

Abstract
Aims
Vitamin D deficiency is associated with cardiac hypertrophy and heart failure, and vitamin D therapy prevents the progression of cardiac hypertrophy in animal models. Here, we examine whether vitamin D therapy prevents progression of pre-existing cardiac hypertrophy and development of heart failure.
Methods and results
When male Dahl salt-sensitive rats were fed a high salt (HS) diet, all rats developed cardiac hypertrophy after 5 weeks. Thereafter, rats were treated with vehicle (V), paricalcitol (PC, an active vitamin D analogue, at 200 ng, IP 3x/week), enalapril (EP, 90 μg/day), and PC + EP. All groups were continued on the HS diet and evaluated after 4 weeks of therapy. The PC and PC + EP groups, but not the V and EP only groups, showed significant prevention of progression of pre-existing cardiac hypertrophy. The signs of decompensated heart failure were evident in the vehicle-treated group; these heart failure parameters significantly improved with PC, EP or PC + EP therapy. The expression of PKCα, which is regulated by Ca2+and known to stimulate cardiac hypertrophy, was significantly increased in the vehicle group, and PC, EP or PC + EP effectively decreased PKCα activation. We also observed normalization of genetic alterations during progression to heart failure with PC treatment.
Conclusion
PC treatment resulted in both the prevention of progression of pre-existing cardiac hypertrophy and the development of heart failure, compared with improvement in progression to heart failure by EP alone. These beneficial findings in heart were associated with inhibition of PKCα activation and reversal of gene alterations.
Keywords: Dahl rats, ACE inhibitor, BNP, Fibrosis, Microarrays
1. Introduction
Accumulating evidence suggests that alterations in vitamin D metabolism are common in patients with congestive heart failure (CHF).1,2 Children with vitamin D deficiency rickets have concomitant cardiomyopathy,3 and vitamin D deficiency and hyperparathyroidism are frequent findings in patients with severe CHF.4 Also, vitamin D deficiency has been linked with increased risk of cardiovascular disease.5 This notion is exemplified in patients with chronic renal failure, who are typically vitamin D deficient because the critical conversion of the storage form to the active form of vitamin D by 1α-hydroxylase (1α-(OH)ase)—which is mainly produced in the kidney—is impaired. In renal failure patients, the prevalence of left ventricular hypertrophy (LVH) and diastolic dysfunction is ∼50–80%, and the rate of cardiovascular-related mortality is 10–20 times higher than in the general population.6,7 We previously reported that vitamin D-treated haemodialysis patients had a reduction in left ventricular (LV) wall thickness and improved diastolic dysfunction parameters by echocardiography compared with untreated patients.8 In addition, haemodialysis patients treated with paricalcitol (PC), a vitamin D analogue, demonstrated improved survival,9 and this improvement was associated with a decrease in cardiovascular mortality.10
To advance this hypothesis, we demonstrated that activated vitamin D therapy using paricalcitol blocks the development of cardiac hypertrophy in the Dahl salt-sensitive (DSS) rat model of cardiac hypertrophy.8 Although these findings demonstrate that vitamin D signalling may have an anti-hypertrophic effect and may prevent the progression of cardiac hypertrophy in the setting of hypertrophic stimulation, it is unknown whether the therapy also prevents progression of established cardiac hypertrophy and development of heart failure. These latter findings would have important clinical and therapeutic implications if translated to humans. Herein we examine whether therapy with an activated vitamin D analogue prevents progression of established cardiac hypertrophy and development of heart failure in the DSS model.
2. Materials and methods
2.1. Animal model
Male DSS rats (Harlan Sprague-Dawley, Somerville, NJ, USA) were bred and fed a normal diet until 6 weeks of age. To generate pressure overload cardiac hypertrophy, they were then fed a high salt (HS) (6%NaCl) diet for the next 5 weeks. Data for baseline hypertrophic group (H) was obtained at the end of 11 weeks (Figure 1A). Rats fed normal diet were used as non-hypertrophic control baseline (N) at the end of 11 weeks. Among H group animals, they were divided as follows and treated for additional 4 weeks: (i) continuation of the HS diet with vehicle injection (H + V), (ii) continuation of the HS diet with paricalcitol (19-nor-1,25-(OH)2 D2) (PC) (200 ng IP 3x/week) injection (H + PC), (iii) continuation of the HS diet with low dose enalapril (EP), an angiotensin-converting enzyme (ACE) inhibitor, infusion via osmotic pump and vehicle injection (H + EP + V), and (iv) continuation of the HS diet with low dose EP infusion via osmotic pump and PC (200 ng IP 3x/week) injection (H + EP + PC; Figure 1A).
Figure 1.

Experimental design and pathologic findings in the DSS rats from different groups. (A) Schematic diagram of experimental design. Thick arrows indicate the time points when the hearts were obtained. NS, normal salt; HS, high salt. (B) Arterial pressure of rats at different escalating doses of enalapril. Study was performed with DSS rats after 5 weeks of HS diet (H group). Arrow indicates the dose used for the study. N = 3. *, P < 0.05. (C) HW/TL ratio and (D) LuW/TL ratio in DSS rats from different groups. N = normal control at 6 weeks of age, H = hypertrophic baseline at 11 weeks of age, V = HS diet with vehicle treatment at 15 weeks of age (H + V group), PC = HS diet with PC treatment at 15 weeks of age (H + PC group), EP = HS diet with EP and vehicle treatments at 15 weeks of age (H + EP + V group), and EP + PC = HS diet with EP and PC treatments at 15 weeks of age (H + EP + PC group). N = 6–10 in each group. *P < 0.05; **P < 0.05 vs. V; †P < 0.05 vs. EP.
