

Abstract
Hyperlipidemia worsens diabetic nephropathy, although the mechanism by which renal lipids accumulate is unknown. We previously demonstrated that renal proteoglycans have high low-density lipoprotein (LDL) binding affinity, suggesting that proteoglycan-mediated LDL retention may contribute to renal lipid accumulation. The aim of this study was to determine the relative effect of diabetes and hyperlipidemia on renal proteoglycan content. Diabetic and non-diabetic LDL receptor–deficient mice were fed diets containing 0% or 0.12% cholesterol for 26 weeks, and then kidneys were analyzed for renal lipid and proteoglycan content. Diabetic mice on the high-cholesterol diet had accelerated development of diabetic nephropathy with elevations in urine albumin excretion, glomerular and renal hypertrophy, and mesangial matrix expansion. Renal lipid accumulation was significantly increased by consumption of the 0.12% cholesterol diet, diabetes, and especially by both. The renal proteoglycans biglycan and decorin were detectable in glomeruli, with a significant increase in renal biglycan content in diabetic mice on the high-cholesterol diet. Renal biglycan and renal apolipoprotein B were colocalized, and regression analyses showed a significant relation between renal biglycan and renal apolipoprotein B content. The increased renal biglycan content in diabetic nephropathy probably contributes to renal lipid accumulation and the development of diabetic nephropathy.
Diabetic nephropathy, the leading cause of end-stage renal disease in the United States, is associated with a dyslipidemia that can exacerbate both the progression of renal disease and the risk of cardiovascular disease. Furthermore, renal disease itself increases the risk of cardiovascular disease, and patients with end-stage renal disease have extremely high mortality rates from cardiovascular disease. Thus, identification of risk factors for and interventions to prevent diabetic nephropathy are of critical public health importance.
Several clinical studies have shown that hyperlipidemia aggravates the progression of renal disease, including diabetic nephropathy.1–3 Glomerular lipid deposition is commonly found on routine biopsies.4 Lipoproteins have relatively free access to the mesangium because of the presence of a fenestrated endothelium without a basement membrane.5 Mesangial and glomerular epithelial cells express low-density lipoprotein (LDL) receptors and are capable of endocytosis of bound LDL.6,7 We and others have demonstrated that hyperlipidemia and diabetes cause accumulation of renal foam cells in animal models,8–10 and we recently demonstrated that lowering of lipid levels via dietary means limits progression of renal injury.10 Increased renal deposition of apolipoproteins (apo) B and/or E is associated with increased mesangial cellularity, increased proteinuria, and increased severity of glomerulosclerosis in a variety of glomerular diseases in humans.1 Thus, mesangial accumulation of lipoproteins may exacerbate and accelerate renal injury (reviewed in Wheeler and Chana,3 Kamanna,11 Abrass,12 and Kamanna et al13).
The mechanisms by which lipid accumulates in the mesangium are unknown. There are numerous commonalities in the pathology of atherosclerosis and nephropathy that suggest common triggers or pathways in the development of these complications. These common features include excess deposition of extracellular matrix, lipid and lipoprotein accumulation, and macrophage infiltration.3 Proteoglycans are a main component of extracellular matrix and participate in the development of atherosclerosis because of their ability to bind and retain lipoproteins.14–16 The main proteoglycans synthesized by mesangial cells are the large chondroitin sulfate proteoglycan versican, the small dermatan sulfate proteoglycans biglycan and decorin, and the heparan sulfate proteoglycan perlecan, and earlier studies have shown altered renal proteoglycan synthesis in diabetes.17–19 Previously, we demonstrated that renal proteoglycans exhibit high-affinity binding to LDL, with affinity constants in the plausible physiological range (Kd 14 ± 5 μg/mL LDL).20 Thus, similar to atherosclerosis, renal lipid accumulation could be mediated, at least in part, via retention by renal proteoglycans. However, it is controversial whether proteoglycans accumulate in the mesangium of diabetic nephropathy.21,22 The aim of this study was to determine the relative effect of diabetes and hypercholesterolemia on renal proteoglycan content during the development of diabetic nephropathy. Hyperlipidemic LDL receptor–deficient (LDLR−/−) mice were selected as the model for this study because we have previously demonstrated that this model develops diabetic nephropathy with overt renal lipid accumulation, which is accelerated in the setting of hypercholesterolemia.10
Materials and Methods
Chemicals and reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise specified.
Murine Studies
LDLR−/− mice (C57BL/6J genetic background; generously provided by Alan Daugherty, Lexington, KY) were selected as the model for this study. Unlike many mouse models, LDLR−/− mice carry their cholesterol in LDL particles, develop further elevations in cholesterol when fed high-cholesterol diets, and are susceptible to renal injury. Female mice were used because male mice can develop a type 2 diabetes–like phenotype on high-fat/high-cholesterol diets.23 Mice were housed in a specific pathogen-free facility with 12-hour light/dark cycles and had free access to food and water. These studies were approved by the Animal Care and Use Committees of the University of Kentucky and the Lexington Veterans Affairs Medical Center.
Insulin-deficient diabetes was induced with repeated low-dose streptozotocin (STZ). Eight-week-old mice received daily i.p. injections of STZ 40 mg/kg for 5 days and then a second series of injections at the age of 10 weeks. Non-diabetic mice received an identical schedule of injections of the citrate buffer. Hyperglycemia was confirmed at age 11 weeks, then mice were started on diets containing either 0% cholesterol (0% diet; 10.8% calories from fat) or 0.12% cholesterol (0.12% diet; 40% calories from fat; TD000241 and TD000242, respectively; Harlan Teklad, Madison WI) diets at age 12 weeks, as previously described.24 Mice were fed the indicated diets for 26 weeks. All mice were weighed weekly. Blood glucose was measured from the tail vein every 4 weeks, when mice lost weight, or when bedding was excessively wet, which indicated significant hyperglycemia and dehydration (Freestyle Flash Complete Blood Glucose Monitoring System; Abbott Laboratories, Abbott Park, IL). Most diabetic mice received insulin in the form of slow-release subcutaneous pellets (insulin release rate 0.1 U/24 hours per implant for >30 days; Linshin Canada Inc., ON, Canada) to avoid or reverse weight loss, but insulin dose was not titrated to achieve euglycemia (see Supplemental Figure S1A at http://ajp.amjpathol.org). Insulin administration (one pellet at a time) was repeated every 2 to 5 weeks as needed. Systolic blood pressure was measured five times per week in conscious mice via tail cuff apparatus (Visitech Systems Inc., Apex, NC) during weeks 8, 16, and 24 after 1 week of acclimation. The blood pressure was measured by the same operator at the same time each day, and daily measurements within each week were averaged.
Metabolic Characterization
Mice were bled before receiving STZ or citrate (baseline) and then during weeks 14 and 26. Levels of cholesterol, triglyceride, and glycated hemoglobin were measured as described previously.24 Plasma TGF-β was measured with the TGF-β1 Emax ImmunoAssay System (Promega, Madison, WI) according to the manufacturer's directions. Each mouse was housed individually for 24 hours in metabolic cages during weeks 9, 17, and 25 for collection of urine. Commercially available kits were used to measure urinary albumin (Exocell, Inc., Philadelphia, PA) and urinary creatinine (R&D Systems, Minneapolis, MN), and data are expressed as milligram of albumin per gram of creatinine.
