHF is gratefully acknowledged. Grant support from NIH (#R01EB003543) to SM is gratefully acknowledged. Technical assistance from DF Smee is gratefully acknowledged.
Footnotes
Disclosure/conflict of interest
None of the authors have any conflict of interest with the information published in this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
S. Hossein Fatemi, Email: fatem002@umn.edu.
Timothy D. Folsom, Email: folso013@umn.edu.
Robert J. Rooney, Email: rrooney@genome-explorations.com.
Susumu Mori, Email: susumu@mri.jhu.edu.
Tess E. Kornfield, Email: kornf002@umn.edu.
Teri J. Reutiman, Email: terireutiman@hotmail.com.
Rachel E. Kneeland, Email: knee0030@umn.edu.
Stephanie B. Liesch, Email: lies0065@umn.edu.
Kegang Hua, Email: khua1@jhu.edu.
John Hsu, Email: hsu@jhu.edu.
Divyen H. Patel, Email: dpatel@genome-explorations.com.
References
- Anitha A, Nakamura K, Yamada K, Suda S, Thanseem I, Tsujii M, Iwayama Y, Hattori E, Toyota T, Miyachi T, Iwata Y, Suzuki K, Matsuzaki H, Kawai M, Sekine Y, Tsuchiya K, Sugihara G, Ouchi Y, Sugiyama T, Koizumi K, Higashida H, Takei N, Yoshikawa T, Mori N. Genetic analyses of roundabout (ROBO) axon guidance receptors in autism. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1019–1027. doi: 10.1002/ajmg.b.30697. [DOI] [PubMed] [Google Scholar]
- Aronsson F, Karlsson H, Ljunggren HG, Kristensson K. Persistence of the influenza A/WSN/33 virus RNA at midbrain levels of immunodefective mice. J Neurovirol. 2001;7:117–124. doi: 10.1080/13550280152058771. [DOI] [PubMed] [Google Scholar]
- Aronsson F, Lannebo C, Paucar M, Brask J, Kristensson K, Karlsson H. Persistence of viral RNA in the brain of offspring to mice infected with influenza A/WSN/33 during pregnancy. J Neurovirol. 2002;8:353–357. doi: 10.1080/13550280290100480. [DOI] [PubMed] [Google Scholar]
- Avishai-Eliner S, Brunson KL, Sandman CA, Baram TZ. Stressed-out, or in (utero)? Trends Neurosci. 2002;25:518–524. doi: 10.1016/s0166-2236(02)02241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacher M, Metz CN, Calandra T, Mayer K, Chesney J, Lohoff M, Gemsa D, Donnelly T, Bucala R. An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc Natl Acad Sci USA. 1996;93:7849–7854. doi: 10.1073/pnas.93.15.7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barman S, Ali A, Hui EK, Adhikary L, Nayak DP. Transport of viral proteins to the apical membranes and interaction of matrix protein with glycoproteins in the assembly of influenza viruses. Virus Res. 2001;77:61–69. doi: 10.1016/s0168-1702(01)00266-0. [DOI] [PubMed] [Google Scholar]
- Beigneux AP, Davies BS, Gin P, Weinstein MM, Farber E, Qiao X, Peale F, Bunting S, Walzem RL, Wong JS, Blaner WS, Ding ZM, Melford K, Wongsiriroj N, Shu X, de Sauvage F, Ryan RO, Fong LG, Bensadoun A, Young SG. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007;5:279–291. doi: 10.1016/j.cmet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benigni F, Atsumi T, Calandra T, Metz C, Echtenacher B, Peng T, Bucala R. The proinflammatory mediator macrophage migration inhibitory factor induces glucose catabolism in muscle. J Clin Invest. 2000;106:1291–1300. doi: 10.1172/JCI9900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitanihirwe BK, Peleg-Raibstein D, Mouttet F, Feldon J, Meyer U. Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology. 2010;35:2462–2478. doi: 10.1038/npp.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boksa P, Luheshi GN. On the use of animal modeling to study maternal infection during pregnancy and prenatal cytokine exposure as risk factors for schizophrenia. Clin Neurosci. 2003;3:339–346. [Google Scholar]