PC was prepared with 95% propylene glycol and 5% ethyl alcohol solution and administered three times a week on Monday, Wednesday, and Friday for four consecutive weeks. Vehicle groups received vehicle injections on the same schedule. Two groups of rats were implanted with pumps to deliver EP for 4 weeks. Since the reduction in blood pressure (BP) from high doses of EP would have effects on cardiac hypertrophy and progression to heart failure11,12 we used low-dose EP at 90 μg/day to study the effects of EP and PC that are independent of BP. In a preliminary experiment, we tested escalating doses of EP by intravenous infusion to find the highest dose that would not have a significant decrease in BP in DSS rats that were fed HS diet for 5 weeks (H group; Figure 1B). We chose 90 μg/day, a maximum dose that did not significantly decrease BP in these rats. Alzet osmotic pump is placed subcutaneously after anaesthesia with isoflurane (initially 4–5% in an induction chamber then 2% via intubation tube) via precise vaporizer. Intraoperative monitoring of adequate anaesthesia is done by toe pinch. A small incision is made in the skin and a small pocket is created in the dorsal subcutaneous space for the osmotic pump insertion. For pain analgesia, meloxicam (5.0 mg/kg; 0.1 mL, sq) is given once just prior to the procedure and then again 24 h later. The investigations conform with the ‘Guide for the Care and Use of Laboratory Animals' (NIH publication no. 85–23, 1996), and were approved by the Institutional Animal Care and Use Committee at Beth Israel Deaconess Medical Center.
2.2. Pathologic, echocardiographic, and BP analyses
Pathological and histological analyses were performed as described previously.13–15 For heart extraction, deep anaesthesia is achieved by overdose of ketamine/xylazine (150 mg/mL, 130 mg of ketamine and 20 mg of xylazine i.p.) given at 0.2cc/100 gm. Then, the chest cavity is opened by incisions made at the left intercostal space and the left lateral chest wall, followed by rapid incision of the heart. Transthoracic echocardiography was performed using an Agilent Sonos 5500 sector scanner ultrasound machine equipped with a 7.5 MHz transducer as described previously at the end of study period on each group of animals.8,13,15 Detailed methods for pathologic and echocardiographic analyses are available in the Supplementary material online. Tail cuff BP was measured in each animal at the end of the study period using the Non-invasive BP (NIBP) measurement system for mouse (AD Instruments, Colorado Springs, CO, USA). Data were recorded using a PowerLab system and Chart 5 software (AD Instruments, Colorado Springs, CO).
2.3. Histology
Hearts were fixed in 10% formalin and paraffin-embedded. Sections were stained with haematoxylin and eosin (H&E) and Masson Trichrome (MT) at the Histology Core facility at Beth Israel Deaconess Medical Center. Fibrosis was quantified at 20X using a calibrated digital camera and software (DP 70 and DPController, Olympus America Inc., Irving, TX, USA) to survey entire heart sections. All areas of fibrosis were scored as either major (MT staining consisting of >10%/visual field) or minor (MT staining in <10%/visual field or presence of perivascular fibrosis) as described previously.16
2.4. Adult cardiomyocyte culture and hypertrophic stimulation
Primary cultures of cardiomyocytes from 6-week-old Sprague–Dawley rats were prepared as described.17–19 To induce hypertrophy, cardiomyocytes were treated with phenylephrine (PE, 100 μM) or endotheline-1 (ET-1, 10 nM) for 24 h. Anti-phospho-PKCα antibodies were used to measure activation of PKCα20 and were obtained from Millipore, Billerica, MA, USA. Detailed analyses are outlined in the Supplementary material online.
2.5. Microarray analysis
Total RNA was extracted from rat ventricle tissue using TRIzol (GibcoBRL, Gaithersburg, MD, USA).8,14 Microarray analysis was performed as described previously.8 Detailed analyses are outlined in the Supplementary material online.
2.6. Plasma brain natriuretic peptide measurement
Tail-vein blood was collected and plasma levels of BNP were measured by using BNP ELISA kit (Assaypro, St. Charles, MO, USA) according to the manufacturer's instructions.8
2.7. Statistical analysis
Data were expressed as means ± SEM. Comparisons between and within groups were conducted with unpaired Student t-tests and repeated-measures ANOVA, respectively. P-values of <0.05 were considered statistically significant.
3. Results
3.1. Effects of PC on established cardiac hypertrophy and heart failure
At 6 weeks of age, the rats were fed either normal or HS diet for 5 weeks. Non-hypertrophic baseline was from rats fed a normal diet for 5 weeks (N) and hypertrophic baseline was from rats fed a HS diet for 5 weeks (H). There was a significant increase in heart weight (HW)/tibial length (TL), but lung weight (LuW)/TL did not significantly changed in the H group compared with the N group, confirming that animals fed a HS diet developed compensated cardiac hypertrophy after 5 weeks (11 weeks of age) (see Supplementary material online, Table S1). Echocardiography demonstrated significant increases in intraventricular septum and posterior wall thickness without significant changes in fractional shortening (FS), again consistent with compensated cardiac hypertrophy (see Supplementary material online, Table S2).