Renal Analyses
After 26 weeks on diet, mice were anesthetized and then perfused at constant, near-physiological pressure through the left ventricle with 10 mL of sterile PBS. The kidneys were removed, decapsulated, and weighed. The right kidney was divided transversely, with one-half embedded in optimum cutting temperature compound and the other half snap frozen in liquid nitrogen. The left kidney was divided transversely, and the halves were fixed in 4% paraformaldehyde then embedded in paraffin. For histologic analyses 4-μm tissue sections were stained with PAS reagent and photographed. Sections were examined by two blinded observers (D.T. and L.R.T.), and matrix accumulation was scored with a semiquantitative scale as previously described.25 Glomerular cross-sectional area was measured in ≥30 glomeruli per mouse in glomeruli located in the outer cortex sectioned through the glomerular tuft, using computer-assisted morphometry (Image Pro; Media Cybernetics Inc., Bethesda, MD). Renal disease was evaluated by our expert renal pathologist (B.M.) who was blinded to group.
Renal Lipid and Proteoglycan Accumulation
Frozen sections (5-μm thick) of optimum cutting temperature compound were stained with oil red O and photographed. Immunohistochemistry for apoB (antibody recognizes the apoB48 and apoB100; BioDesign, Saco, ME), biglycan, decorin (both R&D Systems), versican (Chemicon, Temecula, CA), and perlecan (Lab Vision-NeoMarkers, Fremont, CA) was performed on 4-μm thick paraffin sections as previously described.26 Intensity of staining within individual glomeruli was quantified with computer-assisted morphometry with the use of ImageJ software version 1.42q (NIH, Bethesda, MD). Renal content of proteoglycan, apoB, and TGF-β (G1221; Promega) was also evaluated by Western blot analyses on total protein extracted from frozen kidneys, as previously described.27 Actin was used as the loading control (A2066; Sigma-Aldrich). Blots were scanned, and densitometry was performed with ImageJ software. Data are shown as relative apoB, biglycan, or TGF-β densitometry corrected for actin densitometry. Total renal RNA was isolated with the standard TRIzol method (Invitrogen, Carlsbad, CA), and biglycan expression was evaluated by real-time RT-PCR as previously described28 with the use of forward primer 5′-CACCTGGACCACAACAAAA-3′ and reverse primer 5′-TCCGAATCTGATTGTGACCTA-3′. Data are expressed corrected for 18S expression. Colocalization of apoB with proteoglycans was first evaluated by comparing adjacent sections immunostained for apoB and the various proteoglycans. To validate colocalization single sections were double-stained for apoB and biglycan or for apoB and decorin and analyzed by confocal microscopy with the use of a Leica AOBS TCS SP5 inverted laser scanning confocal microscope (Leica Microsystems Inc. Mannheim, Germany). Negative controls were obtained with isotype-matched irrelevant antibodies, no primary antibody, or no secondary antibody.
Statistical Analyses
Data are presented as mean ± SEM, unless otherwise described. All data were analyzed by two-way analysis of variance with multiple pairwise comparisons with the use of the Holm-Sidak method (SigmaStat Software Inc., San Jose, CA). P values < 0.05 were considered statistically significant.
Results
Metabolic Characterization
Mice were made diabetic by STZ injections, whereas control groups received citrate. All mice that received STZ had elevated blood glucose levels by 2 weeks after the injections (see Supplemental Figure S1A at http://ajp.amjpathol.org). Most diabetic mice required insulin periodically to prevent weight loss, but the usage did not differ between groups. The study protocol called for euthanasia of any mouse that lost body weight and did not respond to insulin treatment. One non-diabetic mouse and 7 of 30 diabetic mice died during the study. To investigate the effects of hypercholesterolemia, diabetic and non-diabetic mice were fed diets containing either 0% or 0.12% cholesterol for 26 weeks.24 At the end of the study diabetic mice on either diet had lower body weight than the non-diabetic mice on the same diet (P < 0.001), but consumption of the high-cholesterol diet increased weight in both non-diabetic and diabetic mice (P = 0.002; Table 1; see also Supplemental Figure S1B at http://ajp.amjpathol.org). Induction of diabetes resulted in significantly higher glycated hemoglobin levels compared with non-diabetic mice (P = 0.002; Table 1), and diabetic mice that consumed the 0.12% diet had the highest glycated hemoglobin levels (P < 0.05; Table 1). Blood glucose levels were recorded every 4 weeks throughout the study and were not affected by diet (see Supplemental Figure S1A at http://ajp.amjpathol.org). As expected, consumption of the 0.12% diet resulted in significantly higher total cholesterol levels in both the non-diabetic and diabetic groups (P < 0.001), but there was no effect of diabetes on plasma cholesterol levels (Table 1).24 Furthermore, the groups did not differ in plasma triglyceride levels regardless of diabetes status or diet consumed (Table 1).
Table 1.
Metabolic Characterization
| 0% Diet |
0.12% Diet |
|||
|---|---|---|---|---|
| Non-diabetic | Diabetic | Non-diabetic | Diabetic | |
| Body weight (g), study end | 23.3 ± 1.9 | 21.8 ± 1.0 | 28.0 ± 2.9⁎ | 23.7 ± 0.7† |
| Glycated hemoglobin (%) | 7.5 ± 0.4 | 8.9 ± 1.1⁎ | 7.0 ± 0.2 | 11.8 ± 1.0⁎†‡ |
| Plasma cholesterol (mg/dL) | 374 ± 19 | 410 ± 53 | 903 ± 154⁎ | 774 ± 51⁎‡ |
| Triglycerides (mg/dL) | 118 ± 54 | 143 ± 42 | 103 ± 25 | 251 ± 58 |
| Plasma TGF-β (pg/mL) | 78 ± 78 | 2936 ± 803⁎ | 1101 ± 361 | 2377 ± 725⁎† |
| Renal weight/ body weight (mg/g) | 5.0 ± 0.5 | 6.0 ± 0.7⁎ | 4.6 ± 0.2 | 6.2 ± 0.3⁎ |
| Glomerular cross sectional area (μm2) | 3954 ± 281 | 3717 ± 233 | 3777 ± 170 | 4946 ± 324⁎†‡ |
| Systolic blood pressure (mmHg) | 120 ± 6 | 120 ± 10 | 112 ± 8 | 114 ± 12 |
Data shown are mean ± SEM for 7 to 14 mice per group as indicated, measured after 26 weeks of the 0% or 0.12% diet and/or diabetes. All analyses were done by two-way analysis of variance with pairwise comparisons by the Holm-Sidak method.
P < 0.05 compared with the non-diabetic group on the 0% diet.
P < 0.05 compared with the non-diabetic group on the 0.12% diet.
P < 0.05 compared with the diabetic group on the 0% diet.