- Boyd JH, Pulver AE, Stewart W. Season of birth: schizophrenia and bipolar disorder. Schizophr Bull. 1986;12:173–186. doi: 10.1093/schbul/12.2.173. [DOI] [PubMed] [Google Scholar]
- Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophr Bull. 2006;32:200–202. doi: 10.1093/schbul/sbj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunn RC, King WD, Winkler MK, Fowlkes JL. Early developmental changes in IGF-1, IGF-II, IGF binding protein-1 and IGF binding protein-3 concentration in the cerebrospinal fluid of children. Pediatr Res. 2005;58:89–93. doi: 10.1203/01.PDR.0000156369.62787.96. [DOI] [PubMed] [Google Scholar]
- Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage-migration inhibitory factor. J Exp Med. 1994;179:1895–1902. doi: 10.1084/jem.179.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter WT, Koenig JI. The evolution of drug development in schizophrenia: past issues and future opportunities. Neuropsychopharmacology. 2008;33:2061–2079. doi: 10.1038/sj.npp.1301639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CJ. Schizophrenia susceptibility genes directly implicated in the life cycles of pathogens: cytomegalovirus, influenza, herpes simplex, rubella, and Toxoplasma gondii. Schizophr Bull. 2009;35:1163–82. doi: 10.1093/schbul/sbn054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, Speck NA. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature. 2009;457:887–891. doi: 10.1038/nature07619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyles DW, Feron F, Cui X, Kesby JP, Harms LH, Ko P, McGrath JJ, Burne TH. Developmental vitamin D deficiency causes abnormal brain development. Psychoneuroendocrinology. 2009;34:S247–S257. doi: 10.1016/j.psyneuen.2009.04.015. [DOI] [PubMed] [Google Scholar]
- Fatemi SH. Prenatal viral infection, brain development and schizophrenia. In: Fatemi SH, editor. Neuropsychiatric Disorders and Infection. Taylor and Francis; London: 2005. pp. 107–130. [Google Scholar]
- Fatemi SH, Emamian ES, Kist D, Sidwell RW, Nakajima K, Akhter P, Shier A, Sheikh S, Bailey K. Defective corticogenesis and reduction in Reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Mol Psychiatry. 1999;4:145–154. doi: 10.1038/sj.mp.4000520. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Earle JA, Kanodia R, Kist D, Emamian ES, Patterson PH, Shi L, Sidwell R. Prenatal viral infection leads to pyramidal cell atrophy and macrocephaly in adulthood: implications for genesis of autism and schizophrenia. Cell Mol Neurobiol. 2002;22:25–33. doi: 10.1023/a:1015337611258. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Pearce DA, Brooks AI, Sidwell RW. Prenatal viral infection in mouse causes differential expression of genes in brains of mouse progeny: a potential animal model for schizophrenia and autism. Synapse. 2005;57:91–99. doi: 10.1002/syn.20162. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Reutiman TJ, Folsom TD, Huang H, Oishi K, Mori S, Smee DF, Pearce DA, Winter C, Sohr R, Juckel G. Maternal infection leads to abnormal gene regulation and brain atrophy in mouse offspring: implications for genesis of neurodevelopmental disorders. Schizophr Res. 2008a;99:56–70. doi: 10.1016/j.schres.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD, Reutiman TJ, Sidwell RW. Viral regulation of aquaporin 4, connexin 43, microcephalin, and nucleolin. Schizophr Res. 2008b;98:163–177. doi: 10.1016/j.schres.2007.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD, Reutiman TJ, Huang H, Oishi K, Mori S. Prenatal viral infection of mice at E16 causes changes in gene expression in the hippocampi of the offspring. Eur Neuropsychopharmacol. 2009a;19:648–653. doi: 10.1016/j.euroneuro.