In groups that were subsequently treated with PC (H + PC and H + EP + PC), cardiac hypertrophy that was established in the hypertrophic baseline (H) group was significantly reduced as shown by a decrease in HW/TL ratios after 4 weeks (Figure 1C). However, H + V and H + EP groups did not show significant reduction in HW/TL. In comparison, there was a significant 73% increase in LuW/TL ratios, which represents left-sided failure, in the H + V group compared with the H group indicating progressive heart failure when compared with the hypertrophic baseline (P < 0.05; Figure 1D). The increase in LuW/TL was significantly reduced in all treated groups. In particular, the group treated with EP + PC showed an additive beneficial effect over the PC or EP treatment alone groups (% reduction vs. H + V: H + PC = −18% and H + EP = −21%, P < 0.05 vs. H + V; H + EP + PC = −56%, P < 0.05 vs. H + PC or H + EP alone).
Echocardiographic analysis, consistent with the pathological findings, revealed significant reductions in intraventricular septum and posterior wall thicknesses in the PC and EP + PC treated groups, but similar reductions were not observed in the EP treated group (Figure 2A and B). Calculation of LV mass using echocardiographic parameters further confirmed that a significant reduction in LV mass occurred only in PC or PC + EP treated groups (% reduction vs. H + V: H + PC = −20% and H + EP + PC = −25%, P < 0.05). Cardiac function as measured by FS revealed significant reduction in the vehicle treated group that was reversed in all three treated groups. Again, in groups that were treated with EP + PC, there was an additive benefit in improvement of FS compared with PC or EP treatment alone (% increase vs. H + V: H + PC = + 30% and H + EP = + 30%, P < 0.05 vs. H + V; H + EP + PC = + 60%, P < 0.05 vs. H + PC or H + EP alone; Figure 2D). These data, in combination, suggest that PC treatment has a significant effect on preventing further increase in cardiac mass in the setting of established cardiac hypertrophy. Furthermore, treatment with PC or EP was able to attenuate the development of heart failure, with the combined treatment with PC and EP having an additive benefit.
Figure 2.

Echocardiographic findings in the DSS rats from different groups. (A) Intraventricular septal wall thickness in diastole (IVSd), (B) posterior wall thickness in diastole (PWd), (C) LV mass, and (D) FS of DSS rats from different groups. N = 6–10 in each group. *P < 0.05; **P < 0.05 vs. V; †P < 0.05 vs. EP.
3.2. Effect of PC on biochemical, haemodynamic, and histologic measures of cardiac dysfunction
As a corollary to the pathophysiologic and echocardiographic findings, we measured serum levels of brain natriuretic peptide (BNP), which rise in the setting of LV wall stress and serve as a marker for decompensated heart failure. There was no significant increase in BNP levels in the hypertrophic baseline (H) group compared with the non-hypertrophic (N) baseline group. However, compared with the baseline hypertrophic control (H), the H + V groups demonstrated approximately a 10-fold increase in plasma BNP level (Figure 3A). All three treatment modalities significantly reversed the BNP increase seen in the vehicle treated group; PC + EP treatment had an additive effect compared with PC or EP treatment alone with even further decrease in BNP nearly to the baseline level (% reduction vs. H + V: H + PC = −76% and H + EP = −79%, P < 0.05 vs. H + V; H + EP + PC = −94%, P < 0.05 vs. H + PC or H + EP alone; Figure 3A).
Figure 3.

Biochemical and haemodynamic findings in DSS rat hearts from different groups. (A) Plasma BNP levels measured by ELISA assay in DSS rats from different groups. N = 6–10 in each group. *P < 0.05; **P < 0.05 vs. V; †P < 0.05 vs. EP. (B) Mean arterial blood pressure (MAP) using tail cuff BP in DSS rats from different groups at the end of experiments. N = 3–10 in each group. *P < 0.05; NS, not significant.
In this model of cardiac hypertrophy and heart failure, there is a significant elevation of BP due to the HS diet.13 To determine whether significant BP changes were associated with the different treatment regimens, we assessed BP via tail cuff pressure at the end of each experiment. Our baseline hypertrophic group had a significant elevation of mean BP compared with the non-hypertrophic control (N) (Figure 3B). BP from the vehicle treated group maintained elevated mean BP, although it was non-significantly lower than the hypertrophic baseline. Treatment with PC, EP, or EP + PC had minimal effect on BP compared with the vehicle treated group.
Previously, vitamin D therapy was shown to have an anti-fibrotic effect21,22 and this may be one of the mechanisms attenuating the development of heart failure.23 To examine this in detail, we performed a histological examination of heart sections. There was an increase in interstitial fibrosis, as determined by increased areas of MT staining, in H + V that was significantly attenuated by the treatment with PC (Figure 4A and B). EP treatment alone attenuated partial, but non-significant, interstitial fibrosis compared with the H + V group. These findings suggest that vitamin D therapy was associated with attenuation of cardiac fibrosis, which may be one mechanism preventing the development of heart failure. These improvements in clinical, biochemical, and histological parameters of cardiac hypertrophy and heart failure occurred in the absence of significant changes in BP.