Development of Diabetic Nephropathy
Despite the relative resistance of the C57BL6 background strain29 to the development of diabetic nephropathy, we found a significant elevation in urine albumin excretion in diabetic LDLR−/− mice fed the 0.12% diet (P < 0.05; Figure 1A). Diabetic mice fed either diet developed increased urine albumin excretion compared with the non-diabetic groups fed the same diets as early as 9 weeks of diabetes (not shown). After 26 weeks of diabetes and diets, the diabetic group fed the 0.12% diet had the highest urinary albumin excretion compared with all other groups. Neither diabetes nor diet altered systolic blood pressure at any time (Table 1; showing blood pressure at week 24). Glomerular cross-sectional area was measured in glomeruli sectioned through the tuft, and glomerular mesangial matrix content was estimated with a semiquantitative score. Only diabetic mice fed the 0.12% diet had a significant increase in glomerular area (P < 0.005; Table 1). Diabetic mice had significant mesangial expansion compared with the non-diabetic mice fed the same diets (P < 0.001; Figure 1, B and C). This extent of renal injury is similar to that observed in the endothelial nitric oxide synthase or decorin-deficient models.30,31
Figure 1.
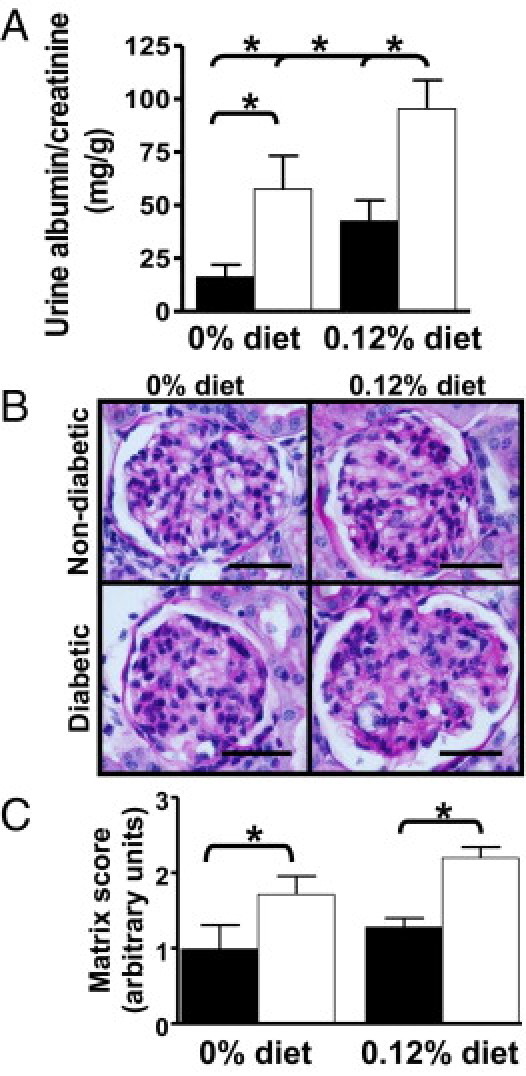
Diabetes and hyperlipidemia cause glomerular injury. A: Urine albumin excretion expressed as milligram of albumin per gram of creatinine was measured on samples collected from 7 to 14 mice per group housed individually for 24 hours in metabolic cages. Closed columns are non-diabetic mice, open columns are diabetic mice. B: Shown are representative sections (from 7 to 14 per group) stained with PAS from non-diabetic or diabetic LDLR−/− mice fed the 0% or 0.12% diet for 26 weeks. Original magnification, ×1000. Scale bar = 25 μm. C: Mesangial matrix accumulation was scored with a semiquantitative scale on ≥30 glomeruli per mouse sectioned through the glomerular tuft by two blinded observers (D.T. and L.R.T.) from 7 to 14 per group. Closed columns are non-diabetic mice, open columns are diabetic mice. *P < 0.05 by Holm-Sidak pairwise comparison.
Diabetes Increases Glomerular Lipid Accumulation
Renal lipid accumulation was evaluated by glomerular staining with the use of the neutral lipid stain oil red O and by immunohistochemistry for apoB. Lipid and lipoprotein accumulation was detectable in glomeruli from diabetic mice fed either diet and from non-diabetic mice fed the 0.12% diet, with the greatest accumulation seen in diabetic mice fed the 0.12% diet (Figure 2, A–C). Minor accumulation of apoB in the interstitium was observed; however, the most intense staining was in the glomeruli (Figure 2B). Western blot analysis of total renal protein similarly showed increased renal apoB content by both diabetes (P = 0.006) and diet (P < 0.001), with the greatest apoB content in diabetic mice fed the 0.12% diet (Figure 2, D and E). The antibody used recognizes both apoB48 and apoB100; however, only apoB100 was seen on Western blot analyses.
Figure 2.
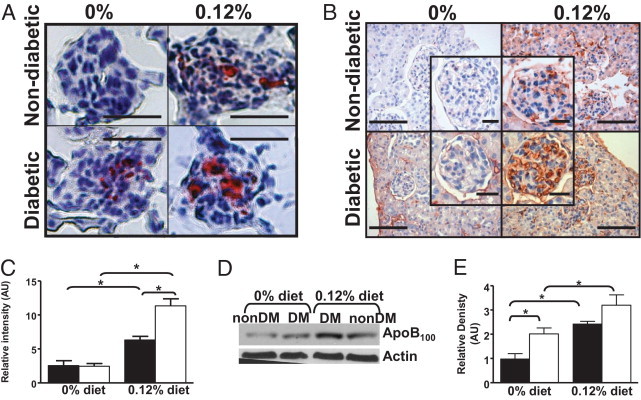
Diabetes and hyperlipidemia cause renal lipid accumulation. A: Shown are representative frozen sections (from 7 to 14 per group) stained with oil red O from non-diabetic or diabetic LDLR−/− mice fed the 0% or 0.12% diet for 26 weeks. Original magnification, ×400. Scale bar = 25 μm B: Shown are representative sections (from 7 to 14 mice per group) immunostained for apoB from non-diabetic or diabetic mice fed the 0% or 0.12% diet for 26 weeks. Original magnification, ×400, with inset showing individual glomeruli. Scale bar = 100 μm with inset scale bars representing 25 μm. C: Intensity of apoB staining in individual glomeruli (≤26 per mouse) was quantified with computer-assisted morphometry for three to four mice per group. D: Total renal protein was analyzed by Western blot analysis for renal apoB content. Each lane shows apoB from one mouse per group, representative of four mice per group. NonDM indicates non-diabetic group, DM indicates diabetic group. Actin was used as the loading control. E: Western blot analyses of renal apoB content were analyzed by densitometry. Closed columns are non-diabetic mice, open columns are diabetic mice. *P < 0.05 by Holm-Sidak pairwise comparison.
