

Abstract
Chronic wasting disease (CWD) is a fatal spongiform encephalopathy that is efficiently transmitted among members of the mammalian family Cervidae, including deer, elk, and moose. Typical of prion diseases, CWD is characterized by the conversion of the native protease-sensitive protein PrPC to a protease-resistant isoform, denoted PrPRES. In native species, spread of the disease likely results from the ingestion of prion-containing excreta, including urine, saliva, or feces. Although cervid prion protein-expressing transgenic [Tg(CerPrP)] mice have been shown to be effective surrogates of natural CWD, uncertainties remain regarding the mechanisms by which CWD prions traffic in vivo, including the manner by which CWD prions traffic from the gastrointestinal tract to the central nervous system. We used elk prion protein-expressing transgenic [Tg(CerPrP-E)] mice, infected by three different routes of inoculation, and tissue-based IHC to elucidate that centripetal and centrifugal CWD prion transit pathways involve cells and fibers of the autonomic nervous systems, including the enteric nervous system and central autonomic network. Moreover, we identified CWD PrPRES associated with the cell bodies and processes of enteric glial cells within the enteric nervous system of CWD-infected Tg(CerPrP-E) mice. The present findings demonstrate the importance of the peripheral and central autonomic networks in CWD neuroinvasion and neuropathogenesis and suggest that enteroglial cells may facilitate the shedding of prions via the intestinal tract.
Chronic wasting disease (CWD) of white-tailed deer (WTD), mule deer, elk, and moose is a prion disease first identified in the Rocky Mountain region of the United States; it is now recognized in 19 states, Canada, and Korea (infected deer imported from Canada) (http://www.cwd-info.org/index.php/fuseaction/about.map, last accessed July 17, 2011).1,2 Typical of all transmissible spongiform encephalopathies, including scrapie, bovine spongiform encephalopathy (BSE), and human Creutzfeldt-Jakob disease, CWD is caused by the conversion of normal protease-sensitive prion protein (PrPC) to a misfolded protease-resistant conformation (PrPRES), which accumulates in the central nervous system (CNS) and the lymphoid system and leads to wasting and spongiform encephalopathy.3–5 The terminal accumulations of PrPRES in the dorsal motor nucleus of the vagus nerve (DMNV), the solitary tract nucleus (NTS), and the neuroendocrine system in infected cervids suggest that the autonomic nervous system (ANS) may be central to the in vivo trafficking of prions.6,7 The pathways of intracranial spread of prions are even more uncertain, but the central autonomic network (CAN) is a prime candidate. The CAN comprises a series of interconnected areas throughout the neuraxis (most notably regions of the cerebral cortex, amygdala, hypothalamus, periaqueductal gray matter, and medulla) and connecting to the ANS.
Although the infectious nature of feces in CWD-afflicted cervids has been repeatedly demonstrated, the manner by which that infectivity is conferred remains uncertain.8–10 One such candidate facilitator of prion infectivity is the enteric nervous system (ENS), which is a series of plexus throughout the intestinal wall consisting of a heterogenous population of specialized neurons and enteric glial cells (EGCs).11,12 Anatomically, the EGCs, through the extension of cellular processes along the length of the intestinal villi, provide a theoretical conduit for the transintestinal trafficking of CWD prions.11,12 Moreover, support for the involvement of the ENS can be found in studies, of both CWD-infected cervids and cervidized mice, that have identified PrPRES in the ENS.7,13
The principal aim of the present investigation focuses on the progressive pattern of PrPRES accumulation in the nervous system of CWD-infected, elk PrP-expressing (Tg[CerPrP-E]) mice, mice with emphasis on the constituents of the ANS: the central autonomic network, the parasympathetic and sympathetic nervous systems, and the enteric nervous system. We studied Tg(CerPrP-E) mice inoculated by one of four routes and mapped the longitudinal accumulation of PrPRES by serial sacrifice and enhanced immunohistochemistry (IHC). We found that cells and fiber tracts of the ANS and CAN serve as major pathway by which prions spread to and within the CNS of CWD-infected mice. Furthermore, to address questions regarding the role of the ENS in CWD infection, we developed a series of single-label and dual-label immunostaining protocols to identify relationships between PrPRES and cells of the ENS. Through this work, we demonstrate that intimate relationships between PrPRES and EGCs may facilitate the transit of CWD prions across the gastrointestinal (GI) mucosal barrier.
Materials and Methods
Generation and Genotyping of 5037 Tg[CerPrP-E] Mice
The transgenic mice expressing the elk PrP coding sequence used in these studies [5037 Tg(CerPrP-E)] were generated and their susceptibility to CWD was established in the laboratory of G.C.T.14 In brief, to generate the elk PrP coding sequence, codon 226 of the deer PrP gene was mutated from Q to E by site-directed mutagenesis. This resultant coding sequence was inserted into the MoPrP.Xho expression vector and the purified transgene was microinjected into FVB/Prnp0/0 oocytes. Transgenic founder mice were identified by PCR screening of genomic DNA and were mated to FVB/Prnp0/0 mice to generate the hemizygous transgenic lines from which the animals used in these studies were derived. The PrP0/0 mice used in the PrPC mapping studies were kindly donated by Dr. Mark Zabel at Colorado State University. The animal experiments were conducted under guidelines developed by the Colorado State University Animal Care and Use Committee.
Inoculum Preparation
The CWD inoculum was prepared as a brain homogenate derived from a terminally ill CWD-positive WTD, which was infected as part of other studies in this laboratory. The negative control inoculum consisted of a brain homogenate from a CWD-negative WTD originating outside of the CWD endemic area (courtesy of David Osborn, University of Georgia, Athens). Both inocula were homogenized in sterile PBS using a reciprocal homogenizer and were diluted to a final concentration of 1% (w/v) using sterile PBS containing penicillin-streptomycin (100 U/mL).
Inoculation Protocol
Four experimental groups of 5037 Tg(CerPrP-E) mice (n = 10 per group) were used in these studies, consisting of 4- to 6-week-old Tg(CerPrP-E) mice of both sexes inoculated with CWD prions via one of four routes: intracerebral, intraperitoneal, intravenous, or oral. For intracerebral inoculation, mice were sedated through the intraperitoneal injection of a mixture of ketamine (120 mg/kg) and xylazine (16 mg/kg). Mice were inoculated with 30 μL of a 1% brain homogenate via a 29-gauge needle in the left parietal lobe of the cerebral cortex. For intraperitoneal inoculation, 100 μL of a 1% brain homogenate was injected via a 29-gauge needle into the right caudal ventral abdomen. For intravenous inoculation, 30 μL of a 1% brain homogenate was injected through either the left or right tail vein. Oral inoculations were performed by oral instillation of 50 μL of a 1% brain homogenate administered on each of two consecutive days. Mice were not sedated for the intraperitoneal, intravenous, or oral inoculations. Negative control mice were inoculated by all four routes (n = 6 per route) with 1% brain homogenate obtained from a CWD-negative WTD.
