

Abstract
The clinical association between hyperlipidemia and renal disease is well established, yet hyperlipidemia as a cause for renal disease is rare. Apolipoprotein E–deficient (ApoE−/−) mice develop hyperlipidemia and are a model for atherosclerosis. Introducing deficiency of inhibitor of differentiation 3 (Id3) in ApoE−/− mice further exacerbates atherosclerosis. ID3 is a transcription regulator expressed in multiple cell types. Id3−/− mice develop antibodies to self-antigens and salivary gland autoimmunity. This study was undertaken to investigate a link between hyperlipidemia, autoimmunity, and renal disease. ApoE−/−, Id3−/−, and ApoE−/−Id3−/− double-knockout (DKO) mice were studied at different ages for renal pathological features and function. Serum samples were analyzed for the presence of autoantibodies. At 16 weeks, DKO mice developed mesangioproliferative glomerulonephritis (GN), leading to severe proteinuria. GN was associated with glomerular deposition of lipids and immune complexes and with macrophage infiltration. DKO mice had high levels of circulating autoantibodies. Although ApoE−/− mice had glomerular lipid deposits and Id3−/− mice had circulating autoantibodies, neither group of age-matched single-knockout mice developed GN. These data provide support for the hypothesis that induction of renal disease in hyperlipidemia is dictated by additional factors. Our study shows that some of these factors are regulated by ID3. Thus, ID3 is a novel risk factor linking cardiovascular and renal disease.
Hyperlipidemia is an established risk factor for atherosclerosis and cardiovascular disease. Despite a strong association between cardiovascular and renal diseases, the role of hyperlipidemia in the induction of renal disease is less understood. Patients with chronic kidney disease show increased rates of disease progression in the presence of lipoprotein abnormalities.1,2 In a prospective study,3 healthy male subjects (n = 4483) were monitored for cholesterol (total, high-density lipoprotein, and non–high-density lipoprotein) levels and renal function over 14 years. Subjects with serum creatinine levels ≥1.5 mg/dL (3%) and a glomerular filtration rate ≤55 mL/minute (5.4%) were identified as developing renal insufficiency. Statistical analyses showed a significant association between an elevated cholesterol level and the risk of developing renal insufficiency. The relative risk of an elevated creatinine level was 1.77 for total cholesterol ≥240 mg/dL and 2.34 for non–high-density lipoprotein cholesterol <40 mg/dL after adjustment for other factors, such as age, smoking, alcohol consumption, diabetes mellitus, and hypertension. These studies suggest a role for hyperlipidemia in the induction and progression of renal disease, although the pathological mechanisms remain unclear. The use of cholesterol-lowering drugs, such as HMG CoA reductase inhibitors or statins, to reduce the severity of renal disease has produced variable results.4,5 The issue is further confounded by the significant anti-inflammatory effects of statins.6 Whether improvement in renal function by statins is through their cholesterol-lowering action or through their anti-inflammatory effects has been difficult to ascertain. Thus, the benefits of lowering serum cholesterol for improvement of renal function in patients with chronic kidney disease are not universally accepted.7
In this study, we describe a mouse model that supports the hypothesis that in hyperlipidemia, additional factors are critical for the development of renal disease.8 Mice deficient in apolipoprotein E (ApoE) become hyperlipidemic (characterized by elevated low-density lipoprotein and triglyceride levels) and spontaneously develop atherosclerosis.9 ApoE is a component of chylomicron remnants and is critical for lipoprotein transport and metabolism. The introduction of a deficiency in the transcriptional regulator, inhibitor of differentiation 3 (Id3−/−), in the ApoE−/− mice led to a significant exacerbation of atherosclerosis.10 ID3 is expressed in many cells and influences the expression of multiple genes.11 It acts by forming heterodimers with transcription factors of the basic helix-loop-helix family and by inhibiting DNA binding and transcription. Specifically, ID3 has modulated immune cell development and vascular smooth muscle cell function.11,12 Yet, ID3-deficient mice have a mild phenotype; they develop autoantibodies and Sjögren's-like disease of salivary and lacrimal glands late in life, from the age of 12 to 16 months.13 Recently, it was reported that Id3−/− mice developed γδ T-cell lymphomas from the age of 33 to 92 weeks.14
Because the ApoE−/−Id3−/− double-knockout (DKO) mice developed severe cardiovascular disease, we investigated the hypothesis that ID3 represents a factor that can act synergistically with hyperlipidemia and ApoE deficiency to induce renal disease. Thus, the goal of the present study was to characterize the renal histopathological and immune features associated with loss of ID3 and ApoE. Our data show that DKO mice maintained on mouse chow spontaneously develop proliferative glomerulonephritis (GN) that leads to renal dysfunction. This is a new mouse model linking cardiovascular and renal disease that will be valuable in understanding the mechanisms connecting these disorders.
Materials and Methods
Mice and Tissues
All mice used in this study are on a C57BL/6 genetic background and were bred in a specific pathogen-free facility at the Department of Comparative Medicine, University of Virginia, Charlottesville. All experiments were approved by the Institutional Animal Care and Use Committee. Mice were given water ad libitum and maintained on a standard chow diet (Harlan Teklad 7012; Harlan Laboratories, Inc., Indianapolis, IN). C57BL/6 mice and ApoE−/− mice were obtained from Jackson Laboratories, Bar Harbor, ME. Id3−/− mice were a gift from Dr. Yuan Zhuang (Duke University, Durham, NC). DKO mice were generated by backcrossing Id3−/− mice with ApoE−/− mice, as previously reported.10 Mice were sacrificed at different ages, and blood and kidneys were harvested. Serum samples were separated and frozen until use for autoantibody analysis. Kidneys were preserved in 10% phosphate-buffered formalin for histopathological examination and in periodate lysine paraformaldehyde fixative for immunostaining, snap frozen in liquid nitrogen for detection of immune complex and C3 deposits, and placed in 2.5% glutaraldehyde with 4% paraformaldehyde in PBS for electron microscopy.
