

Abstract
BACKGROUND
The combination of fish oil-derived docosahexaenoic acid (DHA, 22:6, n-3) and butyrate (4:0), a fiber fermentation product, synergize to enhance colonocyte apoptosis by inducing a p53-independent, oxidation sensitive, mitochondrial Ca2+-dependent (intrinsic) pathway.
METHODS
In this study, we probed the specificity of n-6 and n-3 polyunsaturated fatty acid induction of Ca2+-dependent proapoptotic events in immortalized YAMC colonocytes. We also determined whether combinations of polyunsaturated fatty acid and butyrate trigger endoplasmic stress (ER) stress conditions, thereby promoting mitochondrial Ca2+ overload. Cultures were treated with 0–50 μM of DHA (22:6, n-3), EPA (20:5, n-3), AA (20:4, n-6), LA (18:2, n-6) or OA (18:1, n-9) for a total of 72 h ± RU-360, to inhibit the mitochondrial Ca2+ uniporter, for 30 min prior to butyrate (0 or 5 mM) co-treatment.
RESULTS
DHA and butyrate combination maximally induced apoptosis and mitochondrial-to-cytosolic Ca2+ levels. In comparison, EPA, a precursor to DHA, was minimally effective. Similarly, AA and OA in combination with butyrate had no effect on mitochondrial Ca2+ or apoptosis compared to butyrate alone. DHA ± butyrate co-treatment minimally altered ER stress regulated genes, CHOP and eIF2α.
CONCLUSION
These data indicate that butyrate and DHA, but not EPA, work coordinately to trigger an ER-independent, Ca2+-dependent intrinsic mitochondrial-mediated apoptotic pathway in colonocytes.
Supplemental Key Words: chemoprevention, ER stress, fish oil, dietary fiber, colon cancer, combination chemotherapy, Young adult mouse colon (YAMC) cells
INTRODUCTION
There is substantial experimental, epidemiological and clinical evidence indicating that fish oil-containing diets rich in n-3 polyunsaturated fatty acids (PUFA), e.g. docosahexaenoic acid (DHA, 22:6Δ 4,7,10,13,16,19) and eicosapentaenoic acid (EPA, 20:5Δ 5,8,11,14,17), are protective against colon tumorigenesis (1–9). In a major recent finding, it was demonstrated that EPA reduced rectal polyp number and size in patients with familial adenomatous polyposis (FAP) (10). Most impressive was the fact that fish oil derived EPA suppressed FAP to a degree similar to the selective COX-2 inhibitor celecoxib. Collectively, these data indicate that n-3 PUFA hold promise as a chemoprevention agent for FAP and sporadic colon cancer. In part as a consequence of these studies, fish oil is now being extensively promoted in the unregulated nutraceutical sector. However, some of the fundamental aspects of n-3 PUFA action need to be understood before it can be developed as a clinically viable cancer prevention therapy. These areas pertain to the mechanism of EPA and DHA action, which is still not fully defined in molecular terms. Indeed, it is becoming increasingly clear that these fatty acids have distinct biological properties (11–13).
The balance between proliferation and apoptosis is critical to the maintenance of steady-state number for cell populations in the colon. In general, dysregulation of this mechanism can disrupt homeostasis, resulting in clonal expansion, with the resultant over production of affected cells (14,15). The enhancement of “targeted” apoptosis combined with the reduced formation of DNA adducts during the initiation and late phases of tumorigenesis, in part, accounts for the observed protective effect of n-3 PUFA against experimentally induced colon cancer (16-18). A very exciting outcome of our studies was the demonstration that the pleiotropic bioactive components generated by fermentable fiber (butyrate) and fish oil (DHA) work coordinately to protect against colon tumorigenesis (5,19,20). Although it has been well documented that butyrate is an inhibitor of histone deacetylases and can activate the Fas receptor mediated extrinsic death pathway (21,22), the role of these mechanisms in the induction of colonocyte apoptosis may be a secondary consequence of its ability to promote cellular oxidation (23). With respect to molecular triggers for apoptosis, Ca2+ is one of the most versatile and universal signaling mediators in cells and is required for the activation of many cellular processes. Increasing evidence indicates that alterations in the finely tuned intracellular homeostasis and compartmentalization of Ca2+ can lead to cell death either through apoptosis or necrosis (24). Eukaryotic cells can increase their cytosolic Ca2+ levels via 2 mechanisms: release of Ca2+ from intracellular stores or influx via plasma membrane channels. Although the importance of the endoplasmic reticulum (ER) as the major storage organelle is indisputable, growing evidence indicates that functional compartmentalization of Ca2+ exists within the various cellular organelles. Recent studies have identified the contributions of the mitochondria in maintaining intracellular Ca2+ homeostasis and cellular physiologic function (25). Given the central role of mitochondria in the commitment to apoptosis, we demonstrated that DHA and butyrate interactively promote apoptosis by triggering changes in mitochondrial Ca2+ levels that contribute to caspase activation and colonocyte cell death. For example, isogenic p53 wild type and deficient human colon tumor cell lines as well as an immortalized mouse colonocytes were used to show that DHA modulates intracellular Ca2+ compartmentalization and store-operated channel (SOC) entry to induce colonocyte apoptosis (26). These results corroborate previous observations (19,27) and demonstrate that DHA and butyrate synergistically enhance both mitochondrial Ca2+ accumulation and membrane lipid peroxidation which serve as triggers for apoptosis in a p53-independent manner (28).
In this study, we further probed the specificity of n-6 and n-3 PUFA induction of Ca2+-dependent proapoptotic events in immortalized colonocytes. In addition, since ER stress-dependent apoptotic pathways can be activated by fatty acids (29, 30), we also determined whether combinations of PUFA and butyrate trigger ER stress conditions, thereby promoting mitochondrial Ca2+ overload.