Figure 4.
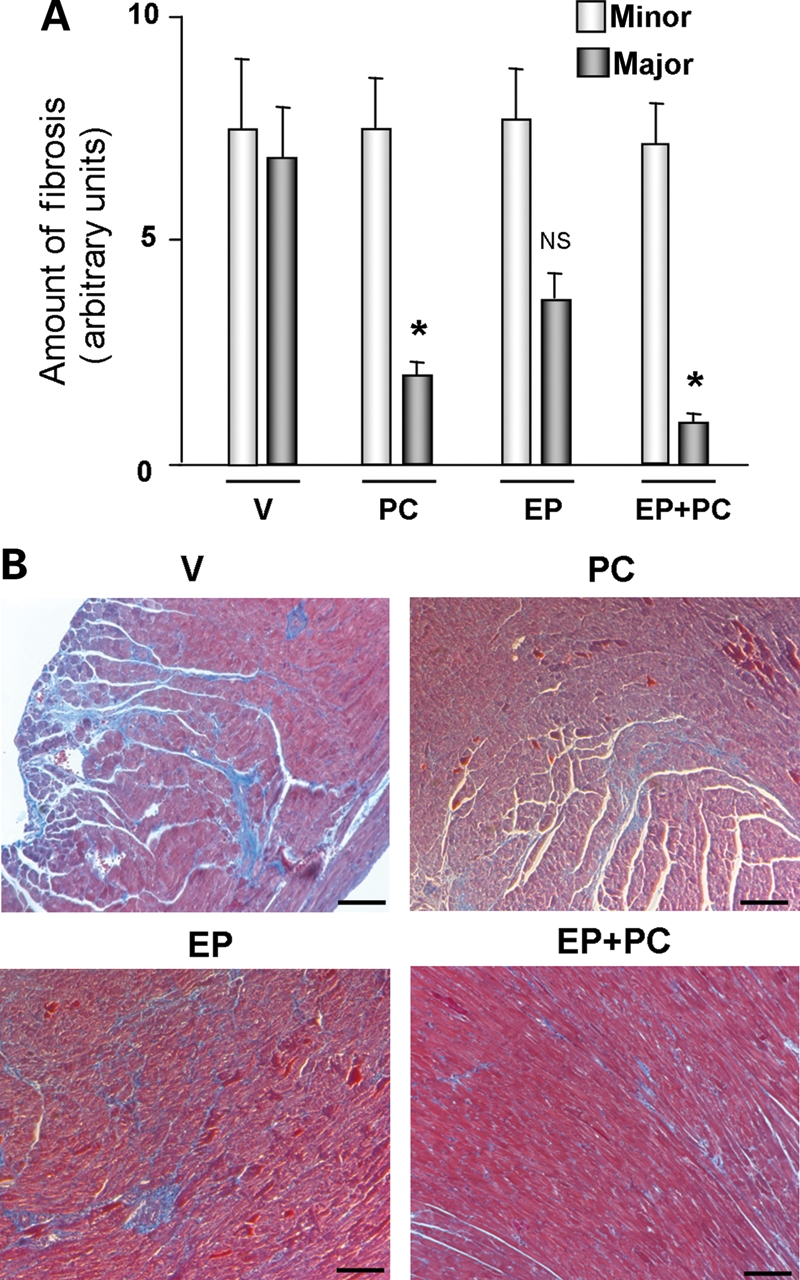
Histological findings of DSS rat hearts from different groups. (A) Quantification of fibrosis in DSS rats from different groups. N = 4 in each group. *P < 0.05; NS, not significant. Major and minor fibrosis are the degrees of fibrosis as defined in the Materials and Methods. (B) Representative Masson Trichrome staining images of DSS rat hearts from different groups. Bar = 100 μm.
3.3. Effect of PC on hypertrophic signalling
Since vitamin D is a critical regulator of maintaining Ca2+homeostasis, we hypothesized that anti-hypertrophic and cardioprotective effects of PC may involve hypertrophic signals that are Ca2+-dependent. One such candidate is protein kinase C signalling, which has previously been implicated in cardiac hypertrophy and heart failure.24–27 We found the expression of classical PKC isoforms, α, which is known to be regulated by Ca2+ and were significantly increased in H and H + V groups compared with the non-hypertrophic control (N) (Figure 5A and B). In contrast, the expression of other PKC isoforms, such as PKCβ and PKCε, were unchanged (Figure 5A, C, and D). Treatment with PC, EP or PC + EP for 4 weeks significantly blocked the activation of PKCα. However, there were no significant effects on PKCβ or PKCɛ expressions in any of the treatment groups. These data suggest that PKCα may be involved in attenuation of hypertrophy and heart failure in this model.
Figure 5.

Expression of PKC isoforms in DSS rat hearts from different groups. (A) Representative western blots analysis of various PKC isoforms in DSS rats from different groups. GAPDH was used as internal loading control. GAPDH, Glyceraldehyde 3-phosphate dehydrogenase. Quantitative analysis of PKCα (B), PKCβ (C), and PKCε (D) in DSS rats from different groups. N = 4–10 in each group. *P < 0.05; **P < 0.05 vs. V; NS, not significant.