Renal Pathological Features by Light and Electron Microscopy
Formalin-fixed kidneys were embedded in paraffin, and sections were stained with H&E and PAS and evaluated for pathological changes, as previously described.15 All slides were evaluated by one of the authors (H.B.), blinded to sex, genotype, and age of the sample. GN was scored on a scale of 0 to 5: 0, no disease; 1, mild focal mesangial involvement; 2 and 3, diffuse glomerular enlargement of intermediate severity; 4, severe mesangial and/or peripheral capillary involvement; and 5, global glomerulosclerosis. Tubular involvement (ie, protein casts, dilation, and atrophy) and interstitial inflammation were also evaluated. Glutaraldehyde and paraformaldehyde fixed tissue was processed for electron microscopy, as previously described.16
Detection of Immune Complex and C3 Complement Deposits
IgG and C3 deposits were detected in snap-frozen kidney sections stained with fluorescein-labeled goat anti-mouse IgG, IgG1, IgG2a, IgG2b, or IgG3 (Southern Biotech Inc., Birmingham, AL) and C3 (ICN/Cappel Laboratories, Aurora, OH), as previously described.15
Immunofluorescence Staining for Immune-Infiltrating Cells and Fibronectin
Kidneys were fixed in periodate lysine paraformaldehyde, followed by 30% sucrose in PBS, and embedded in OCT compound for cryostat sectioning. Serial sections were stained with biotinylated antibodies to macrophages F4/80 (clone BM8) and CD68 (MCA-1957), dendritic cells (CD11c, clone HL3), T cells (anti-CD4, clone GK1.5), and B cells (B220, clone RA3-6B2). The tyramide-biotin system (Perkin Elmer Laboratories, Shelton, CT) was used for detection, according to the manufacturer's directions. Colocalization of major histocompatibility complex class II with macrophage markers was performed as previously described.15 Images were captured on a Zeiss Apotome fluorescence microscope (Carl Zeiss MicroImaging, Inc., New York, NY). All glomeruli in a transverse section of the kidney were studied, and the number of glomeruli with more than three CD68+ cells was considered positive for glomerular macrophage infiltration. Data are represented as percentage of glomeruli with CD68+ cell infiltration.
For fibronectin staining, sections (5-μm thick) of periodate lysine paraformaldehyde fixed kidney tissue were treated with 0.3% Triton X-100 (Sigma-Aldrich, St. Louis, MO) in PBS for 15 minutes; nonspecific binding was blocked with normal goat serum in 3% bovine serum albumin in PBS. The sections were incubated with rabbit anti-fibronectin antibody (Millipore Corporation, Sunnyvale, CA), 1:400 dilution, followed by biotinylated goat anti-rabbit IgG (Southern Biotech). Bound antibody was detected by incubation with Neutravidin Texas Red (Molecular Devices, Sunnyvale) and mounted with Prolong Gold mountant containing DAPI (Invitrogen, Grand Island, NY) for nuclear staining.
Immunohistochemical Staining for Ki-67 Cells
Ki-67+ cells were detected in formalin-fixed paraffin-embedded kidney sections using standard methods.17 Sections were deparaffinized, rehydrated, and boiled in antigen unmasking solution (Vector Laboratories, Burlingame, CA). Endogenous peroxidase activity was quenched with H2O2 in methanol, and nonspecific interaction was blocked with normal goat serum and avidin-biotin blocking solutions (Vector Laboratories). The slides were incubated with antibody to Ki-67 (clone SP6 at 1:100 dilution) at 4°C overnight in a humid chamber. Purified rabbit polyclonal IgG samples at equal concentrations were used as negative controls. The sections were then incubated with biotinylated goat anti-rabbit IgG (Southern Biotech) for 30 minutes at room temperature. Bound antibody was detected using the ABC system (Vector Laboratories) and diaminobenzidine substrate. The slides were counterstained with hematoxylin and mounted in Cytoseal 60 (Richard Allan Scientific, Kalamazoo, MI).
Detection of Lipid Deposits in Kidney
Snap-frozen kidney sections were fixed in cold 10% buffered formalin for 10 minutes and washed with water. The slides were air dried, placed in absolute propylene glycol for 5 minutes, and transferred into an oil red O solution (Electron Microscopy Science, Hatfield, PA) at 60°C for 30 minutes. The slides were differentiated in 85% propylene glycol solution for 2 minutes, followed by washing in water and counterstaining with hematoxylin.
Western Blot for Serum Autoantibodies
Reactivity to different cellular proteins was determined by using Western blot analysis, with mouse WEHI 7.1 cell extract as the substrate, as previously described.18
Estimation of Urinary Albumin and Creatinine
Mice were housed individually in metabolic cages for 24 hours, and urine was collected. Microalbuminuria was estimated using a competitive enzyme-linked immunosorbent assay (ELISA) (Albuwell; Exocell, Philadelphia, PA) using the manufacturer's instructions. The assay was run in duplicate. Briefly, albumin-coated wells were incubated with urine (1:13 dilution) and rabbit anti-murine albumin antibody. This was followed by incubation with horseradish peroxidase–conjugated anti-rabbit antibody. The ELISA was developed with tetramethylbenzidine substrate (Thermo Scientific, Rockford, IL), and the reaction was discontinued with 2N sulfuric acid. A standard curve was generated using different concentrations of albumin (10 to 0.156 μg/mL), and the amount of albumin in the urine samples was estimated. Urine creatinine levels were estimated using the Jaffe method, as previously described,19 and albumin/creatinine ratios were calculated.
Statistical Analyses
Graph Pad Prism software (GraphPad Software, Inc., La Jolla, CA) was used for statistical analyses. The Student's t-test was used to obtain P values. A normality test was performed on each data set. Data sets failing the normality test were analyzed using a Mann-Whitney U-test.
Results
ApoE−/−Id3−/− DKO Mice Spontaneously Develop GN
Female mice genetically deficient in ApoE (ApoE−/−), Id3 (Id3−/−), and both ApoE and Id3 (DKO) on a C57BL/6 genetic background were studied for renal pathological features (Figure 1). Age-matched C57BL/6 wild-type (WT) mice were used as controls. An evaluation of renal pathological features showed enlarged hypercellular glomeruli in DKO female mice (Figure 1D). At 24 weeks, most (80% to 100%) of the glomeruli were affected. Glomeruli showed PAS-positive material in the mesangium (Figure 1H), with some involvement of the peripheral capillary loops. Two of five mice also showed foci of periglomerular inflammation. The tubulointerstitial regions appeared normal and free of inflammatory infiltrates. No significant renal pathological feature was seen in age-matched WT and single-knockout strains (Figure 1, A–C and E–G). The kinetics of glomerular pathological features were studied in cohorts at 16 and 24 weeks (Figure 2). The severity of glomerular pathological features was scored based on percentage glomeruli affected, extent of mesangial expansion, and glomerular hypercellularity. Significant glomerular disease was detected by 16 weeks in DKO mice (P = 0.022); this further worsened at the age of 24 weeks (P < 0.0001) compared with WT controls. The progression of GN was evaluated by measuring proteinuria in an additional cohort of mice. Female DKO mice (aged 38 to 42 weeks) and ApoE−/− mice (aged 43 weeks) were housed in metabolic cages for 24 hours. An analysis of the 24-hour urine sample showed elevated albumin/creatinine ratios (P = 0.0005) in DKO mice (mean ± SEM, 113.3 ± 17.4 mg/g; n = 6) compared with ApoE−/− mice (mean ± SEM, 36.19 ± 8.5 mg/g; n = 10) (Figure 3).