EXPERIMENTAL PROCEDURES
Materials
Two-well Lab-Tek Chambered Coverglassslides were purchased from Nunc, Inc. (Naperville, IL). RPMI 1640 and Hanks' balanced salt solution (HBSS) were from Mediatech (Herndon, VA). Insulin, transferrin, selenium without linoleic acidwas purchased from Collaborative Biomedical Products (Bedford, MA). Fetal bovine serum (FBS) was from Hyclone(Logan, UT). Recombinant mouse interferon-γ (IFN-γ), Glutamax and Leibowitz media werepurchased from GIBCO BRL (Grand Island, NY). Fatty acids were obtained from NuChek (Elysian, MN). Ca2+ sensitive dyes Fluo-4 AM and Rhod-2 AM were from Molecular Probes (Eugene, OR). Soluble recombinant Fas:Fc was from BD Biosciences. Mitochondrial Ca2+ inhibitor RU-360 was from Calbiochem (San Deigo, CA). Cell death detection ELISA kit was obtained from Roche Applied Science (Indianapolis, IN). Sodium butyrate, IgG1 from human serum and all other reagents were obtained from Sigma (St. Louis, MO). Stock solutions of Fluo-4, AM (1 mM) and Rhod-2 (4 mM) were prepared in DMSO and diluted to 3.0 μM and 2.0μM, respectively for cell culture treatment (final concentration of the vehicle DMSO was maintained at 0.1–0.3% in culture). RU-360 (1 mM stock) was prepared in degassed water and diluted in media to a final concentration of 10 μM.
Cell Culture
Conditionally immortalized YAMC cells were originally obtained from R. H. Whitehead, Ludwig Cancer Institute (Melbourne, Australia). These cells are immortalized by virtue of expression of a temperature-sensitive simian virus 40 large (SV40) T antigen (31). YAMC cells were cultured in RPMI 1640 supplemented with 5% FBS, 1% insulin- transferrin-selenium, 2 mM Glutamax and 5,000 units/liter of recombinant IFN-γ. Cells were cultured under permissive (33°C with interferon-γ) or non-permissive conditions (39°C) as previously described (26). For all fluorescence measurements, cells (passages 12–18) were seeded onto borosilicate 2-chambered cover glass slides at a density of 5–7 x 103 to achieve a 50–70% confluence. For apoptosis assays, cells were seeded onto 6 well plates at a density of 35 x103. Following 24 h of plating select cultures were treated with 0-50 μM of Bovine serum albumin (BSA) complexed DHA (22:6, n-3), EPA (20:5, n-3), AA (20:4, n-6), LA (18:2, n-6) or OA (18:1, n-9) for a total of 72 h as previously described (26,28). In select experiments, cultures were pre-incubated with 0 or 10 μM RU-360 in RPMI 1640, to inhibit the mitochondrial uniporter, for 30 min prior to butyrate and fatty acid co-treatment. In other experiments cells were co-incubated with Fas:Fc or IgG1 (3 μg/ml) along with sodium butyrate for the final 24 h of fatty acid pre-treatment following which apoptosis and mitochondrial-to-cytosol Ca2+ ratio were measured.
Analysis of Mitochondrial Ca2+
Following a 24 h fatty acid and butyrate treatment period, cells were washed with Leibowitz media, free of serum and phenol red. Cells were then coincubated with 3 μM Fluo-4 and 2 μM Rhod-2 for 1 h at 33°C. Cultures were subsequently washed with Leibowitz media without serum and the mitochondrial-to-cytosolic Ca2+ ratio was measured. Quantification of Fluo-4 and Rhod-2 fluorescence was performed by excitation at 488 nm and 550 nm, and fluorescence emission was monitored at 530 nm and 580 nm, respectively, as previously described (26,28). The ratio of mitochondrial-to-cytosolic Ca2+ level was calculated. Data from five-twelve areas per well and from at least six to eight wells per treatment group were collected in eachexperiment. Mitochondrial Ca2+ accumulation observed by Rhod-2 fluorescence was corroborated by inhibiting the mitochondrial uniporter using RU-360 (10 μM) for 30 min prior to butyrate co-treatment. In addition, in previous experiments, MitoTracker, a mitochondrial targeting dye, was used to validate the subcellular localization of Rhod-2 fluorescence (26).
Quantitative Reverse Transcriptase PCR
Total RNA was isolated using the RNAqueous 4PCR isolation kit from Ambion (Austin, TX). The isolated total RNA was subsequently treated with DNase to remove contaminating DNA. RNA was reverse-transcribedto cDNA using SuperScript II (Gibco BRL). Real-time PCR was performed using the ABI 7900 (Applied Biosystems, Foster City, CA). Pre developed Taqman gene expression assay for mouse CHOP (NM-008929) was obtained from Applied Biosystems.
Immunoblotting
Cell lysates from treated YAMC cells were immunoblotted with eIF2α or phospho-eIF2α antibody using the method of Davidson et al. (32) to evaluate the protein expression level. Briefly, samples were treated with SDS sample buffer and subjected to electrophoresisin a 4–20% pre-cast Tris–glycine gel. After electrophoresis, proteins were electroblotted onto PVDF membranes using a HoeferMighty Small Transphor Unit (Pharmacia, Piscataway, NJ) at 400mA for 100 min. After transfer, membranes were incubated with rabbit anti-eIF2α (9722) or rabbit anti-phospho-Ser51-eIF2α (9721, Cell Signaling, Danvers, MD) antibody overnight at 4°C, followed by peroxidase labeled goat anti-rabbit IgG incubation for 1 h at room temperature. Bands were detected using SuperSignal West Femto Maximum Sensitivity Substrate (Pierce, Rockford, IL), and the blots were scanned using a Bio-Rad Fluor-S MaxMultiImager System (Hercules, CA).