Diabetes Increases Glomerular Biglycan Accumulation
Glomerular proteoglycan accumulation was evaluated by immunohistochemistry for biglycan, decorin, versican, and perlecan. Biglycan and decorin were the only proteoglycans detectable in any significant amount in any group (Figure 3A), and there was an increase in biglycan staining in glomeruli from diabetic mice fed the 0.12% diet compared with all other groups (Figure 3, B and C). Biglycan showed faint interstitial staining with predominant mesangial and capsular staining. Western blot and densitometry analyses showed increased biglycan content by diabetes and diet; (Figures 3, D and E); however, no significant differences were observed in renal decorin content (Figure 3D) or renal versican content (see Supplemental Figure S2A at http://ajp.amjpathol.org). Biglycan expression measured by real-time PCR was significantly increased by diabetes (P < 0.05; Figure 3F). As expected,32–34 TGF-β levels were significantly higher in the diabetic mice than in the non-diabetic mice (P = 0.005; Table 1). Analysis of renal TGF-β content by Western blot analysis showed a trend toward higher levels in the diabetic mice than in the non-diabetic mice, but it did not reach statistical significance (see Supplemental Figure S2B at http://ajp.amjpathol.org). We and others have previously demonstrated that TGF-β regulates biglycan expression in a variety of tissues.20,27,35 Regression analyses showed that renal biglycan content correlated with renal TGF-β content (see Supplemental Figure S2C at http://ajp.amjpathol.org; r = 0.87, P < 0.001), suggesting that renal biglycan is regulated, at least in part, by renal TGF-β.
Figure 3.
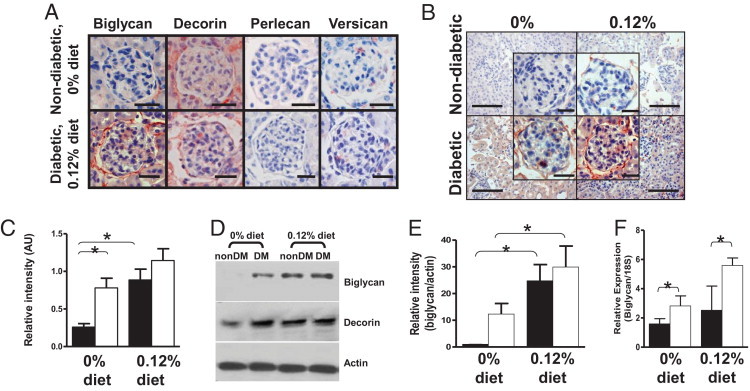
Diabetes and hyperlipidemia increase renal biglycan content. A: Shown are representative sections from a non-diabetic LDLR−/− mouse fed the 0% diet and a diabetic LDLR−/−mouse fed the 0.12% diet (representative of four mice per group) immunostained for the indicated proteoglycans (all red color product). Original magnification, ×400. Scale bar = 25 μm. B: Shown are representative sections (from four per group) immunostained for biglycan (red color product) from non-diabetic or diabetic mice fed the 0% or 0.12% diet for 26 weeks. Original magnification, ×400 with inset showing individual glomeruli. Scale bar = 100 μm with inset scale bars representing 25 μm. C: Intensity of biglycan staining in individual glomeruli (≤34 per mouse) was quantified with computer-assisted morphometry for three to four mice per group. D: Total renal protein was analyzed by Western blot analysis for renal biglycan or decorin content. Each lane shows protein from one mouse per group, representative of four mice per group. NonDM, non-diabetic group; DM, diabetic group. Actin was used as the loading control. E: Western blot analyses of renal biglycan content were analyzed by densitometry. F: Biglycan expression was determined with real-time RT-PCR (normalized to 18S RNA). Shown is mean ± SEM for seven to nine mice per group. Closed columns are non-diabetic mice, open columns are diabetic mice. *P < 0.05 by Holm-Sidak pairwise comparison.
Renal ApoB Colocalizes with Biglycan
To determine whether apoB colocalizes with proteoglycans, adjacent sections were immunostained for apoB and the various proteoglycans. Colocalization was observed only between biglycan and apoB (Figure 4A). To verify the colocalization, single sections were double-labeled for biglycan and apoB or for decorin and apoB, and confocal microscopy showed striking colocalization between apoB and biglycan (Figure 4B) but no colocalization between apoB and decorin (see Supplemental Figure S3 at http://ajp.amjpathol.org). Regression analyses showed that renal apoB content correlated with renal biglycan content (Figure 4C; r = 0.76, P < 0.001). These data suggest that renal apoB accumulation is influenced by renal biglycan content.
Figure 4.
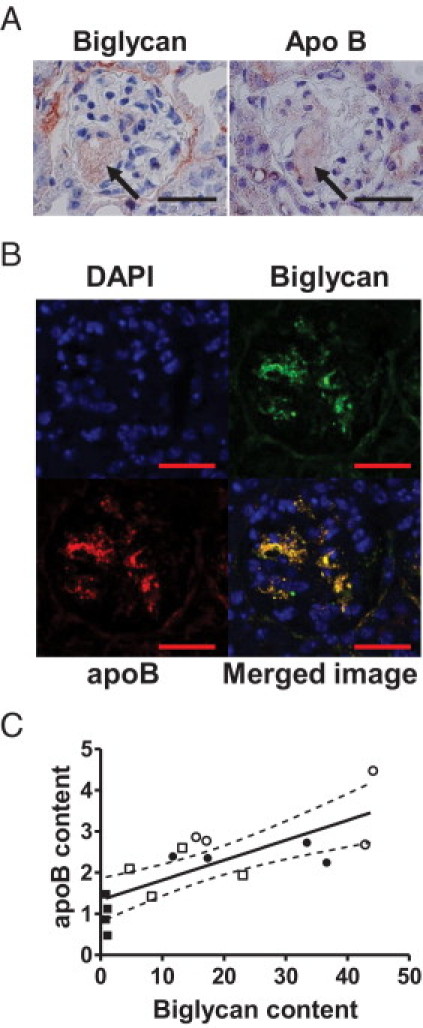
Colocalization of renal biglycan and apoB. A: Shown are representative adjacent sections from a diabetic LDLR−/− mouse fed the 0.12% diet and immunostained for biglycan and apoB (both red color product). The arrows indicate a glomerular foam cell. Scale bar = 25 μm. B: Shown is a representative section from a diabetic LDLR−/− mouse fed the 0.12% diet immunostained for biglycan (green), apoB (red), nuclear DAPI stain (blue), and the merged image (colocalization yellow). Scale bar = 25 μm. C: Renal apoB content and renal biglycan content determined by densitometry were analyzed by linear regression. Solid symbols represent non-diabetic mice, open symbols represent diabetic mice. Squares represent mice on the 0% diet, circles represent mice on the 0.12% diet. Figure depicts four mice per group for all groups; r = 0.76, P < 0.001, and dashed lines represent 95% confidence intervals.
Discussion
We and others have previously demonstrated that hyperlipidemia exacerbates development and progression of renal diseases, including diabetic nephropathy.9,10,36–39 However, the mechanisms leading to renal lipid accumulation are unknown. The retention of lipoproteins by proteoglycans is established as a mechanism leading to atherosclerosis. However, it is not known whether renal lipid accumulation may be similarly attributed to renal proteoglycan-mediated retention. The purpose of this study was to determine the effect of diabetes and hypercholesterolemia on renal proteoglycan content as a mechanism leading to renal lipid accumulation. We selected the LDLR−/− model because it carries most of its cholesterol in the LDL fraction, making it a good model for human dyslipidemia, as opposed to most mice, which have primarily high-density lipoprotein.40 We have previously demonstrated that this model develops modest diabetic nephropathy with increased urinary albumin excretion, expansion of mesangial matrix, and overt renal lipid accumulation.10 We now demonstrate that biglycan, decorin, and versican were all detectable in glomeruli. However, only biglycan content and expression was significantly increased in the diabetic mice fed the 0.12% diet. Biglycan had striking colocalization with renal apoB, and regression analyses showed a significant relation between renal biglycan and renal apoB content. Thus, diabetes and hypercholesterolemia lead to the development of diabetic nephropathy with increased glomerular biglycan. We propose that the increased glomerular biglycan content contributes to the renal lipid accumulation and progression of diabetic nephropathy.