Figure 1.
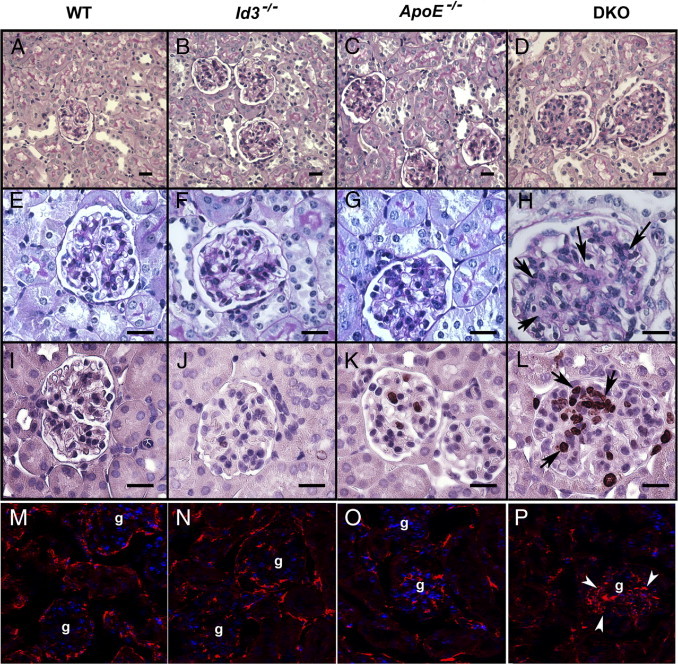
Renal pathological features in B6 WT, Id3−/−, ApoE−/−, and DKO female mice at the age of 24 weeks. Representative photomicrographs of renal histological features showing PAS-stained kidney at low (A–D) and high (E–H) magnification. There were enlarged and hypercellular glomeruli in DKO mice (H). Arrows show areas of PAS-positive material. Immunohistochemical staining for Ki-67 antigen (I–L) in glomeruli. Arrows (L) show positive staining in nuclei. Detection of fibronectin (M–P) by indirect immunofluorescence, showing renal cortex with glomeruli (g). Arrowheads show glomerular fibronectin (P). Nuclei were stained with DAPI. Scale bar = 20 μm (A–L).
Figure 2.
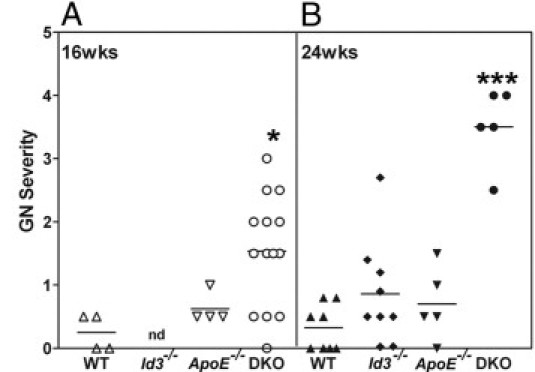
Severity of GN in WT, Id3−/−, ApoE−/−, and DKO female mice at the ages of 16 weeks (A) and 24 weeks (B). H&E-stained kidney sections were evaluated for renal pathological features, as described in Materials and Methods. Each data point represents one mouse. *P = 0.022; ***P < 0.0001 compared with WT. nd, not determined.
Figure 3.
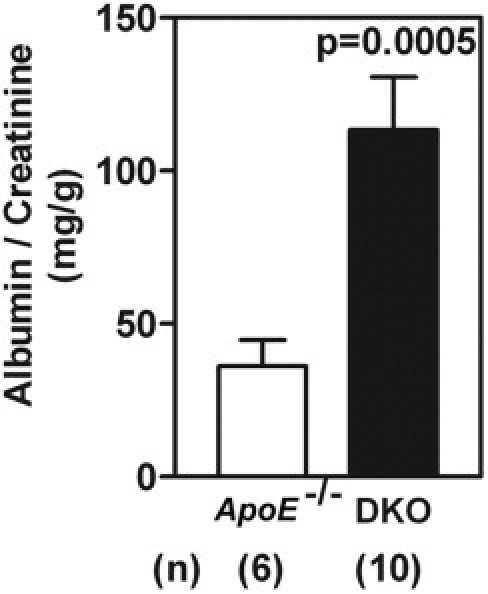
DKO female mice (n = 10) develop significant proteinuria at the age of 38 to 42 weeks, compared with ApoE−/− females (n = 6) at the age of 43 weeks. Mice were housed individually in metabolic cages, and a 24-hour urine sample was assayed for albumin/creatinine ratio. Data are given as the mean ± SEM of urinary albumin/creatinine ratios.
Glomerular hypercellularity at 24 weeks may be attributed to increased glomerular cell division and/or infiltrating immune cells. To investigate cell division, kidney sections were stained with antibody to Ki-67, a nuclear antigen expressed in dividing cells. The increased number of Ki-67+ cells in DKO mice indicates that glomerular cell proliferation contributed to the hypercellularity (Figure 1L). Some dividing cells were also seen in ApoE−/− mice (Figure 1K) but rarely in WT or Id3−/− mice (Figure 1, I and J). For a quantitative analysis of these data, four different fields (using a ×20 objective) from a transverse section of each mouse kidney were captured; the numbers of Ki-67+ cells per glomerulus were counted in 17 to 30 glomeruli per mouse. As shown in Figure 4A, dividing cells were seen in most (mean ± SEM, 93.3% ± 2.7%) glomeruli from DKO mice, significantly higher than in WT and Id3−/− mice (P < 0.0001) and ApoE−/− mice (P = 0.028). In addition, the numbers of Ki-67 cells per glomerulus were also significantly higher in DKO mice (mean ± SEM, 4.0 ± 1.4 cells per glomerulus) compared with WT mice (P = 0.002), Id3−/− mice (P = 0.02), and ApoE−/− mice (P = 0.009) (Figure 4B). Although ApoE−/− mice had some Ki-67+ glomerular cells, the frequencies and numbers did not reach statistical significance compared with WT mice (P = 0.10 and P = 0.11, respectively).