Cell Death Assay
Apoptosis was determined using nucleosomal fragmentation enzyme linked immuno-sorbent (ELISA) (Roche). Floating cells were harvested, washed, lysed and centrifuged to sediment nuclei. The supernatant containing mono and oligonucleosomes was incubated with substrate and analyzed by ELISA. Absorbance values were measured at 405 nm and subsequently normalized to the number of adherent cells per dish as previously described (22). To determine the association between mitochondrial-to-cytosolic Ca2+ ratio and apoptosis, select cultures were pre-incubated with RU-360 (10 μM) for 30 min prior to butyrate exposure. Cells were washed and treated with 5 mM butyrate and apoptosis was measured following a 24 h incubation period. Additionally, the association between the extrinsic pathway and apoptosis was determined by co-incubating select cultures with Fas:Fc or IgG1 with sodium butyrate for the final 24 h. In complimentary apoptosis experiments, adherent and floating cells were harvested, washed, pelleted by centrifugation and lysed.
Statistical Analysis
The effect of independent variables (treatment effects) was assessed using SuperAnova. Differences among means were determined using t/F type tests of contrast. P-values less than 0.05 were considered to be statistically significant.
Results
The findings in Figure 1 indicate that butyrate alone as well as EPA (n-3 PUFA), AA (n-6 PUFA control) and OA (n-9 monounsaturated control) in combination with butyrate significantly (p<0.05) enhanced apoptosis (~80%) compared to untreated control. In comparison, DHA and butyrate maximally enhanced apoptosis (170%) compared to untreated control. Fatty acid treatment alone had no effect on the apoptotic index (Figure 1 inset). The levels of butyrate and fatty acid are considered physiologically relevant because they lie within the range found in the colonic lumen (33) and blood (34), respectively. With regard to the effect of treatment on intracellular calcium levels, the combination of EPA and butyrate modestly (8%) increased (p<0.05) the mitochondrial-to-cytosolic Ca2+ ratio compared to untreated control (Figure 2). In comparison, the combination of DHA and butyrate maximally (47%) induced mitochondrial-to-cytosolic Ca2+ levels compared to untreated control. Results in Figure 2 inset indicate that fatty acid treatment alone had no effect on intracellular Ca2+ homeostasis.
Figure 1. Induction of apoptosis by fatty acid and butyrate co-treatment.

YAMC cells were treated with (50 μM) DHA, EPA, AA or OA for a total of 72 h and 0 or 5 mM butyrate for the final 24 h. Non-adherent cells were harvested and apoptosis was measured by nucleosomal fragmentation assay. C, control- no fatty acid or butyrate; B, butyrate only; DB, DHA and butyrate; EB, EPA and butyrate; AB, AA and butyrate; OB, OA and butyrate. Apoptotic index data are expressed as a percentage of untreated control. Data are from n = 6-12 wells per treatment, n = 2- 4 independent experiments. Mean values ± S.E. not sharing common letters are significantly different, p<0.05. Inset, Apoptotic index measured in cultures treated with fatty acid (50 μM for 72 h) only. D, DHA only; E, EPA only; A, AA only; O, OA only.
Figure 2. Fatty acid and butyrate co-treatment modulate mitochondrial Ca2+ levels.
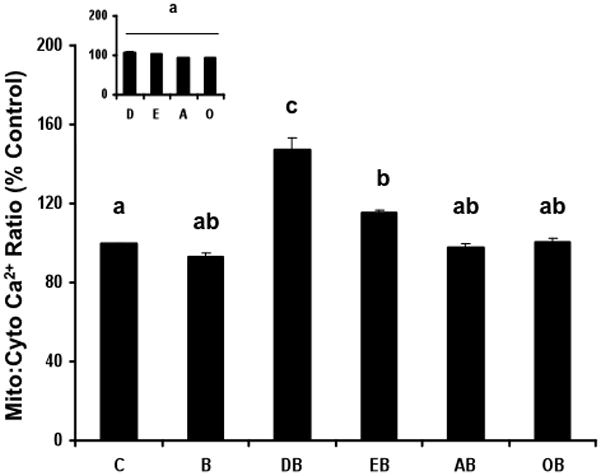
YAMC cells were exposed to 50 μM fatty acid for 72 h in the absence or presence of 5 mM butyrate for the final 24 h. Cells were co-loaded with Fluo-4 (3 μM) and Rhod-2 AM (2 μM) and the ratio of mitochondrial-to-cytosolic Ca2+ was measured as described in the Experimental Procedures Section. Data are means ± S.E., 20–60 cells per treatment, n = 2 independent experiments. Data represent mitochondrial-to-cytosolic Ca2+ levels as a percentage of untreated control. Bars not sharing common letters are significantly different, p< 0.05. Inset, Effect of fatty acid treatment alone on mitochondrial Ca2+ levels. See Figure 1 for legend details. Data are means ± S.E. from a representative experiment, n=2 independent experiments.
Having established that both DHA and EPA when combined with butyrate perturb intracellular Ca2+ homeostasis, we subsequently investigated the ability of an inhibitor of the mitochondrial Ca2+ uniporter, RU360 (28), to suppress the induction of apoptosis. In addition, since Fas receptor signaling transduction is involved in butyrate induction of apoptosis in colonocytes (22), we asked how YAMC cells would respond when cultured with antagonistic soluble Fas:Fc. As shown in Figure 3A, RU-360 partially blocked DHA and butyrate induced apoptosis while Fas:Fc had no effect. The combination of RU-360 + Fas:Fc partially reversed the proapoptotic effect of DHA and butyrate combination (DB). In contrast, Fas:Fc + RU-360 treatment combination (DBRF) did not exhibit any significant difference in apoptosis compared to the RU-360 treatment (DBR) group. In cells not exposed to fatty acid (Figure 3A inset), results indicate that butyrate enhanced apoptosis compared to untreated control cells (C). Fas:Fc alone (BF) and the combination of RU-360 + Fas:Fc (BRF) completely blocked the butyrate-induced increase in apoptosis in the absence of fatty acid treatment. In contrast, RU-360 alone had no effect on butyrate-induced apoptosis. Results in Figure 3B demonstrate the DHA and butyrate co-treatment significantly increased mitochondrial-Ca2+ accumulation compared to untreated control. Fas:Fc (DBF) had no effect on mitochondrial-to-cytosolic Ca2+ levels compared to DHA + butyrate (DB) treated cells. As expected, RU-360 pre-treated cultures (DBR, DBRF) exhibited a significant (p<0.05) reduction in mitochondrial-to-cytosolic Ca2+ levels. In cells not exposed to fatty acid (Figure 3B inset), Fas:Fc treatment (BF, BRF) reduced mitochondrial-to-cytosolic Ca2+ levels. In contrast, RU-360 alone (BR) had no effect on mitochondrial-Ca2+ accumulation, consistent with the lack of an effect of butyrate on intracellular Ca2+ homeostasis (26).