TGF-β has clearly been shown to play a critical role in the development of diabetic nephropathy. Diabetes leads to elevated TGF-β levels in both animal models and human disease,32–34 and inhibition of TGF-β activity protects against diabetic nephropathy.41,42 As expected, we observed elevated TGF-β levels in diabetic mice compared with non-diabetic mice. A robust body of literature reports the effect of TGF-β to up-regulate biglycan expression in a variety of tissues, including the kidney.43–45 We now report a correlation between renal biglycan and renal TGF-β content, suggesting that renal biglycan content is regulated, at least in part, by renal TGF-β activity. Furthermore, we and others have demonstrated that proteoglycans synthesized by cells stimulated with TGF-β have longer glycosaminoglycan chains and increased LDL binding affinity.20,35,46 Thus, we propose that in diabetes the increased systemic and renal TGF-β activity stimulates renal biglycan synthesis with elongated glycosaminoglycan chains. Although all proteoglycans are capable of binding LDL, in our model biglycan is the predominant proteoglycan within the glomerulus. In the setting of hyperlipidemia, this increased renal biglycan content leads to increased LDL retention and the development of renal lipid accumulation. A trend was observed toward increased TGF-β concentrations in non-diabetic mice fed the 0.12% diet (Table 1), which could account for the increase in renal biglycan and apoB content observed in these mice.
The role of proteoglycans in the development of renal diseases is not clear. Previous studies have shown increased biglycan mRNA expression in diabetic nephropathy21,47; however, it has not been clear whether this increased expression is accompanied by increased renal content.21,22 The differences between our study and previous studies could simply be due to differences in the antibody affinity or to the examination of different stages of renal disease. In addition, the functional role or roles of biglycan are unclear; roles have been proposed in the maintenance of the glomerular charge barrier,48,49 in the regulation of mesangial cell growth and survival,50 in the regulation of TGF-β activity,51 in the regulation of inflammation via activation of Toll-like receptors,52 in the regulation of the assembly of connective tissues,53 and in the structural composition of fibrosis,22 among others. Both biglycan and decorin can bind TGF-β, leading to its sequestration and neutralization of activity.54,55 Administration of decorin51 or overexpression of decorin56 has been shown to attenuate the development of renal disease. Thus, it has been proposed that biglycan and decorin are natural inhibitors of TGF-β activity, and their up-regulation by TGF-β may provide a negative feedback loop that limits the adverse effects of TGF-β. In support of this, a recent study reported enhanced diabetic nephropathy with increased mesangial matrix expansion, elevated albuminuria, and increased TGF-β bioactivity in decorin knockout mice compared with decorin wild-type mice.31 However, in the only direct comparison of the TGF-β–neutralizing effects of these two proteoglycans, only decorin, but not biglycan, inhibited fibrosis induced by TGF-β.57 Although previous studies have reported increased renal decorin in diabetic kidneys,21,58 the lack of increased renal decorin found in this study is consistent with the theory that decorin is cleared from the kidney by the vasculature or the urinary tract, possibly in complexes with TGF-β.21 The diabetic mice in our study had both elevations of TGF-β and increased renal biglycan content, as well as increased mesangial matrix accumulation, a major product of increased TGF-β activity. This suggests that the dominant effect of increased renal biglycan content was increased renal lipid retention and not inhibition of TGF-β activity. However, verification of the putative role of biglycan in regulating TGF-β activity and mediating renal lipid retention awaits further studies with the use of the biglycan deficient model.
A potential limitation of our study is the use of our murine model. Genetic susceptibility studies have suggested that mice on the C57BL6 background are resistant to the development of diabetic nephropathy.29,59,60 In addition, the use of STZ to induce diabetes is also a potential confounding feature, because the STZ itself can be nephrotoxic.61 Finally, LDLR−/− mice are considerably more hyperlipidemic than humans, even on the 0% diet. However, unlike most mice that carry their cholesterol primarily in high-density lipoprotein particles, the LDLR−/− mice have significant elevations of LDL and VLDL, with comparatively low high-density lipoprotein levels. This more closely resembles the human lipoprotein profile than most other murine models and was the basis for their use in these experiments. In response to the high-cholesterol diet, the mice developed further elevations in their cholesterol levels, with no other metabolic perturbations: no effect on triglyceride levels, lipoprotein distribution (not shown), or hypertension. Williams et al31 have reported that C57BL6 mice deficient in decorin develop significant features of diabetic nephropathy after 10 months of hyperglycemia. We demonstrate that, in the setting of hyperlipidemia, significant features of diabetic nephropathy are present after only 6 months, further validating this model.
In conclusion, in this murine model we confirm previous reports that hyperlipidemia has adverse effects on the development of diabetic nephropathy. In addition, we demonstrate that diabetes and hypercholesterolemia caused increased renal biglycan content and increased mesangial apoB accumulations. We propose that elevated TGF-β concentrations seen in diabetes caused increased renal biglycan synthesis, which leads to increased renal LDL accumulation, which significantly contributes to the development of glomerular injury. This suggests that strategies to limit TGF-β activity, renal biglycan synthesis, or hyperlipidemia may all be pharmacologic targets in the development of new approaches to intervene in diabetic nephropathy. Although clinical studies that use lipid-lowering medications have been conflicting on their effects on renal function, many studies have either excluded subjects with impaired renal function62–64 or studied subjects with advanced renal failure in which no effect of lipid lowering could reasonably be expected.65 However, given the paucity of clinical treatments for diabetic nephropathy, we encourage studies that evaluate the effect of lipid-lowering medications on the endpoint of changes in renal function in subjects with early stage disease.
Footnotes
Supported in part by grants from the Department of Veterans Affairs (BX000622 to L.R.T.), the American Diabetes Association and the Order of the Amaranth (L.R.T.), the National Institutes of Health (RR020171 to L.R.T.), and Deutsche Forschungsgemeinschaft (SFB 815, project A5 to L.S.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.05.016.
Supplementary data
A: Non-fasting tail vein blood glucose was measured with a Freestyle Flash glucometer at the indicated times. B: Mouse weight was measured at the indicated times. Solid symbols represent non-diabetic mice, open symbols represent diabetic mice. Squares represent mice on the 0% cholesterol diet, circles represent mice on the 0.12% cholesterol diet. Data shown is mean ± SEM from 7 to 14 per group.
A: Renal versican content. B: Renal TGF-β content. Total renal protein was analyzed by Western blot analysis for renal versican or TGF-β content. Each lane shows versican or TGF-β, as indicated, from one mouse per group, representative of 4 mice per group. NonDM indicates non-diabetic group, DM indicates diabetic group. Actin was used as the loading control. C: Renal biglycan and TGF-β content determined by densitometry of Western blot analyses was analyzed by linear regression. Solid symbols represent non-diabetic mice, open symbols represent diabetic mice. Squares represent mice on the 0% cholesterol diet, circles represent mice on the 0.12% cholesterol diet. Figure depicts four per group for all groups. r = 0.873, P < 0.001, dashed lines represent 95% confidence intervals.