Figure 4.
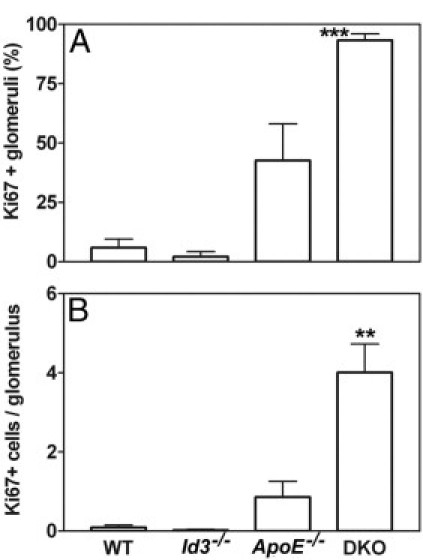
Quantitation of Ki-67+ cells in B6 WT, Id3−/−, ApoE−/−, and DKO female mice at the age of 24 weeks. Glomeruli in one cross section of each kidney were evaluated for frequency of glomeruli with Ki-67+ cells (A) and number of Ki-67+ cells per glomerulus (B). Data are represented as the mean ± SEM in each group (n = 4 per group, except for Id3−/− mice, for which n = 2). ***P < 0.0001, **P = 0.002 compared with WT.
To evaluate extracellular matrix production, kidneys were stained for fibronectin deposits. As shown in Figure 1, M–P, fibronectin was detected in the cortex of all of the strains. However, glomerular fibronectin deposits were greatly increased in DKO mice (Figure 1P), confirming expansion of mesangial matrix compared with WT and single-knockout strains (Figure 1, M–O).
The glomerular pathological features were further confirmed by electron microscopy (Figure 5). DKO mice showed a significantly expanded mesangium (Figure 5B). Podocytes and endothelial cells appeared normal, and no abnormalities were evident in the tubules. Electron micrographs at a lower magnification (Figure 5C) showed that glomerular hypercellularity with mesangial expansion is present throughout the glomerulus in DKO mice. Pathological changes were not seen in control strains. A representative electron micrograph from an ApoE−/− mouse is shown (Figure 5A). Foamy macrophages have been reported in glomerular capillaries of ApoE−/− mice fed a high-fat Western diet.20 These cells were not seen by H&E in our mice maintained on regular chow. The data suggest that at the age of 24 weeks, the disease in DKO mice is predominantly glomerular and affects mesangial cells.
Figure 5.
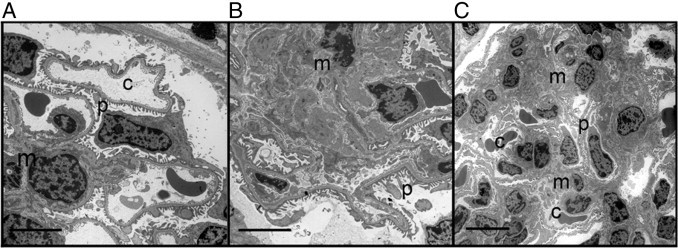
Representative electron micrographs of glomeruli from ApoE−/− and DKO female mice at the age of 24 weeks. ApoE−/− glomerulus (A) showing a glomerular capillary (c) with normal fenestrated endothelium, podocyte foot processes (p), and mesangium (m). B: A DKO glomerulus shows significant mesangial expansion. Scale bar = 5 μm. C: Another example of a DKO glomerulus at a lower power, showing extensive mesangial involvement. Scale bar = 10 μm.
CD68+ Macrophages Are the Dominant Glomerular Infiltrating Inflammatory Cells
To investigate the contribution of immune cells in GN, kidneys were stained for T-cell, B-cell, dendritic cell, and macrophage infiltration using immunofluorescence techniques. T and B cells were rarely seen in the renal cortex. CD11c+ dendritic cells were found in periglomerular infiltrates (data not shown). However, macrophages constituted the major infiltrating cells in the DKO renal cortex. The markers F4/80 and CD68 identified two populations of macrophages: CD68+ cells were seen infiltrating the glomeruli, whereas F4/80+ cells (Figure 5) were predominantly restricted to the periglomerular regions and the cortical interstitium. The frequency of glomeruli with CD68+ cells (more than three cells per glomerulus) in one cross section of the kidney (with 27 to 47 glomeruli per section) was evaluated. As shown in Table 1, the frequency of CD68+ macrophage infiltration in glomeruli of DKO mice was higher than in each of the control strains (P < 0.0001). The macrophages in the renal cortex (F4/80+) and intraglomerular area (CD68+) were activated as shown by the increased expression of major histocompatibility complex class II (Figure 6). Interestingly, relevant to this model, CD68 or macrosialin may be a receptor for uptake of oxidized low-density lipoproteins that are recognized mediators of inflammation.21 In the kidney, CD11c+ dendritic cells also express F4/80 and may contribute to some of the inflammatory infiltrates in the renal cortex.22 Regardless, these cells were rarely seen infiltrating into the glomeruli in DKO mice.
Table 1.
CD68+ Macrophage Infiltration in Glomeruli
| Strain | n | Glomeruli per cross section | CD68+ glomeruli (%) |
|---|---|---|---|
| WT | 3 | 29 ± 2 | 11.3 ± 2.9 |
| Id3−/− | 2 | 31 ± 2 | 21.5 ± 9.5 |
| ApoE−/− | 3 | 33 ± 4 | 30.3 ± 1.7 |
| DKO | 3 | 41 ± 3 | 99.0 ± 1.0⁎ |
Data are given as the mean ± SEM.
P < 0.001 versus the WT, Id3−/−, and ApoE−/− strains.
Figure 6.