To find a direct association between observed beneficial effects of PC and inhibition of PKCα activation, we tested the effect of PC on the activation of PKCα in adult rat cardiomyocytes stimulated with two hypertrophic stimuli: phenylephrine (PE) and endothelin-1 (ET-1). Treatment with these hypertrophic stimuli significantly increased atrial natriuretic factor (ANF) mRNA expression, a reliable marker for cardiac hypertrophy (Figure 6A). Treatment with PC significantly attenuated increased ANF mRNA expression after exposure to PE or ET-1. Using this model, we then tested the effect of PC on the activation of PKCα in adult rat cardiomyocytes stimulated with PE or ET-1. There was a significant activation of PKCα by stimulation with PE or ET-1 as shown by significant increase in phospho-PKCα (p-PKCα, Figure 6B and C). The increase in p-PKCα by PE and ET-1 were significantly reduced by PC treatment.
Figure 6.

PKCα activation by different hypertrophic stimulations is attenuated by PC in adult cardiomyocytes. (A) Representative PCR analysis of ANF in adult cardiomyocytes cultures with or without various hypertrophic stimulations. Twenty-four hours after PC (20 μM) treatment, cells were treated with PE (100 μM), ET-1 (10 nM), or vehicle for 24 h before collecting cells. 18S mRNA was used as an internal loading control. V, vehicle treated; PC, PC treated; PE, phenylephrine; ET-1, endothelin-1; ANF, atrial natriuretic peptide. (B) Representative western blots analysis of phosphorylated PKCα (p-PKCα) and total PKCα in adult cardiomyocytes cultures with or without various hypertrophic stimulations. (C) Quantitative analysis of p-PKCα and PKCα in adult cardiomyocytes cultures with or without various hypertrophic stimulation. N = 4–5 in each group. *P < 0.05 vs. V or PC; †P < 0.05 vs. PE or ET-1.
3.4. Effect of PC on gene expression profiles
We have previously reported that several genes related to cellular signalling, adhesion, and contractile function have been altered, and some of these genes related to hypertrophy have been reversed by PC treatment.8 In order to further examine the effect of various therapies on gene expression, we performed microarray analyses of LV tissue from the respective groups to determine the effect of PC on cardiac hypertrophy. We identified 285 genes that were either up-regulated or down-regulated when the compensated hypertrophic baseline (H) was compared with the decompensated heart failure group (H + V) (Figure 6A). This group of 285 genes was deemed ‘heart failure genes'. Of these 285 genes, PC or EP treatment ‘normalized' 74 and 34, respectively. Interestingly, these two treatments demonstrated distinct patterns of genetic reversal that suggested their potential cardioprotective mechanisms do not entirely overlap. We then compared the genetic changes between EP and EP + PC to determine which genes were renin–angiotensin system (RAS)-independent. We found that a set of distinct genes were up-regulated (135) and down-regulated (109) between EP vs. EP + PC (Figure 6B). These differentially expressed genes include a variety of functional classes (see Supplementary material online, Table S3). For example, α-myosin heavy chain (Myh6) stands out as consistently and significantly up-regulated in EP + V vs. EP + PC. Thus, our study demonstrates that prevention of progression of established cardiac hypertrophy and development of heart failure by PC treatment were associated with transcriptional regulation in heart that are, in part, RAS-independent.
4. Discussion
In this study, we sought to define the effects of active vitamin D therapy in established cardiac hypertrophy and progression to heart failure using the DSS rat model fed a HS diet. We found that only PC had a significant effect on preventing further progression of pre-existing cardiac hypertrophy in DSS animals. In comparison, both PC and EP therapies attenuated the development of clinical and biochemical evidence of heart failure; there was an additive effect on the progression of heart failure in combination therapy with PC and EP. In fact, our gene array data demonstrates that attenuation of heart failure by combination therapy with PC and EP were associated with both common and unique gene expression profiles in heart. Thus, the potential RAS-independent mechanism by PC suggests additive effect of vitamin D therapy that could be explored in the treatment of heart failure. We also found that cardiac structural and functional improvements were coupled to inhibition of PKCα activation. These data are the first to demonstrate the prevention of progression of established cardiac hypertrophy and development of heart failure by an activated vitamin D compound in an animal model. These findings, combined with our previous findings that vitamin D therapy prevents the progression of cardiac hypertrophy8,28 suggests this therapy may be beneficial for those at risk to develop LVH, and those with pre-existing LVH, particularly if they are known to be vitamin D deficient (e.g. chronic renal failure patients).