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD, Reutiman TJ, Abu-Odeh D, Mori S, Huang H, Oishi K. Abnormal expression of myelination genes and alterations in white matter fractional anisotropy following prenatal viral influenza infection at E16 in mice. Schizophr Res. 2009b;112:46–53. doi: 10.1016/j.schres.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes de Abreu DA, Nivet E, Baril N, Khrestchatisky M, Roman F, Féron F. Developmental vitamin D deficiency alters learning in C57Bl/6J mice. Behav Brain Res. 2010;208:603–608. doi: 10.1016/j.bbr.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Gannagé M, Dormann D, Albrecht R, Dengjel J, Torossi T, Rämer PC, Lee M, Strowig T, Arrey F, Conenello G, Pypaert M, Andersen J, García-Sastre A, Münz C. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. 2009;6:367–380. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grecksch G, Rüthrich H, Höllt V, Becker A. Transient prenatal vitamin D deficiency is associated with changes of synaptic plasticity in the dentate gyrus of adult rats. Psychoneuroendocrinology. 2009;34:S258–S264. doi: 10.1016/j.psyneuen.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Gu J, Xie Z, Gao Z, Liu J, Korteweg C, Ye J, Lau LT, Lu J, Gao Z, Zhang B, McNutt MA, Lu M, Anderson VM, Gong E, Yu AC, Lipkin WI. H5N1 infection of the respiratory tract and beyond: a molecular pathology study. Lancet. 2007;370:1137–1145. doi: 10.1016/S0140-6736(07)61515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale BG, Randall RE, Ortin J, Jackson D. The multifunctional NS1 protein of influenza A viruses. J Gen Virol. 2008;89:2359–2376. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- Han VK, Bassett N, Walton J, Challis JR. The expression of insulin-like growth factor (IGF) and IGF-binding protein (IGFBP) genes in the human placenta and membranes: evidence for IGF-IGFBP interations at the feto-maternal interface. J Clin Endocrinol Metab. 1996;81:2680–2693. doi: 10.1210/jcem.81.7.8675597. [DOI] [PubMed] [Google Scholar]
- Hare EH, Price JS, Slater E. Schizophrenia and season of birth. Br J Psychiatry. 1972;120:124–125. doi: 10.1192/bjp.120.554.124-a. [DOI] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Constantini F, Lacy E. A laboratory manual. CSHL Press; New York: 1994. Manipulating the mouse embryo. [Google Scholar]
- Hsing AW, Sakoda LC, Rashid A, Andreotti G, Chen J, Wang BS, Shen MC, Chen BE, Rosenberg PS, Zhang M, Niwa S, Chu L, Welch R, Yeager M, Fraumeni JF, Jr, Gao YT, Chanock SJ. Variants in inflammation genes and the risk of biliary tract cancer and stones: a population-based study in China. Cancer Res. 2008;68:6442–6452. doi: 10.1158/0008-5472.CAN-08-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiung SC, Adlersberg M, Arango V, Mann JJ, Tamir H, Liu KP. Attenuated 5-HT1A receptor signaling in brains of suicide victims: involvement of adenylyl cyclase, phosphatidylinositol 3-kinase, Akt and mitogen-activated protein kinase. J Neurochem. 2003;87:182–194. doi: 10.1046/j.1471-4159.2003.01987.x. [DOI] [PubMed] [Google Scholar]
- Huang I-C, Li W, Sui J, Marasco W, Choe H, Farzan M. Influenza A neuraminidase limits viral superinfection. J Virol. 2008;82:4834–4843. doi: 10.1128/JVI.00079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilyinskii PO, Gabai VL, Sunyaev SR, Thoidis G, Shneider AM. Toxicity of Influenza A virus matrix protein 2 for mammalian cells is associated with its intrinsic proton-channeling activity. Cell Cycle. 2007;6:2043–2047. doi: 10.4161/cc.6.16.4564. [DOI] [PubMed] [Google Scholar]
- Ishiguro H, Saito T, Shibuya H, Toru M, Arinami T. Mutation and association analysis of the Fyn kinase gene with alcoholism and schizophrenia. Am J Med Genet. 2000;96:716–720. [PubMed] [Google Scholar]