Figure 3. Fas-inhibitor and RU-360 block DHA and butyrate-induced changes in apoptosis and mitochondrial Ca2+.

(Panel A) YAMC cells were treated with DHA (50 μM) for 48 h. Subsequently, select cultures were pre-incubated with RU-360 (10 μM) for 30 min. RU-360 treated cells were rinsed and subsequently co-incubated with fatty acid + 5 mM butyrate only (DBR treatment) or DHA + butyrate + Fas-inhibitor (3 ug/ml Fas:Fc) for an additional 24 h (DBRF treatment). Select cultures not pre-treated with RU-360 were co-incubated with fatty acid + butyrate + Fas:Fc (DBF group). Non-adherent cells were harvested and nucleosomal fragmentation was used to quantify apoptosis. Data are means ± S.E., n = 6-12 wells per treatment from 2 separate experiments. Inset, Apoptotic index in cells treated with 5 mM butyrate in the absence of fatty acid (B), butyrate + RU-360 (BR), butyrate + Fas:Fc (BRF) or combination of butyrate + RU-360 + Fas:Fc (BRF). (Panel B) To evaluate mitochondrial-Ca2+ levels, cultures were co-loaded with Fluo-4 (3 μM) and Rhod-2 AM (2 μM), and the mitochondrial-to-cytosolic Ca2+ ratio was measured. Data are means ± S.E., n= 18-90 cells per treatment, n = 3 independent experiments. Inset, Mitochondrial-to-cytosolic Ca2+ levels in cells treated with butyrate, RU-360, Fas:Fc or the combination of RU-360 and Fas:Fc in the absence of fatty acid. Refer to Panel A inset for legend details.
We have previously demonstrated that the immortalized YAMC cell line is a good model system to examine the properties of bioactive chemoprotective with respect to apoptosis (19,22,34). In comparative experiments examining EPA, RU-360 partially blocked EPA and butyrate induced apoptosis while Fas:Fc had no effect (Figure 4A). The combination of RU-360 + Fas:Fc partially reversed the proapoptotic effect of EPA and butyrate combination (EB). In contrast, Fas:Fc + RU-360 treatment combination (EBRF) did not exhibit any significant difference in apoptosis compared to RU-360 treatment (EBR) group. In cells not exposed to fatty acid or butyrate (Figure 4A inset), the addition of cell death modulators alone or in combination (RU-360, Fas:Fc or Ru-360+Fas:Fc) had no effect on apoptosis compared to untreated control cells (C). Analysis of mitochondrial-to-cytosolic Ca2+ ratios following EPA treatment is shown in Figure 4B. EPA + butyrate combination significantly (p<0.05) enhanced mitochondrial-to-cytosolic Ca2+ levels, which was inhibited by RU-360 (EBR) and the combination of Fas:Fc and RU-360 (EBRF). In contrast, Fas:Fc treatment in the absence of RU-360 (EBF) had no effect on mitochondrial-to-cytosolic Ca2+ accumulation. As shown in Figure 4B inset, for cells treated with RU-360 (CR), Fas:Fc (CF) or the combination of RU-360 and Fas:Fc CRF) in the absence of fatty acid and butyrate, only RU-360 and Fas:Fc treatment (CRF) reduced mitochondrial-to-cytosolic Ca2+ levels as compared to untreated control cells.
Figure 4. Fas-inhibitor and RU-360 block EPA and butyrate-induced changes in apoptosis and mitochondrial Ca2+.

(Panel A) YAMC cells were treated with EPA (50 μM) for 48 h after which select cultures were incubated with RU-360 (10 μM) for 30 min to inhibit mitochondrial Ca2+ uptake. RU-360 pre-treated cells were rinsed and subsequently co-incubated with EPA and butyrate (5 mM) only (EBR treatment) or EPA + butyrate + Fas:Fc (3 ug/ml) for the final 24 h (EBRF treatment). Select cultures not pre-treated with RU-360 were co-incubated with EPA + butyrate + Fas:Fc (EBF treatment). Data are means ± S.E., n = 6 wells per treatment. Inset, Mitochondrial-to-cytosolic Ca2+ levels in untreated cells (no fatty acid or butyrate), RU-360, Fas:Fc or the combination of RU-360 and Fas:Fc. C, untreated control cells-no fatty acid or butyrate added; CR, RU-360 pretreatment; CF, Fas:Fc co-treatment; CRF, RU-360 and Fas;Fc co-treatment. (Panel B) Analysis of mitochondrial-to- cytosolic Ca2+ ratios following EPA treatment. Cultures were co-loaded with Fluo-4 (3 μM) and Rhod-2 AM (2 μM), and the mitochondrial-to-cytosolic Ca2+ ratio was measured. See panel A for treatment details. Inset, Mitochondrial-to-cytosolic Ca2+ ratios in untreated control cells-no fatty acid or butyrate added.