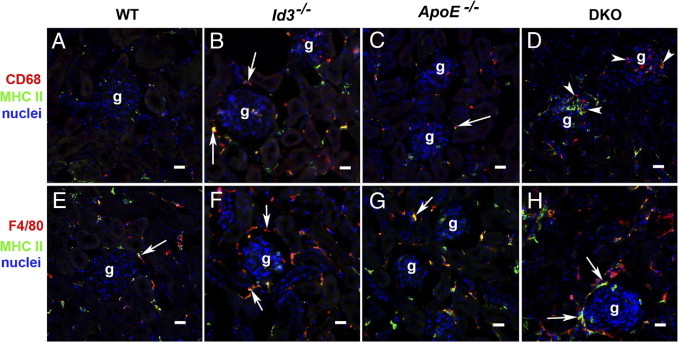
Localization of CD68+ (A–D) and F4/80+ (E–H) macrophages in the renal cortex of WT, Id3−/−, ApoE−/−, and DKO female mice at the age of 24 weeks. Glomeruli (g) showing CD68+ and F4/80+ cells labeled with red fluorescence in the cortical interstitium (arrows). CD68+ cells infiltrating glomerular cells (arrowheads) are seen in DKO mice (D). Colocalization of major histocompatibility complex (MHC) II staining (green) with CD68 or F4/80 markers is indicative of macrophage activation. Nuclei are stained with DAPI. Scale bar = 20 μm.
Glomerular Deposition of Lipids in ApoE−/− and DKO Mice
Monocytes and macrophages play an important role in atherosclerosis and GN. Hyperlipidemic ApoE−/− mice have increased numbers of these cells in circulation.23,24 As described in atherosclerosis, lipid deposition may initiate macrophage infiltration into the tissues. For the detection of renal lipid deposition, kidneys from chow-fed 24-week-old females from all strains were stained with oil red O. Neutral lipid deposition by oil red O staining was not detected in kidneys from WT and Id3−/− mice (Figure 7, A and B). However, both hyperlipidemic strains, ApoE−/− and DKO, have glomerular lipid deposits (Figure 7, C and D). Lipid deposits were restricted to the glomeruli and were not detected in tubules or interstitium. These data from ApoE−/− mice indicate that glomerular lipid deposits alone were insufficient to induce renal pathological features.
Figure 7.
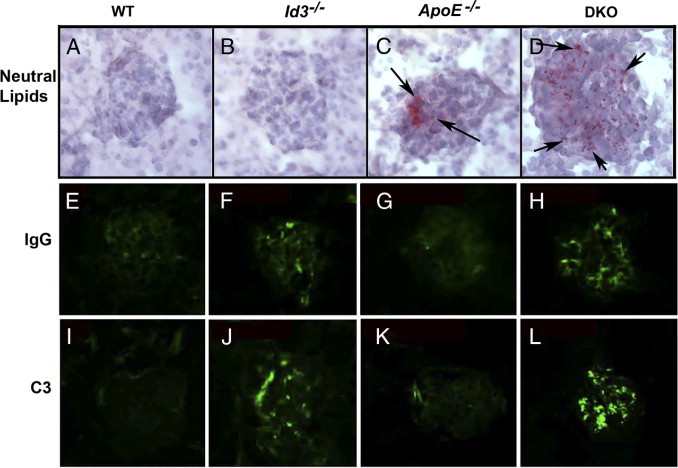
Glomerular deposits of neutral lipids (A–D) by Oil-red O staining, IgG (E–H), and C3 complement (I–L) by direct immunofluorescence in B6 WT, Id3−/−, ApoE−/−, and DKO female mice at the age of 24 weeks. Arrows indicate lipid deposits (C and D).
Glomerular Immune Complex Deposits in DKO Mice
ApoE deficiency renders the mice hyperlipidemic, and ID3 deficiency is associated with autoimmunity.9,13 Therefore, the renal disease in DKO mice may be an outcome of synergistic effect of both characteristics and a consequence of exaggerated autoimmune responses due to hyperlipidemia. To investigate this hypothesis, we stained for immune complex deposits in the kidney at 24 weeks by direct immunofluorescence (Figure 7, E–L). Glomerular deposition of IgG and C3 complement was seen in all of the DKO mice (see Supplemental Figure S1 at http://ajp.amjpathol.org). The immune deposits were of IgG1, IgG2a, and IgG2b isotypes (data not shown). The deposits are predominantly mesangial and rarely extend into the peripheral capillary loops. Fluorescent staining was also seen in Id3−/− and some ApoE−/− mice, although at much lower intensities (Figure 7, F and J). B6 mice at 24 weeks did not show glomerular IgG or C3.
Serum Autoantibody Reactivity to Cellular Antigens
To evaluate the systemic autoimmune response, blood was collected from mice at the ages of 16 and 24 weeks and analyzed using Western blot analyses for antibodies to autoantigens using WEHI 7.1 cell lysates as substrates. Figure 8 shows a representative blot of five mice per group, studied at 1:100 dilution of serum at 24 weeks. Intense reactivity to multiple cellular proteins was seen in serum samples of Id3−/− (four of five) mice and DKO (five of five) mice. ApoE−/− serum samples (two of five mice) also showed strong reactivity to multiple bands. Of the five B6 mice, only one showed reactivity to a single band. Thus, autoantibodies are generated to different levels in all of the experimental mice. A similar trend of reactivity was seen in another cohort of mice studied at 16 weeks, with Id3−/− and DKO mice showing serum autoantibodies to multiple proteins and significantly less antibody in the ApoE−/− mice (data not shown). These results suggest that, in DKO mice, hyperlipidemia in conjunction with autoimmunity, which is exacerbated by ID3 deficiency, contributes to the glomerular injury.
Figure 8.
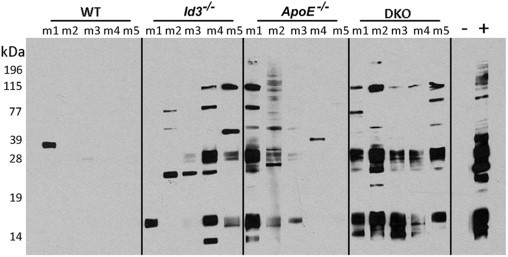
Western blot analysis showing reactivity of serum samples from WT, Id3−/−, ApoE−/−, and DKO female mice at the age of 24 weeks. WEHI 7.1 cell lysates were separated on a 12.5% SDS-PAGE under reducing conditions, transferred to nitrocellulose, and probed with serum samples at 1:100 dilution, followed by goat anti-mouse IgG horseradish peroxidase conjugate. Bound antibody was detected with chemiluminescence, as previously described. Each lane represents serum from one mouse (m). −, negative control (not incubated with serum); +, pooled serum samples from MRL/lpr mice with lupus at 1:1000 dilution.