Animal Evaluation, Euthanasia, and Necropsy
After inoculation, Tg[CerPrP-E] mice were sacrificed at either a predetermined time point or at the onset of terminal neurological disease. The criteria used for the diagnosis of central nervous dysfunction in prion-inoculated mice were as described previously: severe ataxia, difficulty in righting from a supine position, tail rigidity, generalized tremors, and/or severe mental obtundation.15 Tg(CerPrP-E) mice were sacrificed at time points chosen using the previously reported 220-day survival period after intracerebral inoculation,16 as follows: 60 days after inoculation (dpi), 120 dpi, 180 dpi, and 240 dpi or at terminal disease. For mice allowed to progress to terminal disease, time to disease is expressed as mean ± SEM.
At each predetermined sacrifice point, 3 mice per inoculation group were euthanized (2 mice from the CWD-inoculated group and 1 mouse from the sham-inoculated group), for a total of 12 mice at each time point. All animals were perfusion-fixed with paraformaldehyde-lysine-periodate using a commercially available, gravity-feed system (AutoMate Scientific, Berkeley, CA) in a modification of a published protocol, which applies left ventricular fixative injection and right atrial exsanguination,17 and then were necropsied. At necropsy, samples from all tissues were obtained, including those from both nervous (brain and spinal cord) and non-nervous (peripheral) systems. After necropsy, all fixed tissues were immersion postfixed in paraformaldehyde-lysine-periodate fixative for 24 hours before transfer into 70% ethanol for long-term storage. Within 72 to 96 hours of necropsy, tissues were routinely trimmed for histological processing. To standardize the evaluated neuroanatomic regions between mice, brains were identically sectioned using an acryl coronal brain matrix (Stoelting, Wood Dale, IL) to generate slices representing the following five neuroanatomic regions: i) the cerebral cortex; ii) the hippocampus; iii) the diencephalon, including the thalamus; iv) the cerebellum; and v) the medulla oblongata at the level of the obex and the pontine area. These regions have previously been determined useful in mouse models of scrapie.18 In PrPRES detection studies, to ablate and enhance antigen detection, cassetted tissues were immersed in 88% formic acid for 1 hour and then were rinsed in running tap water for 1 hour before histological processing.
Histology and PrPRES IHC
For all experiments, paraffin-embedded tissue sections (4 to 6 μm thick) were mounted on positively charged glass slides and deparaffinized in an oven at 64°C, which was followed by successive immersions in xylene and rehydration through graded ethanol solutions. To enhance detection, tissues were subjected to heat-induced epitope retrieval using an automated antigen-retrieval system (retriever; Electron Microscopy Services, Hatfield, PA) and a proprietary buffer solution (target retrieval solution; DakoCytomation, Carpinteria, CA).
For the detection of PrPRES, an IHC protocol was used that combines a one-step immunostaining procedure with tyramide signal amplification (TSA). Previous work in our laboratory has demonstrated this methodology to be more sensitive in the detection of PrPRES than traditional, two-step indirect IHC protocols.3,19 The TSA-PrPRES protocol used a proprietary TSA-detection kit (PerkinElmer, Waltham, MA) and was performed as follows. After slide rehydration and heat-induced epitope retrieval, endogenous peroxidase activity was quenched with 3% hydrogen peroxide (H2O2) in methanol and sections were blocked with a proprietary protein block (TNB; PerkinElmer) for 60 minutes each. Next, slides were incubated with an optimal dilution of the horseradish peroxidase (HRP)-conjugated anti-prion protein antibody, BAR-224 (HRP-BAR224; Cayman Chemical, Ann Arbor, MI), which is a monoclonal prion protein antibody raised against amino acids 141 to 151 of the ovine prion protein. Between all incubation steps, slides were washed three times (5 minutes each) in TNT wash buffer (0.1 mol/L Tris-HCl, pH 7.5, 0.15 mol/L NaCl, and 0.05% Tween-20). After the application of the antibodies, slides were sequentially incubated with two proprietary TSA reagents: DNP amplification reagent (PerkinElmer) for 10 minutes and anti-DNP-HRP (PerkinElmer) for 30 minutes. Antibody deposition was visualized using the chromogen 3-amino-9-ethylcarbazole (AEC; DakoCytomation), and slides were counterstained with hematoxylin and incubated with a bluing reagent (0.1% sodium bicarbonate). After immunostaining, slides were coverslipped with an aqueous mounting medium (Vector Laboratories, Burlingame, CA).
Cell Phenotype Immunostaining
For single-label immunophenotyping, tissue sections (4 to 6 μm thick) were mounted on positively charged glass slides, deparaffinized, rehydrated, subjected to heat-induced epitope retrieval, quenched of endogenous peroxidase activity, and blocked as described above. Next, slides were incubated with an anti-cell phenotype antibody at defined concentration (see below) for 2 hours, and an anti-rabbit, HRP-conjugated (Bethyl Laboratories, Montgomery, TX) or Alexa Fluor 488 (Invitrogen, Carlsbad, CA) conjugated secondary antibody. Between all incubation steps, slides were washed three times (5 minutes each) in TNT wash buffer. For the detection of GFAP, lymphatic endothelium, and enteric neurons, anti-GFAP antibody (Ab7260; Abcam, Cambridge, MA), anti-lymphatic vessel endothelium (LYVE-1, Ab14917; Abcam), and anti-Hu protein (16A11; Abcam) antibodies, respectively, were used.
Dual-TSA Immunofluorescent Staining
To evaluate for possible relationships between PrPRES and cells of the ENS, selected tissue sections were dual-immunostained using an IHC protocol with two consecutive TSA-based techniques. Tissue sections (4 to 6 μm thick) were mounted on positively charged glass slides, deparaffinized, rehydrated, and subjected to heat-induced epitope retrieval as described above. Next, tissue section endogenous peroxidase activity was quenched using 3% H2O2 for 60 minutes, and sections were blocked with a 1% proprietary blocking reagent (Component D; Invitrogen) for 60 minutes. After blocking, excess block was tapped off and slides were incubated with an anti-cell phenotype antibody for 2 hours. Slides were next incubated with a goat-origin, HRP-conjugated anti-rabbit secondary antibody (Bethyl Laboratories) diluted 1:100 in Component D for 30 minutes followed by diluted Alexa Fluor 488 tyramide amplification solution.
Next, the slides were incubated with HRP-BAR224 (1:500 in Component D) followed by diluted Alexa Fluor 568 tyramide amplification solution (Invitrogen). At the completion of immunostaining, slides were washed, nuclei were labeled with DAPI (Invitrogen), and slides were mounted with fluorescent mounting medium (ProLong gold antifade reagent; Invitrogen). Between all incubation steps, slides were washed three times (5 minutes each) in TNT wash buffer. To confirm the specificity of the immunostaining, and to prevent potential nonspecific immunoreactivity resultant from newly deposited HRP associated with the TSA reagents, after the completion of the first TSA-based protocol endogenous peroxidase activity was requenched with 0.3% H2O2 for 30 minutes. To evaluate for the colocalization of PrPRES with cell phenotype antibodies, dual-labeled sections were examined under a confocal, laser scanning microscope (laser scanning Axiovert confocal microscope, LSM510 meta; Carl Zeiss Microimaging, Oberkochen, Germany).