Since Ca2+ is stored in the ER, and ER stress increases Ca2+ transfer to mitochondria, thereby amplifying the PERK-eIF2α-ATF4-CHOP pathway (36), we further examined the effect of fatty acid and butyrate treatment on ER stress regulated genes. Both butyrate alone and butyrate-fatty acid combinations modestly increased CHOP mRNA expression relative to basal untreated control (Figure 5). Thapsigargin, an inhibitor of sarco/ER Ca2+−ATPase (SERCA) activity, known to induce ER stress, was used as a positive pharmacological control (26,37). Examination of the ER stress protein phosphorylation (phosphorylated alpha-subunit of eukaryotic initiation factor 2 (P-eIF2α)) revealed no induction of the ER stress response (Figure 6). Collectively, these data suggest that the proapoptotic synergy between DHA and butyrate involving mitochondrial Ca2+ loading is not mediated by ER stress.
Figure 5. Butyrate exposure modestly increases CHOP expression.

YAMC cells were treated with butyrate (5 mM), DHA (50 μM), LA (50 μM, n-6 PUFA control) or combinations of butyrate and DHA (But + DHA), butyrate and LA (But + LA), or Thapsigarain (10 μM) (ER stress positive control) for 24 h. Total RNA was isolated and the CHOP mRNA levels were measured using Taqman real-time PCR. Data are means ± SE, expressed as fold of basal CHOP expression, n=3-6 wells. Bars not sharing common letters are significantly different, p<0.05.
Figure 6. Inability of butyrate and fatty acid combination to alter phospho- and total-eIF2α protein levels.

YAMC cells were treated with DHA (50 μM), LA (50 μM), a combination of butyrate (5 mM) and DHA (But + DHA), butyrate and LA (But + LA), or Thapsigargin (10 μM,Th) (ER stress positive control) for 24 h. Cell lysates were prepared and phopho-eIF2α and total eIF2α protein levels were measured by immunoblotting. (A) Representative immunoblots of p-eIF2α and total eIF2α. (B) Quantification of immunoblot data, expressed as percentage of phospho/total eIF2α. Data are means ± SE, n=3-6 wells. Bars not sharing common letters are significantly different, p<0.05.
Discussion
An important outcome of our previous studies was the demonstration that the anti-tumorigenic effect of fish oil (n-3 PUFA) is enhanced when a highly fermentable fiber or its fermentation product-butyrate is added to the diet (4,18–20,38). This chemopreventive effect is mediated in part by the upregulation of targeted apoptosis of DNA adducts during tumor initiation (16,27) and spontaneous apoptosis during promotion (39). With respect to a mechanism of action, pectin is metabolized by bacteria within the lumen of the gut to generate millimolar levels of butyrate and other short chain fatty acids (32). We have recently shown that the combination of DHA and butyrate compared with DHA or butyrate alone further enhances colonocyte apoptosis by inducing a p53-independent, oxidation sensitive, mitochondrial Ca2+−dependent (intrinsic) pathway (26,28). Our results using YAMC cells are consistent with data generated using the human (HCT116) colon cancer cell line as well as mouse and rat azoxymethane in vivo models (4,19,20,26,28). Therefore, similar results are observed when neoplastic cells are used. Collectively, these data indicate that highly fermentable fiber, which generates butyrate in the colon, only has chemotherapeutic value when n-3 PUFA is the lipid source. This critical observation emphasizes the need to examine both the lipid and fiber composition of diets.
In order to further elucidate specifically which dietary n-3 PUFA in combination with fermentable fiber up-regulate apoptosis effector mechanisms in colonocytes, we contrasted the ability of EPA, AA or OA vs DHA in combination with butyrate to synergistically induce apoptosis. Specifically, n-6 and n-3 PUFA were examined with respect to apoptogenic mitochondrial Ca2+ overloading and ER stress. We report that the combination of DHA and butyrate maximally induced apoptosis and mitochondrial-to-cytosolic Ca2+ levels. In comparison, EPA, a precursor to DHA, was minimally effective. Similarly, arachidonic acid (20:4n-6) and oleic acid (18:1n-9) in combination with butyrate had no effect on mitochondrial Ca2+ or apoptosis compared to butyrate alone. RU-360, a mitochondrial-uniporter inhibitor, abrogated mitochondrial-to-cytosolic Ca2+ accumulation and also partially blocked apoptosis in DHA and butyrate cotreated cells. Collectively, these data show that only the combination of DHA and butyrate, compared to fatty acid or butyrate alone, synergistically enhances apoptosis by additionally recruiting a Ca2+-mediated intrinsic mitochondrial pathway. Data from these experiments extend observations in human cell lines and rat colonic crypt cultures (28). Since butyrate is capable of inducing colonocyte apoptosis via a nonmitochondrial, Fas-mediated, extrinsic pathway (22,26), we also examined the effect of a CD95 (Fas receptor) antagonist. As expected, Fas:Fc blocked apoptosis in cultures incubated with butyrate alone. However, this antagonist did not block mitochondrial-to-cytosolic Ca2+ accumulation and apoptosis in DHA plus butyrate treated cells.
Since select fatty acids can regulate cell death via induction of ER stress in a number of cell types (29,30), we examined whether a PUFA/butyrate combination regimen activates ER stress responses in colonocytes. Intriguingly, we found little evidence that DHA ± butyrate cotreatment increases ER stress regulated genes, CHOP and eIF2α. This is in contrast with a previous report where the cytotoxic effect of DHA was associated with signaling pathways involving ER stress (30). In these studies, DHA was added in ethanol at a higher concentration (70 μM) to the colon cancer cell line SW620. Treatment of these cells leads to the accumulation of numerous large lipid droplets, which is likely cytoxic (30). In order to avoid artifacts related to fatty acid oxidation and toxicity, we routinely complex fatty acids to albumin, a physiologically relevant delivery modality for long chain PUFA use in culture (40).