Discussion
This investigation describes an experimental mouse model linking cardiovascular and renal diseases. DKO mice that are ApoE−/− mice and null for Id3 have increased atherosclerosis compared with Id3-sufficient ApoE−/− mice.10 In this study, we demonstrate that the DKO mice spontaneously develop GN, characterized by enlarged hypercellular glomeruli associated with cell proliferation, mesangial expansion, and increased extracellular matrix deposition. Kidneys of DKO mice show glomerular immune complex, C3, and lipid deposition and macrophage infiltration. The progression of these pathological changes was associated with the development of proteinuria.
Although data presented are from female mice, the renal pathological features described herein were also seen in DKO male mice (see Supplemental Figure S2 at http://ajp.amjpathol.org). However, the disease in male mice was less severe and delayed in onset, first seen at the age of 24 weeks.25 Glomeruli were enlarged and hypercellular and were associated with increased fibronectin deposition. DKO males also showed glomerular IgG and C3 deposition. Glomerular infiltration by CD68 macrophages in DKO males was also a characteristic feature.
Cholesterol levels were elevated in both male10 and female ApoE−/− and DKO mice (see Supplemental Table S1 at http://ajp.amjpathol.org). These levels are comparable between the two strains; thus, Id3 does not influence systemic lipid profiles. Both systemic hyperlipidemia and tissue macrophage infiltrates, seen herein, are also common features of atherosclerotic plaques, suggesting the possibility of shared underlying pathological mechanisms.
The role of hyperlipidemia in exacerbating renal disease has been investigated in experimental mouse model systems involving either ApoE or low-density lipoprotein receptor deficiency.6,26 ApoE−/− mice are well-established models of hyperlipidemia and atherosclerosis.9 In addition to affecting lipid levels, ApoE also directly affects mesangial cell growth and survival and, thereby, might influence renal disease in ApoE−/− mouse models.20 Yet, ApoE−/− mice develop renal pathological features only late in life, at approximately the age of 36 weeks; and the pathological features are characterized by mild mesangial proliferation and accumulation of foamy macrophages within the glomerular capillaries.27 These renal pathological features are significantly aggravated by feeding the mice a high-fat diet. This suggests a role for excessive lipid levels in worsening of renal disease.20,28 The introduction of ApoE deficiency into murine models of spontaneous lupus nephritis exacerbated renal pathological features in the mice with lupus.26 Similarly, low-density lipoprotein receptor–deficient mice, also used as models for hyperlipidemia, show evidence for accelerated nephritis, when bred with mice susceptible for the development of lupus.6 The latter two models show that hyperlipidemia can influence renal disease in the presence of other factors, such as spontaneous lupus nephritis. Our study suggests that deficiency of ID3 is an additional susceptibility factor in hyperlipidemia-induced renal pathological features.
ID3 belongs to the class V helix-loop-helix transcription factors that regulate the function of class I and II basic helix-loop-helix transcription factors.11 Specifically, the ID proteins regulate the function of CDK inhibitors that regulate the formation of CDK-cyclin complexes promoting entry into the cell cycle.29 Id3 is up-regulated after stimulation of mitogen-activated protein kinase, suggesting a role for ID3 in promoting cell proliferation.30 Indeed, ID3 facilitates entry into the cell cycle and induces cell growth. In contrast, our data indicating that loss of ID3 significantly enhances Ki-67 immunoreactivity in the glomeruli of ApoE−/− mice suggest that ID3's effects on cellular proliferation may be cell type dependent. In the thymus, ID3 antagonizes E2A, a transcription factor that plays a role in T-cell selection.31 Thus, ID3 acts in a wide range of cell types affecting multiple pathways. ID3 has previously been implicated in vascular smooth muscle function and is a focus of investigation in cardiovascular disease. Recently, a study10 in a diabetes heart patient cohort showed that a single-nucleotide polymorphism variant (rs11574) of Id3 was associated with susceptibility to atherosclerosis. The present study is the first to demonstrate a role for ID3 in hyperlipidemia-associated kidney disease, and our findings should provide an impetus to closely investigate the association of ID3 in human disease.
The precise mechanisms of how renal disease is initiated in DKO mice are not known, but our results provide some clues. The data show that renal disease occurs only in hyperlipidemic mice in the absence of ID3. Thus, in the ID3-intact ApoE−/− mice, ID3 might exert protective effects by influencing the overall autoimmune and inflammatory responses. This is supported by data showing amplified autoantibody responses in both Id3−/− and DKO mice. The development of renal pathological features in DKO mice might then be influenced by glomerular immune complex and lipid deposition. Although the Id3−/− mice show some evidence for some glomerular immune complex deposition, lipid deposits were not detected. In contrast, in ApoE−/− mice, lipid accumulation was readily detected in the kidneys but not immune complexes. Both immune complex and lipid deposits were seen only in the kidneys of DKO mice. Thus, the synergistic effects of inflammatory pathways activated by the deposition of autoantibodies and lipids in the kidneys of DKO mice may be responsible for initiating the renal disease in these mice. Another possibility is that in the DKO mice, hyperlipidemia influences the specificity of autoantibodies, some of which might be reactive with lipid deposits in the kidney. This dictates glomerular immune complex formation and subsequent glomerular inflammation. This hypothesis is indirectly supported by findings of diabetic nephropathy in patients (ie, glomerular deposition of IgG and IgM antibodies to oxidized low-density lipoprotein is associated with worse prognosis).32,33
Our study has significant clinical implications. In DKO mice, glomerular immune complex deposition and glomerular pathological features significantly precede any clinical loss of renal function. This study suggests that individuals who are genetically susceptible to autoimmune diseases may manifest renal disease in the presence of hyperlipidemia. Therefore, the detection of circulating autoantibodies can become an important biomarker to predict the risk of renal disease in individuals with hyperlipidemia. In turn, monitoring autoantibody-positive hyperlipidemic individuals for the development of renal dysfunction can prove valuable for early detection and therapy. Thus, the DKO mouse model can be used to investigate the interaction of autoimmunity and hyperlipidemia in renal disease.
Acknowledgments
We thank Dr. Kenneth S.K. Tung for expert opinion on renal pathological features, Saleh Mohammad for outstanding technical assistance in tissue sectioning and staining, and Jan Redick and the Advanced Electron Microscopy Core for assistance on electron microscopic studies of the kidney.
Footnotes
Supported by the National Institutes of Heath (grants R01DK69769 to H.B., R01 AI079621 and R21DE019883 to U.D., and R01HL0964479 to C.M.) and the Alliance for Lupus Research (grant TIL 113300 to H.B.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.04.029.