Results
Transmission of CWD to 5037 Tg[CerPrP-E] Mice
The inoculation of the 5037 Tg(CerPrP-E) mice with WTD-origin CWD prions resulted in clinical disease in the intracerebrally, intraperitoneally, and intravenously inoculated animals; their survival periods were, respectively, 215 ± 13 dpi, 232 ± 15 dpi, and 220 ± 7 dpi (Table 1). In affected mice, clinical disease was characterized by ataxia, hyperactivity, tail rigidity, and terminal mental obtundation. In contrast, Tg(CerPrP-E) mice inoculated orally with the same 1% CWD brain homogenate remained healthy for >700 dpi, as did all sham-inoculated controls (Table 1).
Table 1.
Susceptibility of 5037 Tg[CerPrP-E] Mice to CWD Prions
| Variable | Route of inoculation |
|||
|---|---|---|---|---|
| IC | i.p. | i.v. | PO, 1% | |
| Time to disease (days)⁎ | 215 ± 13 | 232 ± 15 | 220 ± 7 | ND |
| Ill mice/total mice (n/N) | 4/4 | 4/4 | 4/4 | 0/10 |
For all animals (ie, elk prion protein-expressing transgenic mice), CWD-infectivity was confirmed by IHC. None of the sham-inoculated mice (n = 6 per route) developed clinical neurologic disease nor demonstrated PrPRES accumulations in the brain.
IC, intracerebral; PO, oral.
Data are reported as means ± SEM
Neuropathology in CWD-Infected Tg[CerPrP-E] Mice
Detailed histopathological evaluation of H&E stained brain coronal sections from all CWD-infected mice revealed a profile of neuropathology that varied little among groups and progressively worsened over the course of disease. In affected animals, lesions consisted of a mixture of spongiform degeneration (spongiosis) and plaque formation. Spongiosis manifested as either a discrete form characterized by numerous, variably sized (2 to 20 μm), discrete to coalescing vacuoles within the neuropil and perikaryon (Figure 1A) or as a confluent spongiform change form, characterized by irregular, often regionally extensive bands of coalescing neuropil vacuoles (Figure 1B). In general, the confluent spongiform change form was most commonly seen in animals with terminal disease. Plaques consisted of variably discrete, pale basophilic to eosinophilic irregularly shaped and somewhat fibrillar bodies that immunolabeled for PrPRES (Figure 1B). Although the morphological features of neuropathology were similar among inoculation groups, the anatomical distribution of affected areas varied with route of inoculation.
Figure 1.
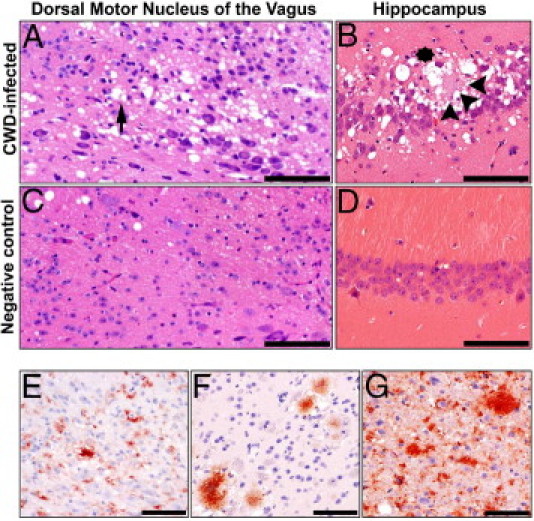
Neuropathology and plaque morphology in CWD-infected Tg[CerPrP-E] mice. H&E stained brain sections from CWD-infected mice revealed two forms of spongiosis: a discrete form with neuropil and perikaryonic (arrow) vacuoles (A) and a confluent form with coalescing neuropil vacuoles (asterisk) (B). Plaques (arrowheads) were most commonly observed in the hippocampal formation. Sham-infected mice (C and D) were devoid of such lesions. PrPRES immunoreactivity (red) manifested in three patterns: a diffuse, granular form within the neuropil and cell cytoplasm (E), a plaque form (F), and a mixed form (G), composed of both the diffuse and plaque forms. Scale bars: 100 μm (A–D); 200 μm (E–G).
In the intracerebrally inoculated animals, the most severe neuropathology was observed within the hippocampal formation, with the Cornu Ammonis (CA) fields most significantly affected. In these areas, confluent spongiform change and plaque formation were most evident (Figure 1B). Contrastingly, in the intravenously and intraperitoneally inoculated animals, regions of the medulla oblongata were mostly affected by discrete spongiosis predominantly in the DMNV and NTS (Figure 1A). Finally, some of the sham-inoculated animals had mild to modest neuropil vacuolization, but no intracellular vacuolization or plaque formation was detected (Figure 1, C and D).
Morphology of PrPRES in CWD-Inoculated Tg[CerPrP-E] Mice
In the CNS, PrPRES immunostaining manifested as a diffuse form (Figure 1E), a plaque form (Figure 1F), or as a combination of the two (Figure 1G). The diffuse form was characterized by finely granular to somewhat clumped deposits, which were either cell-associated or disseminated throughout the neuropil. The plaque form was characterized by larger florid amyloid plaques (often >100 μm in diameter), which ranged from dense plaques with an ill-formed core to those with a more fibrillar and radiating nature. These two patterns of immunoreactivity varied both with the time of sacrifice and between specific neuroanatomic areas.
Early in the course of disease, most notably in those animals sacrificed at 60 dpi, the diffuse form of PrPRES predominated. In these animals, granular PrPRES deposits were seen within and adjacent to single and grouped neurons, within microglia, and as dense punctate deposits scattered in gray matter neuropil. Moreover, this granular pattern of PrPRES immunoreactivity predominated within the cells and neuropil of the locus ceruleus, the substantia nigra, the raphe nuclei, the hippocampal formation, the DMNV, and the NTS. In contrast, the plaque form was more evident later in disease (120 dpi or terminal disease) and was most prominent in the thalamus, hypothalamus, cerebral cortex, forebrain, and cerebellum. The plaques were rarely seen at 60 dpi; notable exceptions include the pyramidal cell layer of the hippocampus in the intracerebrally inoculated mice. Late in disease, however, the PrPRES immunoreactivity took on an increasingly mixed morphology in all three successfully infected groups, with nearly all affected areas containing a mixture of diffuse and plaque forms (Figure 1G).