Acquired resistance to apoptosis is a common defect associated with tumor cells and represents a critical obstacle for therapy (14,41). Therefore, the elucidation of novel apoptotic triggers constitutes an important objective in the effort to develop therapies. It is important to note that chemopreventive interventions using drugs have raised safety concerns. For example, the long-term use of COX-2 selective non-steroidal anti-inflammatory drugs is not feasible due to associated cardiovascular risk (42,43). This has dampened enthusiasm for their extensive use as cancer chemopreventive therapies. Safety problems intrinsic to drugs administered over long periods of time strengthen the interest in diet-related cancer chemoprevention approaches. Therefore, the search for toxicologically innocuous dietary interventions is timely and propitious, and foodstuffs providing both DHA and butyrate-generating fermentable fiber provide an attractive alternative.
In summary, we have demonstrated that the pleiotropic bioactive components generated by fermentable fiber (butyrate) and fish oil (DHA but not EPA) work coordinately to trigger a Ca2+-dependent intrinsic mitochondrial-mediated apoptotic pathway in colonocytes. This is significant because n-3 PUFA (1,6,8) and butyrate (20,41,44,45) reduce colon cancer risk in humans, and appear to be ideally suited as a new generation of nontoxic chemotherapeutics to work either alone or in combination with chemoprotective drugs, e.g., NSAIDs, whose long term use is contraindicated (43). Given the critical nature of apoptosis in colon cancer prevention, and the fact that inhibition of apoptosis is an integral component in the genesis of colon cancer, it is imperative to elucidate the precise mechanisms by which n-3 PUFA and butyrate promote apoptosis in the colon.
Acknowledgments
This work was supported in part by NIH grants CA59034, and USDA 2010-34402-20875, "Designing Foods for Health" through the Vegetable & Fruit Improvement Center.
Abbreviations Used
- AA
arachidonic acid
- BSA
bovine serum albumin
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- ER
endoplasmic reticulum
- FAP
familial adenomatous polyposis
- FBS
fetal bovine serum
- IFNγ
gamma interferon
- LA
linoleic acid
- OA
oleic acid
- SERCA
sarco/ER Ca2+ ATPase
- YAMC
young adult mouse colonocytes
References
- 1.Anti M, Armelao F, Marra G, Percesepe A, Bartoli GM, Palozza P, Parrella P, Canetta C, Gentiloni N, De Vitis I, Gasbarrini G. Effects of Different doses of fish Oil on Rectal Cell Proliferation in Patients with Sporadic Colonic Adenomas. Gastroenterology. 1994;107:1709–1718. doi: 10.1016/0016-5085(94)90811-7. [DOI] [PubMed] [Google Scholar]
- 2.Bartram HP, Gostner A, Scheppach W, Reddy BS, Rao CV, Dusel G, Richter F, Richter A, Kasper H. Effects of Fish Oil on Rectal Cell Proliferation, Mucosal Fatty Acids, and Prostaglandin E2 Release in Healthy Subjects. Gastroenterology. 1993;105:1317–1322. doi: 10.1016/0016-5085(93)90135-y. [DOI] [PubMed] [Google Scholar]
- 3.Caygill CP, Charlett A, Hill MJ. Fat, Fish, fish Oil and Cancer. Br J Cancer. 1996;74:159–164. doi: 10.1038/bjc.1996.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang WCL, Chapkin RS, Lupton JR. Predictive value of proliferation, differentiation and apoptosis as intermediate markers for colon tumorigenesis. Carcinogenesis. 1997;18:721–730. doi: 10.1093/carcin/18.4.721. [DOI] [PubMed] [Google Scholar]
- 5.Chang WC, Chapkin RS, Lupton JR. Fish oil blocks azoxymethane-induced tumorigenesis by increased cell Differentiation and apoptosis rather than decreased cell proliferation. J Nutr. 1998;18:351–357. doi: 10.1093/jn/128.3.491. [DOI] [PubMed] [Google Scholar]
- 6.Cheng J, Ogawa K, Kuriki K, Yokoyama Y, Kamiya T, Seno K, Okuyama H, Wang J, Luo C, Fujii T, Ickikawa H, Shirai T, Tokudome S. Increased intake of n-3 polyunsaturated fatty acids elevates the level of apoptosis in the normal sigmoid colon of patients polypectomized for adenomas/tumors. Cancer Lett. 2003;193:17–24. doi: 10.1016/s0304383502007176. [DOI] [PubMed] [Google Scholar]
- 7.Reddy BS, Patlolla JM, Simi B, Wang SH, Rao CV. Prevention of colon cancer by low doses of Celecoxib, a cyclooxygenase inhibitor, administered in a diet rich in w-3 polyunsaturated fatty acids. Cancer Res. 2005;65:8022–8027. doi: 10.1158/0008-5472.CAN-05-0212. [DOI] [PubMed] [Google Scholar]
- 8.Courtney ED, Matthews S, Finlayson C, Di Pierro D, Belluzzi A, Roda E, Kang JY, Leicester RJ. Eicosapentaenoic acid (EPA) reduces crypt cell proliferation and increases apoptosis in normal colonic mucosa in subjects with a history of colorectal adenomas. Int J Colorectal Dis. 2007;22:765–76. doi: 10.1007/s00384-006-0240-4. [DOI] [PubMed] [Google Scholar]
- 9.Hall MN, Chavarro JE, Lee IM, Willett WC, Ma J. A 22-year prospective study of fish, n-3 fatty acid intake, and colorectal cancer risk in men. Cancer Epidemiol Biomarkers Prev. 2008;17:1136–1143. doi: 10.1158/1055-9965.EPI-07-2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.West NJ, Clark SK, Phillips RK, Hutchinson JM, Leicester RJ, Belluzzi A, Hull MA. Eicosapentaenoic acid reduces rectal polyp number and size in familial adenomatous polyposis. Gut. 2010;59:918–925. doi: 10.1136/gut.2009.200642. [DOI] [PubMed] [Google Scholar]