- Kesby JP, Cui X, O’Loan J, McGrath JJ, Burne TH, Eyles DW. Developmental vitamin D deficiency alters dopamine-mediated behaviors and dopamine transporter function in adult female rats. Psychopharmacology. 2010;208:159–168. doi: 10.1007/s00213-009-1717-y. [DOI] [PubMed] [Google Scholar]
- Kinney DK, Teixeira P, Hsu D, Napoleon SC, Crowley DJ, Miller A, Hyman W, Huang E. Relation of schizophrenia prevalence to latitude, climate, fish consumption, infant mortality, and skin color: a role for prenatal vitamin D deficiency and infections? Schizophr Bull. 2009;35:582–595. doi: 10.1093/schbul/sbp023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochs G, Koerner I, Thiel L, Kothlow S, Kaspers B, Ruggli N, Summerfield A, Pavlovic J, Stech J, Staeheli P. Properties of H7N7 influenza A virus strain SC35M lacking interferon antagonist NS1 in mice and chickens. J Gen Virol. 2007;88:1403–1409. doi: 10.1099/vir.0.82764-0. [DOI] [PubMed] [Google Scholar]
- Larson KC, Lipko M, Dabrowski M, Draper MP. Gng12 is a novel negative regulator of LPS-induced inflammation in the microglial cell line BV-2. Inflamm Res. 2009;58:725. doi: 10.1007/s00011-009-0062-2. [DOI] [PubMed] [Google Scholar]
- Lewis DA. Retroviruses and the pathogenesis of schizophrenia. Proc Nat Acad Sci USA. 2001;94:4293–4294. doi: 10.1073/pnas.081075898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leib DA. Counteraction of interferon-induced antiviral responses by herpes simplex viruses. Curr Top Microbiol Immunol. 2002;269:171–185. doi: 10.1007/978-3-642-59421-2_11. [DOI] [PubMed] [Google Scholar]
- Liu L, Schulz SC, Lee S, Reutiman TJ, Fatemi SH. Hippocampal CA1 pyramidal cell size is reduced in bipolar disorder. Cell Mol Neurobiol. 2007;27:351–358. doi: 10.1007/s10571-006-9128-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lo CK, Geddes JF, Daniels RS, Oxford JS. Lack of detection of influenza genes in archived, formalin-fixed, paraffin wax-embedded brain samples from encephalitis lethargica patients from 1916 to 1920. Virchos Arch. 2003;442:591–596. doi: 10.1007/s00428-003-0795-1. [DOI] [PubMed] [Google Scholar]
- Ma C, Guan SH, Yang M, Liu X, Guo DA. Differential protein expression in mouse splenic mononuclear cells treated with polysaccharides from spores of Ganoderma lucidum. Phytomedicine. 2008;15:268–276. doi: 10.1016/j.phymed.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Machon RA, Mednick SA, Schulsinger F. The interaction of seasonality, place of birth, genetic risk and subsequent schizophrenia in a high risk sample. Br J Psychiatry. 1983;143:383–388. doi: 10.1192/bjp.143.4.383. [DOI] [PubMed] [Google Scholar]
- McBride KL, Varga EA, Pastore MT, Prior TW, Manickam K, Atkin JF, Herman GE. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010;3:137–141. doi: 10.1002/aur.132. [DOI] [PubMed] [Google Scholar]
- McGrath J. Hypothesis: is low prenatal vitamin D a risk-modifying factor for schizophrenia? Schizophr Res. 1999;40:173–177. doi: 10.1016/s0920-9964(99)00052-3. [DOI] [PubMed] [Google Scholar]
- McGrath JJ, Burne TH, Féron F, Mackay-Sim A, Eyles DW. Developmental vitamin D deficiency and risk of schizophrenia: a 10-year update. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26:4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. 2008a;22:469–486. doi: 10.1016/j.bbi.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Meyer U, Murray PJ, Urwyler A, Yee BK, Schedlowski M, Feldon J. Adult behavioral and pharmacological dysfunctions following disruption of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol Psychiatry. 2008b;13:208–221. doi: 10.1038/sj.mp.4002042. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Fatemi SH. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev. 2009;33:1061–1079. doi: 10.1016/j.neubiorev.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J. Epidemiology-driven neurodevelopmental animal models of schizophrenia. Prog Neurobiol. 2010;90:285–326. doi: 10.1016/j.pneurobio.2009.10.018. [DOI] [PubMed] [Google Scholar]