Peripheral Trafficking of PrPRES via ANS-Innervated Tissues in CWD-Infected Tg[CerPrP-E] Mice
In all three successfully infected groups, although there was route of inoculation-dependent variation in the initial site of PrPRES tissue involvement, the systemic progression of PrPRES accumulation was indicative of prion trafficking via the central and peripheral autonomic networks (Figure 2). In the intravenously and intraperitoneally inoculated Tg[CerPrP-E] mice, initial accumulations of PrPRES were seen within the sympathetically innervated spleen and mesenteric lymph nodes at 60 dpi (Figure 2, B and C). At 120 dpi, evidence of ANS-mediated CNS invasion was seen with PrPRES detected in both sympathetic regions of the thoracolumbar spinal cord (intermediolateral column of the spinal cord gray matter; Figure 2, D and E) and in parasympathetic regions of the CAN (DMNV and NTS; Figure 2, F and G). In the intracerebrally inoculated mice, a similar pattern of ANS-mediated CWD prion trafficking was noted, with accumulations of PrPRES identified in the intermediolateral column of the cervical and thoracolumbar spinal cord, spleen, and mesenteric lymph nodes at 120 dpi. Terminally, all successfully infected Tg(CerPrP-E) mice manifested an identical pattern of centrifugal, ANS-mediated spread of CWD prions with the identification of PrPRES in the GI tract, including Peyer's patches, the ENS (Figure 2, H and I), and GI villi (see below).
Figure 2.
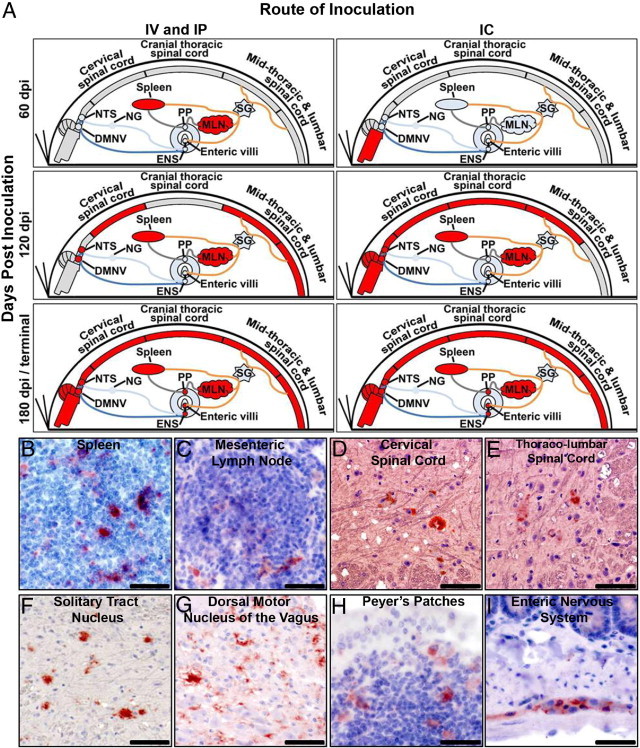
Systemic trafficking of PrPRES via the autonomic nervous system. Independent of route of inoculation, PrPRES immunoreactivity was detected in ANS-innervated peripheral tissues of CWD-infected Tg[CerPrP-E] mice. IHC mapping at 60 dpi, 120 dpi, and 180 dpi or terminal disease revealed centripetal and centrifugal spread of CWD prions via the ANS, presented graphically (A) and immunolabeled (red) (B–I). In A, blue lines represent parasympathetic fiber tracts and orange lines represent sympathetic fiber tracts. PrPRES (red) was detected in the spleen (B), mesenteric lymph nodes (C), cervical spinal cord (D), intermediolateral column of the thoracolumbar spinal cord (E), solitary tract nucleus (F), dorsal motor nucleus of the vagus nerve (G), Peyer's patches (H), and myenteric plexus of the enteric nervous system (I). Scale bars: = 100 μm (A, B, D, and F–H); 150 μm (C); 200 μm (E).
Intracranial trafficking of CWD prions via the CAN was also examined. Within the CNS, although the amount and morphological pattern of PrPRES immunoreactivity varied somewhat between infected mice sacrificed at a given time point, there was general consistency regarding the areas with the most reproducible and intense PrPRES deposition. Moreover, although there was route of inoculation-dependent variation in the initial site of intracranial PrPRES detection, mice from each of the three successfully infected groups demonstrated a progressive pattern of PrPRES accumulation indicative of trafficking via the CAN.
Progressive Transit of CWD Prions via the CAN in Intravenously and Intraperitoneally Infected Tg[CerPrP-E] Mice
In the intravenously and intraperitoneally inoculated animals, the first accumulations of PrPCWD in the CNS were observed at 120 dpi, with immunoreactivity seen in 4/4 mice in the NTS and the DMNV (Figure 3, B and G). Progressive caudal-to-rostral, CAN-mediated spread of CWD prions was identified at the 180 dpi sacrifice time point, with PrPRES detected within the raphe nuclei (4/4 mice), locus ceruleus (2/4), parabrachial nucleus (4/4), periaqueductal gray matter (4/4), paraventricular nuclei of the hypothalamus (4/4), and amygdala (4/4) (Figure 3, C–E, H–J). At terminal disease, further spread of CWD prions via the CAN was evident through the identification of PrPRES in the thalamus (4/4) and ventromedial cerebral cortex (4/4) (Figure 3, F and K).
Figure 3.
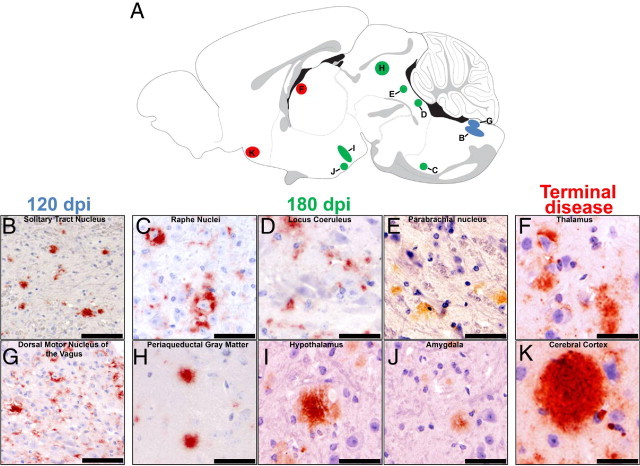
Progressive trafficking of PrPRES via the central autonomic network in intravenously and intraperitoneally inoculated mice. The serial sacrifice of CWD-infected Tg[CerPrP] mice revealed progressive, caudal-to-rostral trafficking of PrPRES (red) via the CAN. Letters marked in the sagittal diagram (A) correspond to coronal localization of PrPRES accumulation in the photomicrographs (B–K), and their colors correspond to the timing of sacrifice. PrPRES (red) was first detected at 120 dpi within the solitary tract nucleus (B) and dorsal motor nucleus of the vagus nerve (G). At 180 dpi, rostral spread of PrPRES (red) was seen, with accumulations noted in the raphe nuclei (C), locus ceruleus (D), parabrachial nucleus (E), periaqueductal gray matter (H), hypothalamus (I), and amygdala (J). At terminal disease, further rostral spread was evident with PrPRES (red) identified in the thalamus (F) and cerebral cortex (K). Scale bars: 200 μm (B); 150 μm (C); 75 μm (G); 40 μm (D–F); and 25 μm (H–K).