A number of experimental studies demonstrate that vitamin D signalling is involved in cardiac hypertrophy. Endothelin-induced cardiomyocyte hypertrophy is blocked by activated vitamin D29 and vitamin D deficiency leads to abnormalities in contraction, proliferation, and collagen and renin gene expression in cardiomyocytes.30,31 It has recently been demonstrated that functional vitamin D receptor (VDR), as well as 1α-(OH)ase and 24-hydroxylase, are present in the ventricular myocardium, and that VDR expression is up-regulated with the induction of cardiac hypertrophy.32 VDR also has been shown to transcriptionally regulate both ANF and BNP expression by interacting with their promoters.32–34 In addition, VDR KO mice exhibit baseline cardiac hypertrophy and concomitant activation of the RAS35,36 and vitamin D therapy blocks the development of cardiac hypertrophy in the DSS rat model of cardiac hypertrophy.8 These findings suggest that the key components required for a functional vitamin D-dependent signalling system are present in the heart, and that vitamin D signalling may have an anti-hypertrophic and cardioprotective effects during the transition to heart failure. In this study, we further demonstrated that the anti-hypertrophic effect can also prevent progression of pre-existing cardiac hypertrophy as well as have a cardioprotective effect during the transition from compensated cardiac hypertrophy to decompensated heart failure. Further studies in other models of cardiac hypertrophy or heart failure, such as aortic banding or myocardial infarction models, would provide additional evidences to confirm if vitamin D supplementation has any cardiovascular beneficial effects. These studies are on going in our laboratory as well as others.
The relationship between vitamin D and PKC signalling is currently not well understood. The PKC family is divided into three subgroups: the classic (α, β1, β2, and γ), the novel (δ, ɛ, η, and θ) and the atypical (ζ, ν, μ, and ι). Of particular interest are the classical PKCs, which are categorized on the basis of their Ca2+-dependent activation. Specifically, PKCα expression has been shown to be up-regulated in animal models of heart failure,24 cardiac specific overexpression of PKCα leads to marked ventricular dysfunction and alterations in Ca2+ homeostasis, and the deletion of PKCα increases cardiac contractility.26 In this study, we found that PKCα activation, which may be involved in the progression to heart failure in this model, was effectively blocked by the PC treatment. These findings suggest that PKCα is an important signalling molecule that regulates cardiac function by sensing intracellular Ca2+. This study is one of the first to demonstrate that vitamin D therapy attenuates PKCα activation in heart.
The effect of vitamin D has been shown to involve RAS activation.35 Li and co-authors found that VDR knockout (KO) mice exhibit increased cardiac renin gene expression and cardiac hypertrophy.35 They also found that kidney renin mRNA and protein levels were markedly increased in both VDR KO mice and 25-hydroxyvitamin D 1α-hydroxylase knockout mice36,37 suggesting that vitamin D signalling may be involved in renin production in the kidney and the heart. Inhibition of ACE by EP showed a significant benefit in progression to heart failure in our study. In comparison, our study also showed that there was an additive benefit of vitamin D therapy in the setting of ACE inhibition, which supports the existence of a RAS-independent effect of vitamin D. However, since we did not use a higher dose of ACE inhibitor, which would have had significant BP effect that could compound the interpretation, we cannot fully rule out the fact that higher dose of ACEI would fully account for the effect of PC. Further studies are needed to fully define the RAS-independent mechanism that may play a role in mediating the anti-hypertrophic and protective role of vitamin D therapy during the progression to heart failure.
Previous studies support the association of vitamin D deficiency with increased risk of developing heart failure. Thus, vitamin D deficiency may be an under-recognized, non-classic risk factor for CHF that is readily correctable. As trials are pursued in patients with cardiac hypertrophy with or at risk for heart failure (e.g. clinicaltrials.gov NCT00497146), there needs to be better understanding of the causal relationship between vitamin D status and cardiac dysfunction, and the mechanism of vitamin D's anti-hypertrophic and cardioprotective effects in heart failure needs to be examined. Nevertheless, vitamin D therapy promises to be potentially novel therapy that might complement currently available therapies for heart failure.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: Paricalcitol is marketed by Abbott Laboratories under the brand name Zemplar. P.M.K. and R.T. received research grants from Abbott Laboratories. Paul Kroeger is employed by Abbott Laboratories.
Funding
This work was supported by the grants from National Institutes of Health RO1 HL091998 (P.M.K.), the World Class University program (R31-20029) from Ministry of Education, Science and Technology, South Korea (P.M.K.), Abbott Laboratories (P.M.K. and R.T.) and the Howard Hughes Medical Institute (S.A.K.).