Supplementary data
Female DKO mice develop glomerular immune complex and C3 complement deposits at 24 weeks. The intensity of glomerular IgG (A) and C3 (B) deposits in chow-fed female mice from B6 WT, Id3−/−, ApoE−/−, and DKO strains at the age of 24 weeks is shown. Scores for glomerular IgG deposits were significantly greater in DKO mice compared with WT (P = 0.0004), Id3−/− (P = 0.04), and ApoE−/− (P = 0.007) mice. Similarly, C3 complement deposits also greater in DKO mice compared with WT (P = 0.0014), Id3−/− (P = 0.009), and ApoE−/− (P = 0.003) mice. Asterisks denote significantly different from wild type.
Male chow-fed DKO mice have GN and glomerular IgG and C3 deposits at the age of 24 weeks. The severity of GN (A), the intensity of glomerular IgG (B), and C3 deposits (C) in chow-fed male mice from B6 WT, Id3−/−, ApoE−/−, and DKO strains at the age of 24 weeks. DKO mice had significantly greater disease severity compared with WT (P < 0.001), Id3−/− (P < 0.004), and ApoE−/− (P < 0.007) mice. Scores for glomerular IgG deposits were also significantly greater in DKO mice compared with WT (P < 0.05), Id3−/− (P < 0.006), and ApoE−/− (P < 0.029) mice. Asterisks denote significantly different from wild type.
References
- 1.Samuelsson O., Mulec H., Knight-Gibson C., Attman P.O., Kron B., Larsson R., Weiss L., Wedel H., Alaupovic P. Lipoprotein abnormalities are associated with increased rate of progression of human chronic renal insufficiency. Nephrol Dial Transplant. 1997;12:1908–1915. doi: 10.1093/ndt/12.9.1908. [DOI] [PubMed] [Google Scholar]
- 2.Kees-Folts D., Diamond J.R. Relationship between hyperlipidemia, lipid mediators, and progressive glomerulosclerosis in the nephrotic syndrome. Am J Nephrol. 1993;13:365–375. doi: 10.1159/000168651. [DOI] [PubMed] [Google Scholar]
- 3.Schaeffner E.S., Kurth T., Curhan G.C., Glynn R.J., Rexrode K.M., Baigent C., Buring J.E., Gaziano J.M. Cholesterol and the risk of renal dysfunction in apparently healthy men. J Am Soc Nephrol. 2003;14:2084–2091. doi: 10.1681/ASN.V1482084. [DOI] [PubMed] [Google Scholar]
- 4.Tonelli M., Moyé L., Sacks F.M., Cole T., Curhan G.C., Cholesterol and Recurrent Events Trial Investigators Effect of pravastatin on loss of renal function in people with moderate chronic renal insufficiency and cardiovascular disease. J Am Soc Nephrol. 2003;14:1605–1613. doi: 10.1097/01.asn.0000068461.45784.2f. [DOI] [PubMed] [Google Scholar]
- 5.Ruggenenti P., Perna A., Tonelli M., Loriga G., Motterlini N., Rubis N., Ledda F., Rota S., Jr, Satta A., Granata A., Battaglia G., Cambareri F., David S., Gaspari F., Stucchi N., Carminati S., Ene-Iordache B., Cravedi P., Remuzzi G., ESPLANADE Study Group Effects of add-on fluvastatin therapy in patients with chronic proteinuric nephropathy on dual renin-angiotensin system blockade: the ESPLANADE trial. Clin J Am Soc Nephrol. 2010;5:1928–1938. doi: 10.2215/CJN.03380410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aprahamian T., Bonegio R., Rizzo J., Perlman H., Lefer D.J., Rifkin I.R., Walsh K. Simvastatin treatment ameliorates autoimmune disease associated with accelerated atherosclerosis in a murine lupus model. J Immunol. 2006;177:3028–3034. doi: 10.4049/jimmunol.177.5.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tonelli M. Do statins protect the kidney as well as the heart? Nephrol Dial Transplant. 2006;21:3005–3006. doi: 10.1093/ndt/gfl505. [DOI] [PubMed] [Google Scholar]
- 8.Saito T. Abnormal lipid metabolism and renal disorders. Tohoku J Exp Med. 1997;181:321–337. doi: 10.1620/tjem.181.321. [DOI] [PubMed] [Google Scholar]
- 9.Jawień J., Nastałek P., Korbut R. Mouse models of experimental atherosclerosis. J Physiol Pharmacol. 2004;55:503–517. [PubMed] [Google Scholar]
- 10.Doran A.C., Lehtinen A.B., Meller N., Lipinski M.J., Slayton R.P., Oldham S.N., Skaflen M.D., Yeboah J., Rich S.S., Bowden D.W., McNamara C.A. Id3 is a novel atheroprotective factor containing a functionally significant single-nucleotide polymorphism associated with intima-media thickness in humans. Circ Res. 2010;106:1303–1311. doi: 10.1161/CIRCRESAHA.109.210294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forrest S., McNamara C. Id family of transcription factors and vascular lesion formation. Arterioscler Thromb Vasc Biol. 2004;24:2014–2020. doi: 10.1161/01.ATV.0000143932.03151.ad. [DOI] [PubMed] [Google Scholar]
- 12.Ueda-Hayakawa I., Mahlios J., Zhuang Y. Id3 restricts the developmental potential of gamma delta lineage during thymopoiesis. J Immunol. 2009;182:5306–5316. doi: 10.4049/jimmunol.0804249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H., Dai M., Zhuang Y. A T cell intrinsic role of Id3 in a mouse model for primary Sjogren's syndrome. Immunity. 2004;21:551–560. doi: 10.1016/j.immuni.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 14.Li J., Maruyama T., Zhang P., Konkel J.E., Hoffman V., Zamarron B., Chen W. Mutation of inhibitory helix-loop-helix protein Id3 causes γδ T-cell lymphoma in mice. Blood. 2010;116:5615–5621. doi: 10.1182/blood-2010-03-274506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bagavant H., Deshmukh U.S., Wang H., Ly T., Fu S.M. Role for nephritogenic T cells in lupus glomerulonephritis: progression to renal failure is accompanied by T cell activation and expansion in regional lymph nodes. J Immunol. 2006;177:8258–8265. doi: 10.4049/jimmunol.177.11.8258. [DOI] [PubMed] [Google Scholar]