Progressive Transit of CWD Prions via the CAN in Intracerebrally Infected Tg[CerPrP-E] Mice
In the intracerebrally inoculated animals, the first accumulations of PrPRES in the CNS were observed at 60 dpi. In both sacrificed mice (2/2), PrPCWD was most consistently identified in the hippocampal formation, most notably within the stratum lucidum, stratum radiatum, and the stratum pyramidale of the CA1, CA2, and CA3 hippocampal fields (Figure 4B). From this initial site of PrPRES accumulation, progressive rostral and caudal spread of CWD prions via the CAN was evident. At 120 dpi, PrPRES was identified in the periaqueductal gray matter (2/2 mice), medial mammillary nuclei (1/2), paraventricular nuclei of the hypothalamus (2/2), amygdala (2/2), cerebral cortex (2/2), and thalamus (2/2) (Figure 4, C–H). Further evidence of prion spread via the CAN was identified through the detection of PrPRES in the locus ceruleus (2/2), NTS (2/2), and DMNV (2/2) (Figure 4, I–K).
Figure 4.

Progressive trafficking of CWD prions in intracerebrally inoculated mice. The serial sacrifice of CWD-infected Tg[CerPrP] mice revealed progressive, caudal-to-rostral trafficking of PrPRES (red) via the CAN. Letters marked in the sagittal diagram (A) correspond to coronal localization of PrPRES accumulation in the photomicrographs (B–K), and their colors correspond to the timing of sacrifice. P PrPRES (red) was first detected at 60 dpi within the hippocampal formation (B). At 120 dpi, spread of CWD prions via the CAN was evident via the detection of PrPRES (red) in the periaqueductal gray matter (C), medial mammillary nuclei (D), paraventricular nuclei of the hypothalamus (E), amygdala (F), cerebral cortex (G), and thalamus (H). At terminal disease, further rostral spread via the CAN was evidenced by detection of PrPRES (red) in the locus ceruleus (I), solitary tract nucleus (J), and dorsal motor nucleus of the vagus nerve (K). Scale bars: 200 μm (H); 100 μm (A); 90 μm (E); 75 μm (B–D, F, and G); 40 μm (J); and 25 μm (I).
In addition to its identification in structures of the CAN, PrPRES was also identified in a number of non-CAN areas of the brain, including the lateral, third, and fourth ventricles, the subfornical organ, the gigantocellular nucleus, the hippocampal formation, the central canal, the medial habenular nuclei, the caudoputamen, the accumbens nucleus, and the cerebellum.
PrPRES Associates with EGCs in the GI Tract of CWD-Infected Tg[CerPrP-E] Mice
Accumulations of PrPRES were identified in the myenteric and submucosal plexus of mice in the intravenously (2/4 mice affected), intraperitoneally (3/4), and intracerebrally (1/2) inoculated groups at 180 dpi and at terminal disease. In these animals, PrPRES immunoreactivity, which was most prominent in the caudal jejunum and ileum and was identified in 10% to 20% of plexus, was often unevenly distributed throughout the affected cells, with a finely granular pattern dominating within the cytoplasm and a denser, clumped to aggregate pattern within the perinuclear area (Figure 5A). Additionally, in the submucosa of 2% to 5% of intestinal villi from mice with CWD-affected plexus, a linear, homogenous to somewhat clumped pattern of PrPRES immunoreactivity was detected segmentally, extending up the length of the long axis of the villus (Figure 5B). PrPRES immunoreactivity was not observed in any intestinal area, including the ENS or villi, in any of the sham-inoculated or orally inoculated mice.
Figure 5.
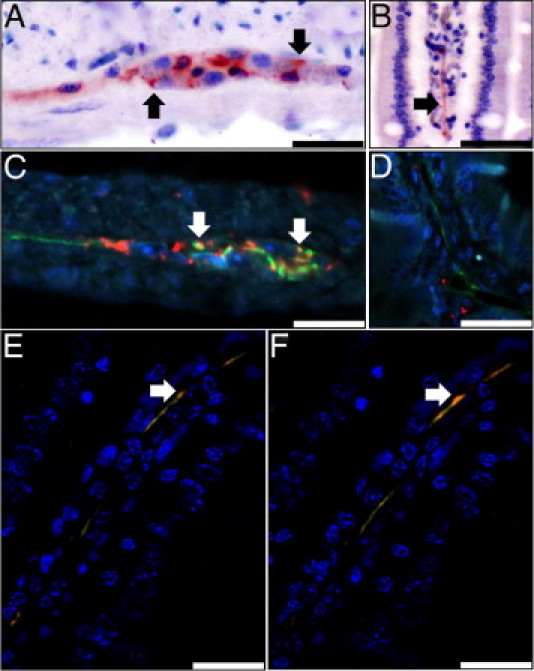
CWD prions in the enteric nervous system associated with EGCS in CWD-infected Tg[CerPrP] mice. PrPRES immunoreactivity (red) was detected in a finely granular to aggregate pattern in the myenteric (A) and submucosal plexus (not shown) and within the lamina propria of intestinal villi (B). In all infected mice, PrPRES (red) colocalized (yellow) (arrows) with GFAP+ (green) EGC cytoplasm and processes within myenteric plexus (C), but PrPRES was distinct from LYVE-1-positive lined lymphatic lacteals (green) (D). Confocal analysis of Z-stack images demonstrates colocalization (yellow) of linear PrPRES immunoreactivity (red) with the GFAP-positive (green) processes of EGCs (E and F); nuclei are stained blue (DAPI). Scale bars: 200 μm (B and D); 75 μm (E and F); and 25 μm (A and C).
To evaluate for possible associations between PrPRES deposits in the GI tract and the cell bodies and processes of EGCs and the lumina of intestinal lymphatics, affected sections were fluorescently dual-immunostained with an anti-PrP and either an anti-GFAP or anti-LYVE1 primary antibody and were evaluated using fluorescent and confocal microscopy. In the enteric plexus of dual-stained sections, there were scattered areas of colocalization between the GFAP+ EGCs and aggregates of PrPRES (Figure 5C). Two to three dual-stained sections from each of the six affected mice were evaluated and, although the degree of colocalization varied between animals, concurrent GFAP-PrPRES positivity was seen in 5% to 10% of the PrPRES+ ENS cells, with the majority of the PrPRES+ cells not demonstrating simultaneous GFAP+ positivity. Moreover, PrPRES was not observed within the lumina of any LYVE1+ endothelial-lined intestinal lymphatics (Figure 5D). Unfortunately, no anti-Hu immunoreactivity was observed in any positive control sections, and this was not used in further investigations (data not shown). Within the affected GI villi, similar areas of PrPRES-GFAP colocalization were identified using confocal microscopy (Figure 5, E and F). These areas of colocalization indicate that, in part, CWD prions associate with the cell bodies and processes of GFAP+ EGCs within the GI tract of terminally ill, infected Tg[CerPrP-E] mice.
Discussion
Here we have reported on the utility of elk prion protein-expressing [Tg(CerPrP-E)] mice in the study of CWD through studies documenting i) the susceptibility of Tg(CerPrP-E) mice to CWD prions after intracerebral, intraperitoneal, and intravenous inoculation; ii) the ANS-mediated trafficking of CWD prions; and iii) the identification of PrPRES in intimate association with the processes and cell bodies of EGCs of the ENS.