Shown is a representative section from a diabetic LDLR−/− mouse fed the 0.12% cholesterol diet immunostained for decorin (red), apoB (green), nuclear DAPI stain (blue), and the merged image (colocalization yellow).
References
- 1.Sato H., Suzuki S., Kobayashi H., Ogino S., Inomata A., Arakawa M. Immunohistological localization of lipoproteins in the glomeruli in renal disease: specifically apoB and apoE. Clin Nephrol. 1991;36:127–133. [PubMed] [Google Scholar]
- 2.Takemura T., Yoshioka K., Aya N., Murakami K., Matumoto A., Itakura H., Kodama T., Suzuki H., Maki S. lipoproteins and lipoprotein receptors in glomeruli in human kidney diseases. Kidney Int. 1993;43:918–927. doi: 10.1038/ki.1993.129. [DOI] [PubMed] [Google Scholar]
- 3.Wheeler D.C., Chana R.S. Interactions between lipoproteins, glomerular cells and matrix. Miner Electrolyte Metab. 1993;19:149–164. [PubMed] [Google Scholar]
- 4.Lee H.S., Lee J.S., Koh H.I., Ko K.W. Intraglomerular lipid deposition in routine biopsies. Clin Nephrol. 1991;36:67–75. [PubMed] [Google Scholar]
- 5.Michael A.F., Keane W.F., Raij L., Vernier R.L., Mauer S.M. The glomerular mesangium. Kidney Int. 1980;17:141–154. doi: 10.1038/ki.1980.18. [DOI] [PubMed] [Google Scholar]
- 6.Wasserman J., Santiago A., Rifici V., Holthofer H., Scharschmidt L., Epstein M., Schlondorff D. Interactions of low density lipoprotein with rat mesangial cells. Kidney Int. 1989;35:1168–1174. doi: 10.1038/ki.1989.106. [DOI] [PubMed] [Google Scholar]
- 7.Coritsidis G., Rifici V., Gupta S., Rie J., Shan Z.H., Neugarten J., Schlondorff D. Preferential binding of oxidized LDL to rat glomeruli in vivo and cultured mesangial cells in vitro. Kidney Int. 1991;39:858–866. doi: 10.1038/ki.1991.108. [DOI] [PubMed] [Google Scholar]
- 8.Sano J., Shirakura S., Oda S., Hara T., Ishihara T. Foam cells generated by a combination of hyperglycemia and hyperlipemia in rats. Pathol Int. 2004;54:904–913. doi: 10.1111/j.1440-1827.2004.01778.x. [DOI] [PubMed] [Google Scholar]
- 9.Spencer M.W., Muhlfeld A.S., Segerer S., Hudkins K.L., Kirk E., LeBoeuf R.C., Alpers C.E. Hyperglycemia and hyperlipidemia act synergistically to induce renal disease in LDL receptor-deficient BALB mice. Am J Nephrol. 2004;24:20–31. doi: 10.1159/000075362. [DOI] [PubMed] [Google Scholar]
- 10.Taneja D., Thompson J., Wilson P., Brandewie K., Schaefer L., Mitchell B., Tannock L.R. Reversibility of renal injury with cholesterol lowering in hyperlipidemic diabetic mice. J Lipid Res. 2010;51:1464–1470. doi: 10.1194/jlr.M002972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamanna V.S. Low density lipoproteins and mitogenic signal transduction processes: role in the pathogenesis of renal disease. Histol Histopathol. 2002;17:497–505. doi: 10.14670/HH-17.497. [DOI] [PubMed] [Google Scholar]
- 12.Abrass C.K. Cellular lipid metabolism and the role of lipids in progressive renal disease. Am J Nephrol. 2004;24:46–53. doi: 10.1159/000075925. [DOI] [PubMed] [Google Scholar]
- 13.Kamanna V.S., Roh D.D., Kirschenbaum M.A. Hyperlipidemia and kidney disease: concepts derived from histopathology and cell biology of the glomerulus. Histol Histopathol. 1998;13:169–179. doi: 10.14670/HH-13.169. [DOI] [PubMed] [Google Scholar]
- 14.Williams K.J., Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–561. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tabas I., Williams K.J., Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 16.Tannock L.R., King V.L. Proteoglycan mediated lipoprotein retention: a mechanism of diabetic atherosclerosis. Rev Endocr Metab Disord. 2008;9:289–300. doi: 10.1007/s11154-008-9078-0. [DOI] [PubMed] [Google Scholar]
- 17.Hadad S.J., Michelacci Y.M., Schor N. Proteoglycans and glycosaminoglycans synthesized in vitro by mesangial cells from normal and diabetic rats. Biochim Biophys Acta. 1996;1290:18–28. doi: 10.1016/0304-4165(95)00183-2. [DOI] [PubMed] [Google Scholar]
- 18.Silbiger S., Schlondorff D., Crowley S., Rosenberg L., Choi H., Hatcher V., Gordon P. The effect of glucose on proteoglycans produced by cultured mesangial cells. Diabetes. 1993;42:1815–1822. doi: 10.2337/diab.42.12.1815. [DOI] [PubMed] [Google Scholar]
- 19.Ziyadeh F.N. The extracellular matrix in diabetic nephropathy. Am J Kidney Dis. 1993;22:736–744. doi: 10.1016/s0272-6386(12)80440-9. [DOI] [PubMed] [Google Scholar]
- 20.Tannock L.R. Proteoglycans can mediate renal lipoprotein retention. Diabetologia. 2006;49:1115–1116. doi: 10.1007/s00125-006-0181-z. [DOI] [PubMed] [Google Scholar]
- 21.Schaefer L., Raslik I., Grone H.J., Schonherr E., Macakova K., Ugorcakova J., Budny S., Schaefer R.M., Kresse H. Small proteoglycans in human diabetic nephropathy: discrepancy between glomerular expression and protein accumulation of decorin, biglycan, lumican, and fibromodulin. FASEB J. 2001;15:559–561. doi: 10.1096/fj.00-0493fje. [DOI] [PubMed] [Google Scholar]
- 22.Stokes M.B., Holler S., Cui Y., Hudkins K.L., Eitner F., Fogo A., Alpers C.E. Expression of decorin, biglycan, and collagen type I in human renal fibrosing disease. Kidney Int. 2000;57:487–498. doi: 10.1046/j.1523-1755.2000.00868.x. [DOI] [PubMed] [Google Scholar]
- 23.Merat S., Casanada F., Sutphin M., Palinski W., Reaven P.D. Western-type diets induce insulin resistance and hyperinsulinemia in LDL receptor-deficient mice but do not increase aortic atherosclerosis compared with normoinsulinemic mice in which similar plasma cholesterol levels are achieved by a fructose-rich diet. Arterioscler Thromb Vasc Biol. 1999;19:1223–1230. doi: 10.1161/01.atv.19.5.1223. [DOI] [PubMed] [Google Scholar]
- 24.Renard C.B., Kramer F., Johansson F., Lamharzi N., Tannock L.R., Herrath M.G., Chait A., Bornfeldt K.E. Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. J Clin Invest. 2004;114:659–668. doi: 10.1172/JCI17867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taneda S., Pippin J.W., Sage E.H., Hudkins K.L., Takeuchi Y., Couser W.G., Alpers C.E. Amelioration of diabetic nephropathy in SPARC-null mice. J Am Soc Nephrol. 2003;14:968–980. doi: 10.1097/01.asn.0000054498.83125.90. [DOI] [PubMed] [Google Scholar]