- Morgane PJ, Austin-LaFrance RJ, Bronzino JD, Tonkiss J, Galler JR. Malnutrition and the developing central nervous system. In: Isaacson RL, Jensen RF, editors. The vulnerable brain and environmental risks, volume 1: Malnutrition and hazard assessment. Plenum Press; New York: 1992. pp. 3–44. [Google Scholar]
- Mori S, Ito R, Zhang J, Kaufmann WE, van Zijl PC, Solaiyappan M, Yarowski P. Diffusion tensor imaging of the developing mouse brain. Magn Reson Med. 2001;46:18–23. doi: 10.1002/mrm.1155. [DOI] [PubMed] [Google Scholar]
- Muzumdar RH, Ma X, Fishman S, Yang X, Atzmon G, Vuguin P, Einstein FH, Hwang D, Cohen P, Barzilai N. Central and opposing effects of IGF-1 and IGF-binding protein-3 on systemic insulin action. Diabetes. 2006;55:2788–2796. doi: 10.2337/db06-0318. [DOI] [PubMed] [Google Scholar]
- Nakatani N, Hattori E, Ohnishi T, Dean B, Iwayama Y, Matsumoto I, Kato T, Osumi N, Higuchi T, Niwa S, Yoshikawa T. Genome-wide expression analysis detects eight genes with robust alterations specific to bipolar I disorder: relevance to neuronal network perturbation. Hum Mol Genet. 2006;15:1949–1962. doi: 10.1093/hmg/ddl118. [DOI] [PubMed] [Google Scholar]
- Nelson WG, De Marzo AM, DeWeese TL, Isaacs WB. The role of inflammation in the pathogenesis of prostate cancer. J Urol. 2002;172:S6–S11. doi: 10.1097/01.ju.0000142058.99614.ff. [DOI] [PubMed] [Google Scholar]
- O’Connor JA, Hemby SE. Elevated Gria expression in Layer II/III and V pyramidal cells in DLPFC in schizophrenia. Schizophr Res. 2007;97:277–288. doi: 10.1016/j.schres.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offenbacher S, Riche EL, Barros SP, Bobetsis YA, Lin D, Beck JD. Effects of maternal Campylobacter rectus infection on murine placenta, fetal and neonatal survival, and brain development. J Periodontol. 2005;76:2133–2143. doi: 10.1902/jop.2005.76.11-S.2133. [DOI] [PubMed] [Google Scholar]
- Ohnuma T, Kato H, Arai H, McKenna PJ, Emson PC. Expression of Fyn, a non-receptor tyrosine kinase in prefrontal cortex from patients with schizophrenia and its correlation with clinical onset. Brain Res Mol Brain Res. 2003;112:90–94. doi: 10.1016/s0169-328x(03)00051-2. [DOI] [PubMed] [Google Scholar]
- Okuda K, Ihata A, Watabe S, Okada E, Yamakawa T, Hamajima K, Yang J, Ishii N, Nakazawa M, Okuda K, Ohnari K, Nakajima K, Xin KQ. Protective immunity against influenza A virus induced by immunization with DNA plasmid containing influenza M gene. Vaccine. 2001;19:3681–3691. doi: 10.1016/s0264-410x(01)00078-0. [DOI] [PubMed] [Google Scholar]
- Pallast EG, Jongbloet PH, Straatman HM. Excess seasonality of births among patients with schizophrenia and seasonal ovopathy. Schizophr Bull. 1994;20:269–276. doi: 10.1093/schbul/20.2.269. [DOI] [PubMed] [Google Scholar]
- Parker-Athill E, Luo D, Bailey A, Giunta B, Tian J, Shytle RD, Murphy T, Legradi G, Tan J. Flavonoids, a prenatal prophalaxis targeting JAK2/STAT3 signaling to oppose IL-6/MIA associated autism. J Neuroimmunol. 2009;217:20–27. doi: 10.1016/j.jneuroim.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbeck TR, Rennert H, Walker AH, Panossian S, Tran T, Walker K, Spangler E, Patacsil-Coomes M, Sachdeva R, Wein AJ, Malkowicz SB, Zeigler-Johnson C. Joint effects of inflammation and androgen metabolism on prostate cancer severity. Int J Cancer. 2008;123:1385–1389. doi: 10.1002/ijc.23687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehn AE, Van den Buuse M, Copolov D, Briscoe T, Lambert G, Rees S. An animal model of chronic placental insufficiency: relevance to neurodevelopmental disorders including schizophrenia. Neuroscience. 2004;129:381–391. doi: 10.1016/j.neuroscience.2004.07.047. [DOI] [PubMed] [Google Scholar]