In the IHC experiments reported here, we used protocols combining tissue fixation with paraformaldehyde-lysine-periodate and two 88% formic acid immersion steps and signal amplification with TSA (PrPRES). The benefits provided by the choice of paraformaldehyde-lysine-periodate as a fixative are uncertain, but it has been shown to react with sugars to stabilize proteins, which may explain its ability to enhance the detection of both isoforms of the sialoglycoprotein PrP, compared with either formalin-fixed or glutaraldehyde-fixed tissues.19–24 The utility of formic acid in IHC studies of prions is twofold: it abolishes PrPC immunoreactivity, thus allowing for specific detection of the PrPCWD isoform, and it greatly enhances the immunoreactivity of PrPCWD in paraffin-embedded tissue sections.23–26 The incorporation of TSA into anti-PrPCWD IHC protocols results in a dramatic increase in visually detectable signal, similar to what has been reported in previous cervid and transgenic mice CWD studies.3,19
Although the Tg(CerPrP-E) mice used in these studies have previously been shown susceptible to prions derived from elk and mule deer, this is the first study to demonstrate the susceptibility of these mice to prions derived from WTD.14 The disease incubation periods reported here are similar to those reported for a separate line of mule-deer PrP-expressing Tg(CerPrP) mice inoculated with WTD-origin CWD prions, although there was a slight prolongation of each of the three route-specific incubation periods.19 Such a prolongation is presumably the result of one or both of the following factors: differences in infectious titer resulting from the different inocula used in the two studies, or a species mismatch between the cervid species from which the CWD+ inocula were derived (WTD) and the cervid PrP coding sequence used to generate the Tg study mice (elk). Additionally, the failure of the orally inoculated mice to demonstrate susceptibility to a low dose of CWD inoculum was not unexpected, given the results of previous work in Tg[CerPrP] mice, which emphasized the importance of dosage.19
Neuropathologic evaluation of the CWD-infected mice revealed two distinct patterns of spongiosis and PrPRES plaque morphology and immunoreactivity. Spongiosis of the neuropil is considered to be one of the hallmark features of prion disease, and in the successfully infected Tg(CerPrP-E) mice in these studies two patterns were observed: a discrete form and a confluent form. Given that the discrete form was generally observed in mice sacrificed early in the course of disease, and confluent spongiform change was most commonly seen in mice sacrificed later, it seems likely that the latter form represents a more severe manifestation of the former and that these two patterns of spongiosis are points along a severity spectrum.
In contrast, PrPCWD morphology manifested as granular and plaque forms, with variations depending on inoculation route, degree of disease progression, and neuroanatomic location. The granular pattern i) was most commonly seen in mice inoculated by either the intravenous or intraperitoneal route; ii) was observed largely in animals sacrificed early (60 dpi); iii) was largely restricted to the gray matter neuropil in particular neuroanatomic areas (most notably within the medulla, pons, and stratum lucidum and radiatum of the hippocampal formation); and iv) was the most common manifestation when cell-associated PrPCWD was detected. In contrast, the plaque form i) was most common in intracerebrally inoculated animals (irrespective of time point); ii) was frequently observed in animals late in disease progression (120 dpi and later); iii) was identified in areas with heavy PrPCWD immunoreactivity (including the stratum pyramidale of the hippocampal formation, as well as the cerebral cortex, thalamus, and cerebellum); and iv) was largely restricted to the extracellular neuropil. The reason for the variations in PrPCWD immunoreactivity morphology is uncertain, but such patterns substantiate suggestions that intrinsic differences in cellular subpopulations within distinct microanatomic regions, including differential patterns of prion protein glycosylation, may be the cause.27
Although there is an ever increasing number of reports documenting successful CWD infection of cervidized transgenic mice,9,16,19,28–32 none have effectively demonstrated the extra- and intracranial patterns of CWD prion trafficking by mapping the progressive pattern of PrPRES accumulation in the lymphoid system of the peripheral and central nervous systems, which is an approach that has proven invaluable in seminal work discerning the pathogenic features of scrapie and other transmissible spongiform encephalopathies.6,25,33,34 In the infected Tg[CerPrP-E] mice in these studies, despite route of inoculation-dependent variations in its progressive temporal-spatial pattern of accumulation, the overall pattern of PrPRES accumulation is indicative of prion trafficking via the peripheral and central circuitry of the ANS, as evidenced, in intravenously and intraperitoneally infected mice, by early accumulations of PrPRES in the spleen and mesenteric lymph nodes with progressive accumulation in the DMNV, NTS, and the thoracolumbar spinal cord segments. Similarly, in the intracerebrally infected mice, progressive accumulations of PrPRES were found in the spleen, mesenteric lymph node, Peyer's patches, and ENS. These findings implicate fibers of the parasympathetic and sympathetic nervous systems (notably the afferent and efferent fibers of the vagal and splanchnic tracts, respectively) as conduits of prion neuroinvasion, and further validate these mice as an accurate model system of CWD by mimicking of peripheral prion pathogenesis as reported in CWD-infected cervids, scrapie-infected sheep, and rodent models of Creutzfeldt-Jakob disease and scrapie.7,25,33,35,36
Subsequent to successful neuroinvasion, CNS trafficking of CWD prions in infected Tg(CerPrP-E) mice occurred via the CAN. Although there was route of inoculation-dependent variation, progressive, rostral and caudal, CAN-mediated prion spread was evident from the identification of PrPRES in a number of structures within, and connected to, the CAN, including the DMNV, NTS, raphe nuclei, gigantocellular reticular nucleus, locus ceruleus, parabrachial nucleus, PAGM, dorsomedial and ventromedial nuclei of the hypothalamus, numerous thalamic nuclei, and the anterior cingulate and ventromedial cortices.37
The demonstration of PrPRES in the ENS confirms the utility of the Tg[CerPrP-E] mice as effective surrogates of CWD GI pathogenesis, as reported in infected mule deer.7,36 In the successfully infected mice, PrPRES was identified only in the distal jejunum and ileum, which is similar to scrapie-infected animals, and may be the result of delayed ileal transit time or increased local expression of Peyer's patch-associated M cells.38,39 More noteworthy was the identification of PrPRES within the lamina propria of affected GI segments and the colocalization between PrPRES and the cell bodies and processes of GFAP+ EGCs. These findings suggest a possible role for EGCs as conferrers of prion fecal infectivity.
The presence of infectious prions in the feces of CWD-infected animals has been demonstrated in both natural and experimental hosts, but the manner in which such infectivity is conferred is still uncertain.9,10 It has been hypothesized that the alimentary excretion of prions is the result of the shedding of prion-infected cells or cell fragments, although this has yet to be clearly demonstrated.8 The EGCs (the resident glial cell of the ENS) reside throughout the entire intestinal tract. EGCs show morphological and functional similarities to CNS astrocytes, and their cellular processes lie in close proximity to intestinal epithelial cells, thus providing an anatomical roadmap by which EGCs might facilitate GI prion shedding.11,12,40 In such a scenario, which is bolstered by the IHC and longitudinal PrPRES accumulation data presented here, the shedding of prions from the GI tract is mediated by the cells and processes of neurons and EGCs of the ENS and occurs late in disease, only after the PrPCWD can be identified in the CNS, and is resultant of centrifugal spread.