- 26.Huang F., Thompson J.C., Wilson P.G., Aung H.H., Rutledge J.C., Tannock L.R. Angiotensin II increases vascular proteoglycan content preceding and contributing to atherosclerosis development. J Lipid Res. 2008;49:521–530. doi: 10.1194/jlr.M700329-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Schonherr E., Jarvelainen H.T., Kinsella M.G., Sandell L.J., Wight T.N. Platelet derived growth factor and transforming growth factor-beta1 differentially affect the synthesis of biglycan and decorin by monkey arterial smooth muscle cells. Arterioscler Thromb. 1993;13:1026–1036. doi: 10.1161/01.atv.13.7.1026. [DOI] [PubMed] [Google Scholar]
- 28.Cai L., Ji A., de Beer F.C., Tannock L.R., van der Westhuyzen D.R. SR-BI protects against endotoxemia in mice through its roles in glucocorticoid production and hepatic clearance. J Clin Invest. 2007;118:364–375. doi: 10.1172/JCI31539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brosius F.C., 3rd, Alpers C.E., Bottinger E.P., Breyer M.D., Coffman T.M., Gurley S.B., Harris R.C., Kakoki M., Kretzler M., Leiter E.H., Levi M., McIndoe R.A., Sharma K., Smithies O., Susztak K., Takahashi N., Takahashi T. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20:2503–2512. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakagawa T., Sato W., Glushakova O., Heinig M., Clarke T., Campbell-Thompson M., Yuzawa Y., Atkinson M.A., Johnson R.J., Croker B. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol. 2007;18:539–550. doi: 10.1681/ASN.2006050459. [DOI] [PubMed] [Google Scholar]
- 31.Williams K.J., Qiu G., Usui H.K., Dunn S.R., McCue P., Bottinger E., Iozzo R.V., Sharma K. Decorin deficiency enhances progressive nephropathy in diabetic mice. Am J Pathol. 2007;171:1441–1450. doi: 10.2353/ajpath.2007.070079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shankland S.J., Scholey J.W., Ly H., Thai K. Expression of transforming growth factor-beta 1 during diabetic renal hypertrophy. Kidney Int. 1994;46:430–442. doi: 10.1038/ki.1994.291. [DOI] [PubMed] [Google Scholar]
- 33.Sharma K., Ziyadeh F.N. Renal hypertrophy is associated with upregulation of TGF-beta 1 gene expression in diabetic BB rat and NOD mouse. Am J Physiol. 1994;267 doi: 10.1152/ajprenal.1994.267.6.F1094. F1094-F1001. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto T., Nakamura T., Noble N.A., Ruoslahti E., Border W.A. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci U S A. 1993;90:1814–1818. doi: 10.1073/pnas.90.5.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Little P.J., Tannock L., Olin K.L., Chait A., Wight T.N. Proteoglycans synthesized by arterial smooth muscle cells in the presence of transforming growth factor-beta1 exhibit increased binding to LDLs. Arterioscler Thromb Vasc Biol. 2002;22:55–60. doi: 10.1161/hq0102.101100. [DOI] [PubMed] [Google Scholar]
- 36.Muhlfeld A.S., Spencer M.W., Hudkins K.L., Kirk E., LeBoeuf R.C., Alpers C.E. Hyperlipidemia aggravates renal disease in B6.ROP Os/+ mice. Kidney Int. 2004;66:1393–1402. doi: 10.1111/j.1523-1755.2004.00854.x. [DOI] [PubMed] [Google Scholar]
- 37.Ravid M., Brosh D., Ravid-Safran D., Levy Z., Rachmani R. Main risk factors for nephropathy in type 2 diabetes mellitus are plasma cholesterol levels, mean blood pressure, and hyperglycemia. Arch Intern Med. 1998;158:998–1004. doi: 10.1001/archinte.158.9.998. [DOI] [PubMed] [Google Scholar]
- 38.Samuelsson O., Mulec H., Knight-Gibson C., Attman P.O., Kron B., Larsson R., Weiss L., Wedel H., Alaupovic P. Lipoprotein abnormalities are associated with increased rate of progression of human chronic renal insufficiency. Nephrol Dial Transplant. 1997;12:1908–1915. doi: 10.1093/ndt/12.9.1908. [DOI] [PubMed] [Google Scholar]
- 39.Wen M., Segerer S., Dantas M., Brown P.A., Hudkins K.L., Goodpaster T., Kirk E., LeBoeuf R.C., Alpers C.E. Renal injury in lipoprotein E-deficient mice. Lab Invest. 2002;82:999–1006. doi: 10.1097/01.lab.0000022222.03120.d4. [DOI] [PubMed] [Google Scholar]
- 40.Fazio S., Linton M.F. Mouse models of hyperlipidemia and atherosclerosis. Front Biosci. 2001;6:D515–D525. doi: 10.2741/fazio. [DOI] [PubMed] [Google Scholar]
- 41.Sharma K., Jin Y., Guo J., Ziyadeh F.N. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes. 1996;45:522–530. doi: 10.2337/diab.45.4.522. [DOI] [PubMed] [Google Scholar]
- 42.Ziyadeh F.N., Hoffman B.B., Han D.C., Iglesias-De La Cruz M.C., Hong S.W., Isono M., Chen S., McGowan T.A., Sharma K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A. 2000;97:8015–8020. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Border W.A., Okuda S., Languino L.R., Ruoslahti E. Transforming growth factor-beta regulates production of proteoglycans by mesangial cells. Kidney Int. 1990;37:689–695. doi: 10.1038/ki.1990.35. [DOI] [PubMed] [Google Scholar]
- 44.Mozes M.M., Bottinger E.P., Jacot T.A., Kopp J.B. Renal expression of fibrotic matrix proteins and of transforming growth factor-beta (TGF-beta) isoforms in TGF-beta transgenic mice. J Am Soc Nephrol. 1999;10:271–280. doi: 10.1681/ASN.V102271. [DOI] [PubMed] [Google Scholar]
- 45.Ungefroren H., Lenschow W., Chen W.B., Faendrich F., Kalthoff H. Regulation of biglycan gene expression by transforming growth factor-beta requires MKK6-p38 mitogen-activated protein Kinase signaling downstream of Smad signaling. J Biol Chem. 2003;278:11041–11049. doi: 10.1074/jbc.M300035200. [DOI] [PubMed] [Google Scholar]
- 46.Wilson P., Drennon K., Tannock L.R. Regulation of vascular proteoglycan synthesis by metabolic factors associated with diabetes. J Investig Med. 2007;55:18–25. doi: 10.2310/6650.2007.05067. [DOI] [PubMed] [Google Scholar]
- 47.Page R.A., Malik A.N. Elevated levels of beta defensin-1 mRNA in diabetic kidneys of GK rats. Biochem Biophys Res Commun. 2003;310:513–521. doi: 10.1016/j.bbrc.2003.09.034. [DOI] [PubMed] [Google Scholar]
- 48.Bjornson A., Moses J., Ingemansson A., Haraldsson B., Sorensson J. Primary human glomerular endothelial cells produce proteoglycans, and puromycin affects their posttranslational modification. Am J Physiol Renal Physiol. 2005;288:F748–F756. doi: 10.1152/ajprenal.00202.2004. [DOI] [PubMed] [Google Scholar]