- 11.Verlengia R, Gorjao R, Kanunfre CC, Bordin S, De Lima TM, Martins EF, Curi R. Comparative effects of eicosapentaenoic acid and docosahexaenoic acid on proliferation, cytokine production, and pleiotropic gene expression in Jurkat cells. J Nutr Biochem. 2004;15:657–664. doi: 10.1016/j.jnutbio.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 12.Rahman MD, Bhattacharya A, Fernandez G. Docosahexaenoic acid is more potent inhibitor of osteoclast differentiation in RAW 264.7 cells than eicosapentaenoic acid. J Cell Physiol. 2008;214:201–209. doi: 10.1002/jcp.21188. [DOI] [PubMed] [Google Scholar]
- 13.Chapkin RS, Seo J, McMurray DN, Lupton JR. Mechanisms by which docosahexaeonic acid and related fatty acids reduce colon cancer risk and inflammatory disorders of the intestine. Chemistry Phys Lipids. 2008;153:14–23. doi: 10.1016/j.chemphyslip.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bedi A, Pasricha PJ, Akhtar AJ, Barber JP, Bedi GC, Giairdiello FM, Zehnbauer BA, Hamilton SR, Jones RJ. Inhibition of apoptosis during development of colorectal cancer. Cancer Res. 1995;55:1811–1816. [PubMed] [Google Scholar]
- 15.Siniscrope FA, Ruan SB, Cleary KR, Stephens LC, Lee JJ, Levin B. bcl-2 and p53 oncoprotein expression during colorectal tumorigenesis. Cancer Res. 1995;55:237–241. [PubMed] [Google Scholar]
- 16.Hong MY, Lupton JR, Morris JS, Wang N, Carroll RJ, Davidson LA, Elder RH, Chapkin RS. Dietary fish oil reduces DNA adduct levels in rat colon in part by increasing apoptosis during tumor initiation. Cancer Epidemiology Biomarkers and Prev. 2000;9:819–826. [PubMed] [Google Scholar]
- 17.Hong MY, Bancroft LK, Turner ND, Davidson LA, Murphy ME, Carroll RJ, Chapkin RS, Lupton JR. Fish oil decreases oxidative DNA damage by enhancing apoptosis in rat colon. Nutrition & Cancer. 2005;52:166–175. doi: 10.1207/s15327914nc5202_7. [DOI] [PubMed] [Google Scholar]
- 18.Chapkin RS, McMurray DN, Lupton JR. Colon cancer, fatty acids and anti-inflammatory compounds. Curr Opin Gastroenterol. 2007;23:48–54. doi: 10.1097/MOG.0b013e32801145d7. [DOI] [PubMed] [Google Scholar]
- 19.Ng Y, Barhoumi R, Tjalkens RB, Fan YY, Kolar S, Wang N, Lupton JR, Chapkin RS. The role of docosahexaenoic acid mediating mitochondrial membrane lipid oxidation and apoptosis in colonocytes. Carcinogenesis. 2005;26:1914–1921. doi: 10.1093/carcin/bgi163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crim KC, Sanders L, Hong MY, Taddeo S, Turner ND, Chapkin RS, Lupton JR. Upregulation of p21waf1/cip1 expression in vivo by butyrate administration can be chemoprotective or chemopromotive depending on the lipid component of the diet. Carcinogenesis. 2008;29:1415–1420. doi: 10.1093/carcin/bgn144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith JG, Yokoyama WH, German JB. Butyric acid from the diet: actions at the level of gene expression. Crit Rev Food Sci. 1998;38:259–297. doi: 10.1080/10408699891274200. [DOI] [PubMed] [Google Scholar]
- 22.Fan YY, Zhang J, Barhoumi R, Burghardt RC, Turner ND, Lupton JR, Chapkin RS. Antagonism of CD95 signaling blocks butyrate induction of apoptosis in young adult mouse colonic cells. Am J Physiol. 1999;277(2 Pt 1):C310–319. doi: 10.1152/ajpcell.1999.277.2.C310. [DOI] [PubMed] [Google Scholar]
- 23.Fan YY, Tian Y, Davidson LA, Zhou L, Callaway E, Weeks BR, Toyokuni S, Lupton JR, Chapkin RS. Proapoptotic effects of n-3 fatty acids are enhanced in SOD2 knockout mouse colon. J Nutr. 2009;139:1328–1332. doi: 10.3945/jn.109.106203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signaling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 25.Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007;47:143–183. doi: 10.1146/annurev.pharmtox.47.120505.105122. [DOI] [PubMed] [Google Scholar]
- 26.Kolar SS, Barhoumi R, Lupton JR, Chapkin RS. Docosahexaenoic acid and butyrate synergistically induce colonocyte apoptosis by enhancing mitochondrial Ca2+ accumulation. Cancer Res. 2007;67:5561–5568. doi: 10.1158/0008-5472.CAN-06-4716. [DOI] [PubMed] [Google Scholar]
- 27.Hong MY, Chapkin RS, Barhoumi R, Burghardt RC, Turner ND, Henderson CE, Sanders LM, Fan YY, Davidson LA, Murphy ME, Spinka CM, Carroll RJ, Lupton JR. Fish oil increases mitochondrial phospholipid unsaturation, upregulating reactive oxygen species and apoptosis in rat colonocytes. Carcinogenesis. 2002;23:1919–1925. doi: 10.1093/carcin/23.11.1919. [DOI] [PubMed] [Google Scholar]
- 28.Kolar SS, Barhoumi R, Callaway ES, Fan YY, Wang N, Lupton JR, Chapkin RS. Synergy between docosahexaenoic acid and butyrate elicits p53-independent apoptosis via mitochondrial Ca2+ accumulation in human colon cancer cells and primary cultures of rat colonic crypts. Am J Physiology: GI and Liver Physiology. 2007;293:G935–G943. doi: 10.1152/ajpgi.00312.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diakogiannaki E, Morgan NG. Differential regulation of the ER stress response by long-chain fatty acids in the pancreatic b-cell. Biochem Soc Trans. 2008;36:959–962. doi: 10.1042/BST0360959. [DOI] [PubMed] [Google Scholar]