Within PrPRES-positive intestinal sections, anti-prion immunoreactivity was found to be, in part, associated with the GFAP+ EGC cell bodies and processes; however, there was a substantial amount of non-EGC-associated PrPRES immunoreactivity (Figure 5C), suggesting that EGCs are not the only ENS constituent capable of prion accumulation. Unfortunately, further attempts to clarify the association between PrPCWD and other cell types was confounded by a failure to effectively label enteric neurons with the pan-neuronal anti-Hu antibody (probably because of the tissue fixation used).41
In summary, the findings reported here for CWD-infected Tg(CerPrP) mice provide strong circumstantial evidence for i) the trafficking of CWD prions via the parasympathetic and sympathetic branches of the autonomic nervous system during the process of central nervous system neuroinvasion, ii) the trafficking of CWD prions in the central nervous system along fibers and nuclei of the CAN, and iii) the involvement of the enteric glial cells and enteric nervous system in the pathogenesis of CWD and, possibly, in the shedding of prions via the intestinal tract. These observations, although consistent with previous reports in experimental and natural prion disease, offer new insights into the possible pathways of prion trafficking and shedding in CWD and potentially other prion diseases.
Acknowledgments
We thank Jan Langeveld for his generous gift of anti-prion antibody R505.5; Jeanette Hayes-Klug, Heather Bender, Kelly Anderson, Erin McNulty, and Kate Bruner for their excellent care and management of the Tg mice used in these studies; and Mark Zabel for his helpful comments on this work.
Footnotes
Supported by NIH contract NIAID-N01-AI-25491 (E.A.H.) and training grant NCRR-T32-RR07072 (D.M.S.).
References
- 1.Sigurdson C.J. A prion disease of cervids: chronic wasting disease. Vet Res. 2008;39:41–53. doi: 10.1051/vetres:2008018. [DOI] [PubMed] [Google Scholar]
- 2.Williams E.S., Young S. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis. 1980;16:89–98. doi: 10.7589/0090-3558-16.1.89. [DOI] [PubMed] [Google Scholar]
- 3.Sigurdson C.J., Barillas-Mury C., Miller M.W., Oesch B., van Keulen L.J., Langeveld J.P., Hoover E.A. PrP(CWD) lymphoid cell targets in early and advanced chronic wasting disease of mule deer. J Gen Virol. 2002;83:2617–2628. doi: 10.1099/0022-1317-83-10-2617. [DOI] [PubMed] [Google Scholar]
- 4.Sigurdson C.J., Williams E.S., Miller M.W., Spraker T.R., O'Rourke K.I., Hoover E.A. Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus) J Gen Virol. 1999;80:2757–2764. doi: 10.1099/0022-1317-80-10-2757. [DOI] [PubMed] [Google Scholar]
- 5.Spraker T.R., Zink R.R., Cummings B.A., Sigurdson C.J., Miller M.W., O'Rourke K.I. Distribution of protease-resistant prion protein and spongiform encephalopathy in free-ranging mule deer (Odocoileus hemionus) with chronic wasting disease. Vet Pathol. 2002;39:546–556. doi: 10.1354/vp.39-5-546. [DOI] [PubMed] [Google Scholar]
- 6.Beekes M., McBride P.A., Baldauf E. Cerebral targeting indicates vagal spread of infection in hamsters fed with scrapie. J Gen Virol. 1998;79:601–607. doi: 10.1099/0022-1317-79-3-601. [DOI] [PubMed] [Google Scholar]
- 7.Fox K.A., Jewell J.E., Williams E.S., Miller M.W. Patterns of PrPCWD accumulation during the course of chronic wasting disease infection in orally inoculated mule deer (Odocoileus hemionus) J Gen Virol. 2006;87:3451–3461. doi: 10.1099/vir.0.81999-0. [DOI] [PubMed] [Google Scholar]
- 8.Safar J.G., Lessard P., Tamgüney G., Freyman Y., Deering C., Letessier F., Dearmond S.J., Prusiner S.B. Transmission and detection of prions in feces. J Infect Dis. 2008;198:81–89. doi: 10.1086/588193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haley N.J., Mathiason C.K., Zabel M.D., Telling G.C., Hoover E.A. Detection of sub-clinical CWD infection in conventional test-negative deer long after oral exposure to urine and feces from CWD+ deer. PLoS One. 2009;4:e7990. doi: 10.1371/journal.pone.0007990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tamgüney G., Miller M.W., Wolfe L.L., Sirochman T.M., Glidden D.V., Palmer C., Lemus A., DeArmond S.J., Prusiner S.B. Asymptomatic deer excrete infectious prions in faeces [Erratum appeared in Nature 2010, 466:652 (dosage error in article text)] Nature. 2009;461:529–532. doi: 10.1038/nature08289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Savidge T.C., Sofroniew M.V., Neunlist M. Starring roles for astroglia in barrier pathologies of gut and brain. Lab Invest. 2007;87:731–736. doi: 10.1038/labinvest.3700600. [DOI] [PubMed] [Google Scholar]
- 12.Rühl A. Glial cells in the gut. Neurogastroenterol Motil. 2005;17:777–790. doi: 10.1111/j.1365-2982.2005.00687.x. [DOI] [PubMed] [Google Scholar]
- 13.Trifilo M.J., Ying G., Teng C., Oldstone M.B. Chronic wasting disease of deer and elk in transgenic mice: oral transmission and pathobiology. Virology. 2007;365:136–143. doi: 10.1016/j.virol.2007.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Angers R.C., Seward T.S., Napier D., Green M., Hoover E., Spraker T., O'Rourke K., Balachandran A., Telling G.C. Chronic wasting disease prions in elk antler velvet. Emerg Infect Dis. 2009;15:696–703. doi: 10.3201/eid1505.081458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carlson G.A., Kingsbury D.T., Goodman P.A., Coleman S., Marshall S.T., DeArmond S., Westaway D., Prusiner S.B. Linkage of prion protein and scrapie incubation time genes. Cell. 1986;46:503–511. doi: 10.1016/0092-8674(86)90875-5. [DOI] [PubMed] [Google Scholar]
- 16.Browning S.R., Mason G.L., Seward T., Green M., Eliason G.A., Mathiason C., Miller M.W., Williams E.S., Hoover E., Telling G.C. Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. J Virol. 2004;78:13345–13350. doi: 10.1128/JVI.78.23.13345-13350.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Symour R., Ichiki T., Mikaelian I., BGoggess D., Silva K.A., Sundber J.P. Necropsy Methods. In: Hedrich H.J., editor. The Laboratory Mouse. Elsevier Academic Press; London: 2004. pp. 495–516. [Google Scholar]