- 16.Waters S.T., McDuffie M., Bagavant H., Deshmukh U.S., Gaskin F., Jiang C., Tung K.S., Fu S.M. Breaking tolerance to double stranded DNA, nucleosome, and other nuclear antigens is not required for the pathogenesis of lupus glomerulonephritis. J Exp Med. 2004;199:255–264. doi: 10.1084/jem.20031519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deshmukh U.S., Nandula S.R., Thimmalapura P.R., Scindia Y.M., Bagavant H. Activation of innate immune responses through Toll-like receptor 3 causes a rapid loss of salivary gland function. J Oral Pathol Med. 2009;38:42–47. doi: 10.1111/j.1600-0714.2008.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deshmukh U.S., Bagavant H., Sim D., Pidiyar V., Fu S.M. A SmD peptide induces better antibody responses to other proteins within the small nuclear ribonucleoprotein complex than to SmD protein via intermolecular epitope spreading. J Immunol. 2007;178:2565–2571. doi: 10.4049/jimmunol.178.4.2565. [DOI] [PubMed] [Google Scholar]
- 19.Kinsey G.R., Sharma R., Huang L., Li L., Vergis A.L., Ye H., Ju S.T., Okusa M.D. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J Am Soc Nephrol. 2009;8:1744–1753. doi: 10.1681/ASN.2008111160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wen M., Segerer S., Dantas M., Brown P.A., Hudkins K.L., Goodpaster T., Kirk E., LeBoeuf R.C., Alpers C.E. Renal injury in apolipoprotein E-deficient mice. Lab Invest. 2002;82:999–1006. doi: 10.1097/01.lab.0000022222.03120.d4. [DOI] [PubMed] [Google Scholar]
- 21.Ramprasad M.P., Terpstra V., Kondratenko N., Quehenberger O., Steinberg D. Cell surface expression of mouse macrosialin and human CD68 and their role as macrophage receptors for oxidized low density lipoprotein. Proc Natl Acad Sci U S A. 1996;93:14833–14838. doi: 10.1073/pnas.93.25.14833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li L., Huang L., Sung S.S., Vergis A.L., Rosin D.L., Rose C.E., Jr, Lobo P.I., Okusa M.D. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int. 2008;74:1526–1537. doi: 10.1038/ki.2008.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swirski F.K., Libby P., Aikawa E., Alcaide P., Luscinskas F.W., Weissleder R., Pittet M.J. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gordon S. Macrophage heterogeneity and tissue lipids. J Clin Invest. 2007;117:89–93. doi: 10.1172/JCI30992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bagavant H, Scindia Y, Doran A, Nandula S, Cutchins A, Oldham S, McNamara C, Deshmukh U: ApoE−/− and Id3−/− double knockout mice: a murine model for atherosclerosis and glomerulonephritis. Presented at the American Society of Nephrology Meeting, 2009, October 29–November 1, San Diego, CA. Abstract PO1939.
- 26.Feng X., Li H., Rumbin A.A., Wang X., La Cava A., Brechtelsbauer K., Castellani L.W., Witztum J.L., Lusis A.J., Tsao B.P. ApoE−/−Fas−/− C57BL/6 mice: a novel murine model simultaneously exhibits lupus nephritis, atherosclerosis, and osteopenia. J Lipid Res. 2007;48:794–805. doi: 10.1194/jlr.M600512-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Chen G., Paka L., Kako Y., Singhal P., Duan W., Pillarisetti S. A protective role for kidney apolipoprotein E: regulation of mesangial cell proliferation and matrix expansion. J Biol Chem. 2001;276:49142–49147. doi: 10.1074/jbc.M104879200. [DOI] [PubMed] [Google Scholar]
- 28.Liberopoulos E., Siamopoulos K., Elisaf M. Apolipoprotein E and renal disease. Am J Kidney Dis. 2004;43:223–233. doi: 10.1053/j.ajkd.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 29.Forrest S.T., Barringhaus K.G., Perlegas D., Hammarskjold M.L., McNamara C.A. Intron retention generates a novel Id3 isoform that inhibits vascular lesion formation. J Biol Chem. 2004;279:32897–32903. doi: 10.1074/jbc.M404882200. [DOI] [PubMed] [Google Scholar]
- 30.Sherr C.J., Roberts J.M. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 31.Rivera R.R., Johns C.P., Quan J., Johnson R.S., Murre C. Thymocyte selection is regulated by the helix-loop-helix inhibitor protein, Id3. Immunity. 2000;12:17–26. doi: 10.1016/s1074-7613(00)80155-7. [DOI] [PubMed] [Google Scholar]
- 32.Atchley D.H., Lopes-Virella M.F., Zheng D., Kenny D., Virella G. Oxidized LDL-anti-oxidized LDL immune complexes and diabetic nephropathy. Diabetologia. 2002;45:1562–1571. doi: 10.1007/s00125-002-0962-y. [DOI] [PubMed] [Google Scholar]
- 33.Virella G., Carter R.E., Saad A., Crosswell E.G., Game B.A., DCCT/EDIC Study Group. Lopes-Virella M.F. Distribution of IgM and IgG antibodies to oxidized LDL in immune complexes isolated from patients with type 1 diabetes and its relationship with nephropathy. Clin Immunol. 2008;127:394–400. doi: 10.1016/j.clim.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Female DKO mice develop glomerular immune complex and C3 complement deposits at 24 weeks. The intensity of glomerular IgG (A) and C3 (B) deposits in chow-fed female mice from B6 WT, Id3−/−, ApoE−/−, and DKO strains at the age of 24 weeks is shown. Scores for glomerular IgG deposits were significantly greater in DKO mice compared with WT (P = 0.0004), Id3−/− (P = 0.04), and ApoE−/− (P = 0.007) mice. Similarly, C3 complement deposits also greater in DKO mice compared with WT (P = 0.0014), Id3−/− (P = 0.009), and ApoE−/− (P = 0.003) mice. Asterisks denote significantly different from wild type.
Male chow-fed DKO mice have GN and glomerular IgG and C3 deposits at the age of 24 weeks. The severity of GN (A), the intensity of glomerular IgG (B), and C3 deposits (C) in chow-fed male mice from B6 WT, Id3−/−, ApoE−/−, and DKO strains at the age of 24 weeks. DKO mice had significantly greater disease severity compared with WT (P < 0.001), Id3−/− (P < 0.004), and ApoE−/− (P < 0.007) mice. Scores for glomerular IgG deposits were also significantly greater in DKO mice compared with WT (P < 0.05), Id3−/− (P < 0.006), and ApoE−/− (P < 0.029) mice. Asterisks denote significantly different from wild type.