- Rodier PM. Chronology of neuron development: animal studies and their clinical implications. Develop Med Child Neuro. 1980;22:525–545. doi: 10.1111/j.1469-8749.1980.tb04363.x. [DOI] [PubMed] [Google Scholar]
- Rybakowski JK, Borkowska A, Skibinska M, Hauser J. Polymorphisms of the Fyn kinase gene and a performance on the Wisconsin Card Sorting Test in schizophrenia. Psychiatr Genet. 2007;17:201–204. doi: 10.1097/YPG.0b013e3280991219. [DOI] [PubMed] [Google Scholar]
- Schaubach B, Wen HY, Kellems RE. Regulation of murine ADA gene expression in the placenta by transcription factor RUNX1. Placenta. 2006;27:269–277. doi: 10.1016/j.placenta.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Tu N, Patterson PH. Maternal influenza infection is likely to alter fetal brain development indirectly: the virus is not detected in fetus. Int J Dev Neurosci. 2005;23:299–305. doi: 10.1016/j.ijdevneu.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Shi L, Smith SE, Malkova N, Tse D, Su Y, Patterson PH. Activation of maternal immune system alters cerebellar development in the offspring. Brain Behav Immun. 2009;23:116–123. doi: 10.1016/j.bbi.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short SJ, Lubach GR, Karasin AI, Olsen CW, Styner M, Knickmeyer RC, Gilmore JH, Coe CL. Maternal influenza infection during pregnancy impacts postnatal brain development in the rhesus monkey. Biol Psychiatry. 2010;67:965–973. doi: 10.1016/j.biopsych.2009.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susser ES, Brown AS, Gorman JM. Prenatal Exposures in Schizophrenia. Psychiatric Press; Washington D.C: 1999. [Google Scholar]
- Szczepankiewicz A, Rybakowski JK, Skibinska M, Dmitrzak-Weglarz M, Leszczynska-Rodziewicz A, Wilkosc M, Hauser J. FYN kinase gene: another glutamatergic gene associated with bipolar disorder? Neuropsychobiology. 2009;59:178–183. doi: 10.1159/000219305. [DOI] [PubMed] [Google Scholar]
- Thalmeier A, Dickmann M, Giegling I, Schneider BM, Hartmann A, Maurer K, Schnabel A, Kauert G, Möller HJ, Rujescu D. Gene expression profiling of post-mortem orbitofrontal cortex in violent suicide victims. Int J Neuropsychopharmacol. 2008;11:217–228. doi: 10.1017/S1461145707007894. [DOI] [PubMed] [Google Scholar]
- Theriault FM, Roy P, Stifani S. AML1/Runx1 is important for the development of hindbrain cholinergic branchiovisceral motor neurons and selected cranial sensory neurons. Proc Natl Acad Sci USA. 2004;101:10343–10348. doi: 10.1073/pnas.0400768101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian T, Li Y, Zhao M, Stefansson K. Interferon-γ-induced oliogendrocyte cell death: implications for the pathogenesis of multiple sclerosis. Mol Med. 1995;1:732–743. [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Singh U, Shi W, Konno T, Soares MJ, Geyer R, Fundele R. Inflence of murine maternal diabetes on placental morphology, gene expression, and function. Arch Physiol Biochem. 2008;114:99–110. doi: 10.1080/13813450802033776. [DOI] [PubMed] [Google Scholar]
- Zambon MC. The pathogenesis of influenza in humans. Rev Med Virol. 2001;11:227–241. doi: 10.1002/rmv.319. [DOI] [PubMed] [Google Scholar]
- Zuckerman L, Weiner I. Post-pubertal emergence of disrupted latent inhibition following prenatal immune activation. Psychopharmacology. 2003;169:308–313. doi: 10.1007/s00213-003-1461-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.