- 18.Castilla J., Gutiérrez-Adán A., Brun A., Doyle D., Pintado B., Ramírez M.A., Salguero F.J., Parra B., Segundo F.D., Sánchez-Vizcaíno J.M., Rogers M., Torres J.M. Subclinical bovine spongiform encephalopathy infection in transgenic mice expressing porcine prion protein. J Neurosci. 2004;24:5063–5069. doi: 10.1523/JNEUROSCI.5400-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seelig D.M., Mason G.L., Telling G.C., Hoover E.A. Pathogenesis of chronic wasting disease in cervidized transgenic mice. Am J Pathol. 2010;176:2785–2797. doi: 10.2353/ajpath.2010.090710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McLean I.W., Nakane P.K. Periodate-lysine-paraformaldehyde fixative: A new fixation for immunoelectron microscopy. J Histochem Cytochem. 1974;22:1077–1083. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- 21.Wiley C.A., Burrola P.G., Buchmeier M.J., Wooddell M.K., Barry R.A., Prusiner S.B., Lampert P.W. Immuno-gold localization of prion filaments in scrapie-infected hamster brains. Lab Invest. 1987;57:646–656. [PubMed] [Google Scholar]
- 22.McBride P.A., Bruce M.E., Fraser H. Immunostaining of scrapie cerebral amyloid plaques with antisera raised to scrapie-associated fibrils (SAF) Neuropathol Appl Neurobiol. 1988;14:325–336. doi: 10.1111/j.1365-2990.1988.tb00892.x. [DOI] [PubMed] [Google Scholar]
- 23.Liu W.G., Brown D.A., Fraser J.R. Immunohistochemical comparison of anti-prion protein (PrP) antibodies in the CNS of mice infected with scrapie. J Histochem Cytochem. 2003;51:1065–1071. doi: 10.1177/002215540305100810. [DOI] [PubMed] [Google Scholar]
- 24.Bruce M.E., McBride P.A., Farquhar C.F. Precise targeting of the pathology of the sialoglycoprotein, PrP, and vacuolar degeneration in mouse scrapie. Neurosci Lett. 1989;102:1–6. doi: 10.1016/0304-3940(89)90298-x. [DOI] [PubMed] [Google Scholar]
- 25.Muramoto T., Kitamoto T., Tateishi J., Goto I. The sequential development of abnormal prion protein accumulation in mice with Creutzfeldt-Jakob disease. Am J Pathol. 1992;140:1411–1420. [PMC free article] [PubMed] [Google Scholar]
- 26.Kitamoto T., Ogomori K., Tateishi J., Prusiner S.B. Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest. 1987;57:230–236. [PubMed] [Google Scholar]
- 27.DeArmond S.J., Qiu Y., Sánchez H., Spilman P.R., Ninchak-Casey A., Alonso D., Daggett V. PrPc glycoform heterogeneity as a function of brain region: implications for selective targeting of neurons by prion strains. J Neuropathol Exp Neurol. 1999;58:1000–1009. doi: 10.1097/00005072-199909000-00010. [DOI] [PubMed] [Google Scholar]
- 28.Haley N.J., Seelig D.M., Zabel M.D., Telling G.C., Hoover E.A. Detection of CWD prions in urine and saliva of deer by transgenic mouse bioassay. PLoS One. 2009;4(3):e4848. doi: 10.1371/journal.pone.0004848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kong Q., Huang S., Zou W., Vanegas D., Wang M., Wu D., Yuan J., Zheng M., Bai H., Deng H., Chen K., Jenny A.L., O'Rourke K., Belay E.D., Schonberger L.B., Petersen R.B., Sy M.S., Chen S.G., Gambetti P. Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci. 2005;25:7944–7949. doi: 10.1523/JNEUROSCI.2467-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LaFauci G., Carp R.I., Meeker H.C., Ye X., Kim J.I., Natelli M., Cedeno M., Petersen R.B., Kascsak R., Rubenstein R. Passage of chronic wasting disease prion into transgenic mice expressing Rocky Mountain elk (Cervus elaphus nelsoni) PrPC. J Gen Virol. 2006;87:3773–3780. doi: 10.1099/vir.0.82137-0. [DOI] [PubMed] [Google Scholar]
- 31.Tamgüney G., Giles K., Bouzamondo-Bernstein E., Bosque P.J., Miller M.W., Safar J., DeArmond S.J., Prusiner S.B. Transmission of elk and deer prions to transgenic mice. J Virol. 2006;80:9104–9114. doi: 10.1128/JVI.00098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Angers R.C., Browning S.R., Seward T.S., Sigurdson C.J., Miller M.W., Hoover E.A., Telling G.C. Prions in skeletal muscles of deer with chronic wasting disease. Science. 2006;311:1117. doi: 10.1126/science.1122864. [DOI] [PubMed] [Google Scholar]
- 33.Kimberlin R.H., Field H.J., Walker C.A. Pathogenesis of mouse scrapie: evidence for spread of infection from central to peripheral nervous system. J Gen Virol. 1983;64(Pt 3):713–716. doi: 10.1099/0022-1317-64-3-713. [DOI] [PubMed] [Google Scholar]
- 34.Muramoto T., Kitamoto T., Tateishi J., Goto I. Accumulation of abnormal prion protein in mice infected with Creutzfeldt-Jakob disease via intraperitoneal route: a sequential study. Am J Pathol. 1993;143:1470–1479. [PMC free article] [PubMed] [Google Scholar]
- 35.van Keulen L.J., Schreuder B.E., Vromans M.E., Langeveld J.P., Smits M.A. Pathogenesis of natural scrapie in sheep. Arch Virol Suppl. 2000;(16):57–71. doi: 10.1007/978-3-7091-6308-5_5. [DOI] [PubMed] [Google Scholar]
- 36.Sigurdson C.J., Spraker T.R., Miller M.W., Oesch B., Hoover E.A. PrP(CWD) in the myenteric plexus, vagosympathetic trunk and endocrine glands of deer with chronic wasting disease. J Gen Virol. 2001;82:2327–2334. doi: 10.1099/0022-1317-82-10-2327. [DOI] [PubMed] [Google Scholar]
- 37.Benarroch E.E. Functional anatomy of the central autonomic nervous system. In: Appenzeller O., editor. Handbook of Clinical Neurology. Elsevier Science B.V; Amsterdam: 1999. pp. 53–86. [Google Scholar]
- 38.Krüger D., Thomzig A., Lenz G., Kampf K., McBride P., Beekes M. Faecal shedding, alimentary clearance and intestinal spread of prions in hamsters fed with scrapie. Vet Res. 2009;40:4. doi: 10.1051/vetres:2008042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holgate A.M., Read N.W. Relationship between small bowel transit time and absorption of a solid meal: Influence of metoclopramide, magnesium sulfate, and lactulose. Dig Dis Sci. 1983;28:812–819. doi: 10.1007/BF01296904. [DOI] [PubMed] [Google Scholar]
- 40.Savidge T.C., Newman P., Pothoulakis C., Ruhl A., Neunlist M., Bourreille A., Hurst R., Sofroniew M.V. Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology. 2007;132:1344–1358. doi: 10.1053/j.gastro.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 41.Qu Z.D., Thacker M., Castelucci P., Bagyánszki M., Epstein M.L., Furness J.B. Immunohistochemical analysis of neuron types in the mouse small intestine. Cell Tissue Res. 2008;334:147–161. doi: 10.1007/s00441-008-0684-7. [DOI] [PubMed] [Google Scholar]