Supplementary Material
References
- 1.Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–281. doi: 10.1056/NEJMra070553. doi:10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- 2.Lee W, Kang PM. Vitamin D deficiency and cardiovascular disease: is there a role for vitamin D therapy in heart failure? Curr Opin Investig Drugs. 2010;11:309–314. [PubMed] [Google Scholar]
- 3.Uysal S, Kalayci AG, Baysal K. Cardiac functions in children with vitamin D deficiency rickets. Pediatr Cardiol. 1999;20:283–286. doi: 10.1007/s002469900464. doi:10.1007/s002469900464. [DOI] [PubMed] [Google Scholar]
- 4.Shane E, Mancini D, Aaronson K, Silverberg SJ, Seibel MJ, Addesso V, et al. Bone mass, vitamin D deficiency, and hyperparathyroidism in congestive heart failure. Am J Med. 1997;103:197–207. doi: 10.1016/s0002-9343(97)00142-3. doi:10.1016/S0002-9343(97)00142-3. [DOI] [PubMed] [Google Scholar]
- 5.Wang TJ, Pencina MJ, Booth SL, Jacques PF, Ingelsson E, Lanier K, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation. 2008;117:503–511. doi: 10.1161/CIRCULATIONAHA.107.706127. doi:10.1161/CIRCULATIONAHA.107.706127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.US Renal Data Systems. Causes of death in ESRD. Am J Kidney Dis. 1999;34:S87–94. doi: 10.1053/AJKD034s00087. [DOI] [PubMed] [Google Scholar]
- 7.Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis. 1998;32:S112–119. doi: 10.1053/ajkd.1998.v32.pm9820470. doi:10.1053/ajkd.1998.v32.pm9820470. [DOI] [PubMed] [Google Scholar]
- 8.Bodyak N, Ayus JC, Achinger S, Shivalingappa V, Ke Q, Chen YS, et al. Activated vitamin D attenuates left ventricular abnormalities induced by dietary sodium in Dahl salt-sensitive animals. Proc Natl Acad Sci USA. 2007;104:16810–16815. doi: 10.1073/pnas.0611202104. doi:10.1073/pnas.0611202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teng M, Wolf M, Lowrie E, Ofsthun N, Lazarus JM, Thadhani R. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N Engl J Med. 2003;349:446–456. doi: 10.1056/NEJMoa022536. doi:10.1056/NEJMoa022536. [DOI] [PubMed] [Google Scholar]
- 10.Teng M, Wolf M, Ofsthun MN, Lazarus JM, Hernan MA, Camargo CA, Jr, et al. Activated injectable vitamin D and hemodialysis survival: a historical cohort study. J Am Soc Nephrol. 2005;16:1115–1125. doi: 10.1681/ASN.2004070573. doi:10.1681/ASN.2004070573. [DOI] [PubMed] [Google Scholar]
- 11.Sharma JN, Fernandez PG, Kim BK, Idikio H, Triggle CR. Cardiac regression and blood pressure control in the Dahl rat treated with either enalapril maleate (MK 421, an angiotensin converting enzyme inhibitor) or hydrochlorothiazide. J Hypertens. 1983;1:251–256. doi: 10.1097/00004872-198310000-00009. doi:10.1097/00004872-198310000-00009. [DOI] [PubMed] [Google Scholar]
- 12.Kim-Mitsuyama S, Izumi Y, Izumiya Y, Yoshida K, Yoshiyama M, Iwao H. Additive beneficial effects of the combination of a calcium channel blocker and an angiotensin blocker on a hypertensive rat-heart failure model. Hypertens Res. 2004;27:771–779. doi: 10.1291/hypres.27.771. doi:10.1291/hypres.27.771. [DOI] [PubMed] [Google Scholar]
- 13.Kang PM, Yue P, Liu Z, Tarnavski O, Bodyak N, Izumo S. Alterations in apoptosis regulatory factors during hypertrophy and heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H72–80. doi: 10.1152/ajpheart.00556.2003. doi:10.1152/ajpheart.00556.2003. [DOI] [PubMed] [Google Scholar]
- 14.Kong SW, Bodyak N, Yue P, Liu Z, Brown J, Izumo S, et al. Genetic expression profiles during physiologic and pathologic cardiac hypertrophy and heart failure in rats. Physiol Genomics. 2005;21:34–42. doi: 10.1152/physiolgenomics.00226.2004. [DOI] [PubMed] [Google Scholar]
- 15.Siu PM, Bae S, Bodyak N, Rigor DL, Kang PM. Response of caspase-independent apoptotic factors to high salt diet-induced heart failure. J Mol Cell Cardiol. 2007;42:678–686. doi: 10.1016/j.yjmcc.2007.01.001. doi:10.1016/j.yjmcc.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rigor DL, Bodyak N, Bae S, Choi JH, Zhang L, Ter-Ovanesyan D, et al. Phosphoinositide 3-kinase Akt signaling pathway interacts with protein kinase Cbeta2 in the regulation of physiologic developmental hypertrophy and heart function. Am J Physiol Heart Circ Physiol. 2009;296:H566–572. doi: 10.1152/ajpheart.00562.2008. doi:10.1152/ajpheart.00562.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choudhury S, Bae S, Kumar SR, Ke Q, Yalamarti B, Choi JH, et al. Role of AIF in cardiac apoptosis in hypertrophic cardiomyocytes from Dahl salt-sensitive rats. Cardiovasc Res. 2010;85:28–37. doi: 10.1093/cvr/cvp261. doi:10.1093/cvr/cvp261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han Y, Chen YS, Liu Z, Bodyak N, Rigor D, Bisping E, et al. Overexpression of HAX-1 protects cardiac myocytes from apoptosis through caspase-9 inhibition. Circ Res. 2006;99:415–423. doi: 10.1161/01.RES.0000237387.05259.a5. doi:10.1161/01.RES.0000237387.05259.a5. [DOI] [PubMed] [Google Scholar]