- 49.Bjornson Granqvist A., Ebefors K., Saleem M.A., Mathieson P.W., Haraldsson B., Nystrom J.S. Podocyte proteoglycan synthesis is involved in the development of nephrotic syndrome. Am J Physiol Renal Physiol. 2006;291:F722–F730. doi: 10.1152/ajprenal.00433.2005. [DOI] [PubMed] [Google Scholar]
- 50.Schaefer L., Beck K.F., Raslik I., Walpen S., Mihalik D., Micegova M., Macakova K., Schonherr E., Seidler D.G., Varga G., Schaefer R.M., Kresse H., Pfeilschifter J. Biglycan, a nitric oxide-regulated gene, affects adhesion, growth, and survival of mesangial cells. J Biol Chem. 2003;278:26227–26237. doi: 10.1074/jbc.M210574200. [DOI] [PubMed] [Google Scholar]
- 51.Border W.A., Noble N.A., Yamamoto T., Harper J.R., Yamaguchi Y., Pierschbacher M.D., Ruoslahti E. Natural inhibitor of transforming growth factor-beta protects against scarring in experimental kidney disease. Nature. 1992;360:361–364. doi: 10.1038/360361a0. [DOI] [PubMed] [Google Scholar]
- 52.Babelova A., Moreth K., Tsalastra-Greul W., Zeng-Brouwers J., Eickelberg O., Young M.F., Bruckner P., Pfeilschifter J., Schaefer R.M., Grone H.J., Schaefer L. Biglycan, a danger signal that activates the NLRP3 inflammasome via Toll-like and P2X receptors. J Biol Chem. 2009;284:24035–24048. doi: 10.1074/jbc.M109.014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schaefer L., Mihalik D., Babelova A., Krzyzankova M., Grone H.J., Iozzo R.V., Young M.F., Seidler D.G., Lin G., Reinhardt D.P., Schaefer R.M. Regulation of fibrillin-1 by biglycan and decorin is important for tissue preservation in the kidney during pressure-induced injury. Am J Pathol. 2004;165:383–396. doi: 10.1016/S0002-9440(10)63305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hildebrand A., Romaris M., Rasmussen L.M., Heinegard D., Twardzik D.R., Border W.A., Ruoslahti E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem J. 1994;302(Pt 2):527–534. doi: 10.1042/bj3020527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamaguchi Y., Mann D.M., Ruoslahti E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature. 1990;346:281–284. doi: 10.1038/346281a0. [DOI] [PubMed] [Google Scholar]
- 56.Huijun W., Long C., Zhigang Z., Feng J., Muyi G. Ex vivo transfer of the decorin gene into rat glomerulus via a mesangial cell vector suppressed extracellular matrix accumulation in experimental glomerulonephritis. Exp Mol Pathol. 2005;78:17–24. doi: 10.1016/j.yexmp.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 57.Kolb M., Margetts P.J., Sime P.J., Gauldie J. Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1327–L1334. doi: 10.1152/ajplung.2001.280.6.L1327. [DOI] [PubMed] [Google Scholar]
- 58.Mogyorosi A., Ziyadeh F.N. Increased decorin mRNA in diabetic mouse kidney and in mesangial and tubular cells cultured in high glucose. Am J Physiol. 1998;275:F827–F832. doi: 10.1152/ajprenal.1998.275.5.F827. [DOI] [PubMed] [Google Scholar]
- 59.Gurley S.B., Clare S.E., Snow K.P., Hu A., Meyer T.W., Coffman T.M. Impact of genetic background on nephropathy in diabetic mice. Am J Physiol Renal Physiol. 2006;290:F214–F222. doi: 10.1152/ajprenal.00204.2005. [DOI] [PubMed] [Google Scholar]
- 60.Qi Z., Fujita H., Jin J., Davis L.S., Wang Y., Fogo A.B., Breyer M.D. Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes. 2005;54:2628–2637. doi: 10.2337/diabetes.54.9.2628. [DOI] [PubMed] [Google Scholar]
- 61.Tay Y.C., Wang Y., Kairaitis L., Rangan G.K., Zhang C., Harris D.C. Can murine diabetic nephropathy be separated from superimposed acute renal failure? Kidney Int. 2005;68:391–398. doi: 10.1111/j.1523-1755.2005.00405.x. [DOI] [PubMed] [Google Scholar]
- 62.Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels: The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med. 1998;339:1349–1357. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- 63.MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22. doi: 10.1016/S0140-6736(02)09327-3. [DOI] [PubMed] [Google Scholar]
- 64.Major outcomes in moderately hypercholesterolemic, hypertensive patients randomized to pravastatin vs usual care: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT-LLT) JAMA. 2002;288:2998–3007. doi: 10.1001/jama.288.23.2998. [DOI] [PubMed] [Google Scholar]
- 65.Fellstrom B.C., Jardine A.G., Schmieder R.E., Holdaas H., Bannister K., Beutler J., Chae D.W., Chevaile A., Cobbe S.M., Gronhagen-Riska C., De Lima J.J., Lins R., Mayer G., McMahon A.W., Parving H.H., Remuzzi G., Samuelsson O., Sonkodi S., Sci D., Suleymanlar G., Tsakiris D., Tesar V., Todorov V., Wiecek A., Wuthrich R.P., Gottlow M., Johnsson E., Zannad F. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N Engl J Med. 2009;360:1395–1407. doi: 10.1056/NEJMoa0810177. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A: Non-fasting tail vein blood glucose was measured with a Freestyle Flash glucometer at the indicated times. B: Mouse weight was measured at the indicated times. Solid symbols represent non-diabetic mice, open symbols represent diabetic mice. Squares represent mice on the 0% cholesterol diet, circles represent mice on the 0.12% cholesterol diet. Data shown is mean ± SEM from 7 to 14 per group.
A: Renal versican content. B: Renal TGF-β content. Total renal protein was analyzed by Western blot analysis for renal versican or TGF-β content. Each lane shows versican or TGF-β, as indicated, from one mouse per group, representative of 4 mice per group. NonDM indicates non-diabetic group, DM indicates diabetic group. Actin was used as the loading control. C: Renal biglycan and TGF-β content determined by densitometry of Western blot analyses was analyzed by linear regression. Solid symbols represent non-diabetic mice, open symbols represent diabetic mice. Squares represent mice on the 0% cholesterol diet, circles represent mice on the 0.12% cholesterol diet. Figure depicts four per group for all groups. r = 0.873, P < 0.001, dashed lines represent 95% confidence intervals.
Shown is a representative section from a diabetic LDLR−/− mouse fed the 0.12% cholesterol diet immunostained for decorin (red), apoB (green), nuclear DAPI stain (blue), and the merged image (colocalization yellow).