- 30.Jacobsen CH, Storvold GL, Bremseth H, Follestad T, Sand K, Mack M, Olsen KS, Lundemo AG, Iverson JG, Krokan HE, Schonberg SA. DHA induces ER stress and growth arrest in human colon cancer cells: associations with cholesterol and calcium homeostasis. J Lipid Res. 2008;49:2089–2100. doi: 10.1194/jlr.M700389-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitehead RH, VanEeden PE, Noble MD, Ataliotis P, Jat PS. Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H-2Kb-tsA58 transgenic mice. Proc Natl Acad Sci. 1993;90:587–591. doi: 10.1073/pnas.90.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davidson LA, Lupton JR, Jiang YH, Chapkin RS. Carcinogen and dietary lipid regulate ras expression and localization in rat colon without affecting farnesylation kinetics. Carcinogenesis. 1999;20:785–791. doi: 10.1093/carcin/20.5.785. [DOI] [PubMed] [Google Scholar]
- 33.Zoran DL, Turner ND, Taddeo SS, Chapkin RS, Lupton JR. Wheat bran reduces tumor incidence in a rodent model of colon cancer independent of effects on distal luminal butyrate concentrations. J Nutr. 1997;127:2217–2225. doi: 10.1093/jn/127.11.2217. [DOI] [PubMed] [Google Scholar]
- 34.Conquer JA, Holub BJ. Effect of supplementation with different doses of DHA on the levels of circulating DHA as non-esterified fatty acid in subjects of Asian Indian background. J Lipid Res. 1998;39:286–292. [PubMed] [Google Scholar]
- 35.Turk HF, Kolar SS, Fan YY, Cozby CA, Lupton JR, Chapkin RS. Linoleic acid and butyrate synergize to increase bcl-2 in colonocytes. Inter J Cancer. 2011;128:63–71. doi: 10.1002/ijc.25323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chami M, Oules B, Szabadkai G, Tacine R, Rizzuto R, Paterlini-Brechot P. Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol Cell. 2008;32:641–651. doi: 10.1016/j.molcel.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao P, Xiao X, Kim AS, Leite MF, Xu J, Zhu X, Ren J, Li J. c-Jun inhibits thapsigargin-induced ER stress through up-regulation of DSCR1/Adapt78. Exp Biol Med. 2008;233:1289–1300. doi: 10.3181/0803-RM-84. [DOI] [PubMed] [Google Scholar]
- 38.Sanders LM, Henderson CE, Hong MY, Barhoumi R, Burghardt RC, Wang N, Spinka CM, Carroll RJ, Turner ND, Chapkin RS, Lupton JR. Enhancement of reactive oxygen species by dietary fish oil and attenuation of antioxidant defenses by dietary pectin coordinately heightens apoptosis in rat. J Nutr. 2004;134:3233–3238. doi: 10.1093/jn/134.12.3233. [DOI] [PubMed] [Google Scholar]
- 39.Davidson LA, Nguyen DV, Hokanson RM, Callaway ES, Isett RB, Turner ND, Dougherty ER, Wang N, Lupton JR, Carroll RJ, Chapkin RS. Chemopreventive n-3 polyunsaturated fatty acids reprogram genetic signatures during colon cancer initiation and progression in the rat. Cancer Res. 2004;64:6797–6804. doi: 10.1158/0008-5472.CAN-04-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim W, McMurray DN, Chapkin RS. n-3 polyunsaturated fatty acids – physiological relevance and dose. Prostaglandins Leukotrienes & Essential Fatty Acids. 2010;82:155–158. doi: 10.1016/j.plefa.2010.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lopez de Silanes N, Olmo J, Turnay G, Gonzalez de Buitrago P, Perez-Ramos A, Guzman-Aranguez M, Garcia-Diez E, Lecona M, Gorospe M, Lizarbe MA. Acquisition of resistance to butyrate enhances survival after stress and induces malignancy of human colon carcinoma cells. Cancer Res. 2004;64:4593–4600. doi: 10.1158/0008-5472.CAN-04-0711. [DOI] [PubMed] [Google Scholar]
- 42.Graham DJ. Cox-2 inhibitors, other NSAIDs, and cardiovascular risk. JAMA. 2006;296:1653–1656. doi: 10.1001/jama.296.13.jed60058. [DOI] [PubMed] [Google Scholar]
- 43.Bresalier RS. Chemoprevention of colorectal neoplasia: advances and controversies (theCOX-2 story) Curr Opin Gastroenterol. 2007;23:44–47. doi: 10.1097/MOG.0b013e328011c482. [DOI] [PubMed] [Google Scholar]
- 44.Bingham SA, Day NE, Luben R, Ferrari P, Slimani N, Norat T, Clavel-Chapelon F, Kesse E, Nieters A, Boeing H, Tjonneland A, Overvad K, Martinez C, Dorronsoro M, Gonzalez CA, Key TJ, Trichopoulou A, Naska A, Vineis P, Tumino R, Krogh V, Bueno-de-Mesquita HB, Peeters PH, Berglund G, Hallmans G, Lund E, Skeie G, Kaaks R, Riboli E. European Prospective Investigation into Cancer and Nutrition. Dietary fibre in food and protection against colorectal cancer in the European Prospective investigation into cancer and nutrition (EPIC): an observational study. Lancet. 2003;361:1496–1501. doi: 10.1016/s0140-6736(03)13174-1. [DOI] [PubMed] [Google Scholar]
- 45.Gupta N, Martin PM, Prasad PD, Ganapathy V. SLC5A8 (SMCT1)-mediated transport of butyrate forms the basis for the tumor suppressive function of the transporter. Life Sci. 2006;78:2419–2425. doi: 10.1016/j.lfs.2005.10.028. [DOI] [PubMed] [Google Scholar]