- 19.Kang PM, Haunstetter A, Aoki H, Usheva A, Izumo S. Morphological and molecular characterization of adult cardiomyocyte apoptosis during hypoxia and reoxygenation. Circ Res. 2000;87:118–125. doi: 10.1161/01.res.87.2.118. [DOI] [PubMed] [Google Scholar]
- 20.Jeong D, Cha H, Kim E, Kang M, Yang DK, Kim JM, et al. PICOT inhibits cardiac hypertrophy and enhances ventricular function and cardiomyocyte contractility. Circ Res. 2006;99:307–314. doi: 10.1161/01.RES.0000234780.06115.2c. doi:10.1161/01.RES.0000234780.06115.2c. [DOI] [PubMed] [Google Scholar]
- 21.Simpson RU, Hershey SH, Nibbelink KA. Characterization of heart size and blood pressure in the vitamin D receptor knockout mouse. J Steroid Biochem Mol Biol. 2007;103:521–524. doi: 10.1016/j.jsbmb.2006.12.098. doi:10.1016/j.jsbmb.2006.12.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Spataro BC, Yang J, Dai C, Liu Y. 1,25-dihydroxyvitamin D inhibits renal interstitial myofibroblast activation by inducing hepatocyte growth factor expression. Kidney Int. 2005;68:1500–1510. doi: 10.1111/j.1523-1755.2005.00562.x. doi:10.1111/j.1523-1755.2005.00562.x. [DOI] [PubMed] [Google Scholar]
- 23.Wright JW, Mizutani S, Harding JW. Pathways involved in the transition from hypertension to hypertrophy to heart failure. Treatment strategies. Heart Fail Rev. 2008;13:367–375. doi: 10.1007/s10741-007-9060-z. doi:10.1007/s10741-007-9060-z. [DOI] [PubMed] [Google Scholar]
- 24.Belin RJ, Sumandea MP, Allen EJ, Schoenfelt K, Wang H, Solaro RJ, et al. Augmented protein kinase C-alpha-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ Res. 2007;101:195–204. doi: 10.1161/CIRCRESAHA.107.148288. doi:10.1161/CIRCRESAHA.107.148288. [DOI] [PubMed] [Google Scholar]
- 25.Bowman JC, Steinberg SF, Jiang T, Geenen DL, Fishman GI, Buttrick PM. Expression of protein kinase C beta in the heart causes hypertrophy in adult mice and sudden death in neonates. J Clin Invest. 1997;100:2189–2195. doi: 10.1172/JCI119755. doi:10.1172/JCI119755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, et al. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;10:248–254. doi: 10.1038/nm1000. doi:10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 27.Wakasaki H, Koya D, Schoen FJ, Jirousek MR, Ways DK, Hoit BD, et al. Targeted overexpression of protein kinase C beta2 isoform in myocardium causes cardiomyopathy. Proc Natl Acad Sci USA. 1997;94:9320–9325. doi: 10.1073/pnas.94.17.9320. doi:10.1073/pnas.94.17.9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong J, Kim GH, Wei M, Sun T, Li G, Liu SQ, et al. Therapeutic effects of vitamin D analogs on cardiac hypertrophy in spontaneously hypertensive rats. Am J Pathol. 2010;177:622–631. doi: 10.2353/ajpath.2010.091292. doi:10.2353/ajpath.2010.091292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest. 1996;97:1577–1588. doi: 10.1172/JCI118582. doi:10.1172/JCI118582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O'Connell TD, Berry JE, Jarvis AK, Somerman MJ, Simpson RU. 1,25-Dihydroxyvitamin D3 regulation of cardiac myocyte proliferation and hypertrophy. Am J Physiol. 1997;272:H1751–1758. doi: 10.1152/ajpheart.1997.272.4.H1751. [DOI] [PubMed] [Google Scholar]
- 31.O'Connell TD, Weishaar RE, Simpson RU. Regulation of myosin isozyme expression by vitamin D3 deficiency and 1,25-dihydroxyvitamin D3 in the rat heart. Endocrinology. 1994;134:899–905. doi: 10.1210/endo.134.2.8299585. doi:10.1210/en.134.2.899. [DOI] [PubMed] [Google Scholar]
- 32.Chen S, Glenn DJ, Ni W, Grigsby CL, Olsen K, Nishimoto M, et al. Expression of the vitamin d receptor is increased in the hypertrophic heart. Hypertension. 2008;52:1106–1112. doi: 10.1161/HYPERTENSIONAHA.108.119602. doi:10.1161/HYPERTENSIONAHA.108.119602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)(2)-vitamin D(3) actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol. 2007;103:533–537. doi: 10.1016/j.jsbmb.2006.12.099. doi:10.1016/j.jsbmb.2006.12.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen S, Wu J, Hsieh JC, Whitfield GK, Jurutka PW, Haussler MR, et al. Suppression of ANP gene transcription by liganded vitamin D receptor: involvement of specific receptor domains. Hypertension. 1998;31:1338–1342. doi: 10.1161/01.hyp.31.6.1338. [DOI] [PubMed] [Google Scholar]
- 35.Xiang W, Kong J, Chen S, Cao LP, Qiao G, Zheng W, et al. Cardiac hypertrophy in vitamin D receptor knockout mice: role of the systemic and cardiac renin-angiotensin systems. Am J Physiol Endocrinol Metab. 2005;288:E125–132. doi: 10.1152/ajpendo.00224.2004. doi:10.1152/ajpendo.00224.2004. [DOI] [PubMed] [Google Scholar]
- 36.Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110:229–238. doi: 10.1172/JCI15219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li YC. Vitamin D regulation of the renin-angiotensin system. J Cell Biochem. 2003;88:327–331. doi: 10.1002/jcb.10343. doi:10.1002/jcb.10343. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.