

Abstract
Niemann-Pick disease type C (NPC) is a severe neurovisceral lysosomal storage disorder caused by defects in NPC1 or NPC2 proteins. Although numerous studies support the primacy of cholesterol storage, neurons of double-mutant mice lacking both NPC1 and an enzyme required for synthesis of all complex gangliosides (β1,4GalNAc transferase) have been reported to exhibit dramatically reduced cholesterol sequestration. Here we show that NPC2-deficient mice lacking this enzyme also exhibit reduced cholesterol, but that genetically restricting synthesis to only a-series gangliosides fully restores neuronal cholesterol storage to typical disease levels. Examining the subcellular locations of sequestered compounds in neurons lacking NPC1 or NPC2 by confocal microscopy revealed that cholesterol and the two principal storage gangliosides (GM2 and GM3) were not consistently co-localized within the same intracellular vesicles. To determine whether the lack of GM2 and GM3 co-localization was due to differences in synthetic versus degradative pathway expression, we generated mice lacking both NPC1 and lysosomal β-galactosidase, and therefore unable to generate GM2 and GM3 in lysosomes. Double mutants lacked both gangliosides, indicating that each is the product of endosomal/lysosomal processing. Unexpectedly, GM1 accumulation in double mutants increased compared to single mutants consistent with a direct role for NPC1 in ganglioside salvage. These studies provide further evidence that NPC1 and NPC2 proteins participate in endosomal/lysosomal processing of both sphingolipids and cholesterol.
Niemann-Pick disease type C (NPC) is a progressive and ultimately fatal neurovisceral lysosomal disorder caused by genetic defects in either NPC1 or NPC2 proteins. In the brain, the disease is characterized by intraneuronal storage of unesterified cholesterol and sphingolipids, including GM2 and GM3 gangliosides, glucosylceramide, and lactosylceramide.1–3 The NPC1 protein is a large transmembrane protein found principally in late endosomes (LEs). It contains a sterol sensing domain, exhibits homology with proteins involved in cholesterol homeostasis4 and was recently shown to bind cholesterol and oxysterols.5–7 The NPC2 protein is a soluble protein which also binds cholesterol and is located in LEs and lysosomes (LYs).8–11 Recent studies indicating that NPC1 and NPC2 proteins play a coordinated role in cholesterol processing in LEs,12 coupled with the striking similarities in clinicopathological features of NPC1 or NPC2 deficiencies in humans2 and mice13, provide compelling arguments for these two proteins functioning in a common metabolic pathway. In concert, these findings support the widely-held view that NPC disease results in accumulation of unesterified cholesterol in cells secondary to a block in retroendocytic trafficking.14
Yet a wide variety of lysosomal diseases with primary or secondary defects in ganglioside catabolism (eg, the gangliosidoses and mucopolysaccharidoses, respectively, also exhibit significant intraneuronal sequestration of cholesterol.3,15,16 In mucopolysaccharidoses IIIA disease, for example, defective sulfamidase function causes heparan sulfate accumulation followed by intraneuronal storage of GM2 and GM3 gangliosides and cholesterol.15 There is evidence that sequestration of gangliosides and cholesterol in NPC disease may follow a similar pattern. That is, mice lacking both NPC1 and β−1,4-N-acetylgalactosaminyl-transferase (β1,4GalNAcT; GalNAcT or GM2/GD2 synthase; B4galnt), an enzyme required for synthesis of all complex gangliosides belonging to the sialo-ganglio series (Figure 1A),17 exhibit dramatically diminished cholesterol accumulation consistent with the degree of ganglioside storage in neurons influencing that of cholesterol.18,19
Figure 1.

Schematic illustration of the synthetic and degradative paths involving glycosphingolipids. A: Synthesis of glycosphingolipids occurs in step-wise fashion in the Golgi/TGN through the action of a series of glycosyltransferases.17 Following synthesis, higher order gangliosides are believed trafficked to the plasmalemma where they reside in membrane microdomains rich in cholesterol.37 Absence of GalNAcT (GM2/GD2 synthase) (1) secondary to ablation of the B4galnt gene blocks the ability of cells to generate gangliosides beyond GM3, GD3, and GT3, whereas ablation of St8sia1 (2) and loss of GD3 synthase activity blocks synthesis of gangliosides other than those of the a-series. B: Glycosphingolipids in the plasmalemma are endocytosed and degraded in the E/L system through actions of a series of acid hydrolases. Ablation of lysosomal β-gal (3) blocks the ability of the cell to degrade GM1 ganglioside to generate simpler components, including GM2, GM3 and lactosylceramide.17
In the current study, we have further explored the relationship between gangliosides and cholesterol in NPC disease. Confocal microscopy used to analyze subcellular storage locations revealed largely independent patterns of intraneuronal ganglioside and cholesterol accumulation in Npc1 and Npc2 mice that was remarkably similar to mucopolysaccharidose disease. Analysis of Npc1 and Npc2 mice crossed with knockouts for specific ganglioside synthetic enzymes revealed dependency on a-series gangliosides for cholesterol sequestration in most CNS neurons. Double-mutant mice lacking NPC1 and the ability to degrade GM1 ganglioside further showed that GM2 and GM3 storage compounds in NPC disease are both derived from lysosomal GM1 degradation. These double-mutant mice also exhibited more GM1 accumulation than either single mutant consistent with NPC1 facilitating the retroendocytic trafficking of simple gangliosides. Together, these studies reveal a complex, interdependent relationship between gangliosides and cholesterol in NPC disease and suggest a role for NPC1 and NPC2 proteins in ganglioside salvage and homeostatic control similar to that reported for cholesterol.
Materials and Methods
Antibodies and Reagents
Anti-GM2 ganglioside mAb (mouse IgM, cell culture supernatant) (mab 10–11) was generously provided by Progenics Pharmaceuticals, Inc. (Tarrytown, NY). Anti-GM3 ganglioside mAb (DH2, mouse IgG3, cell culture supernatant) was generously provided by Dr. S. Hakomori (Pacific Northwest Research Laboratory, Seattle, WA) or purchased (10–011) from GlycoTech Corp. (Gaithersburg, MD). Anti-GM1 ganglioside pAb (IgG, serum) (G2006-11) was purchased from US Biological (Swampscott, MA). Anti-GD3 ganglioside mAb (R-24, mouse IgG, cell culture supernatant) (#1977) was purchased from BIOTREND Chemicals LLC (Destin, FL). Anti-lysosomal membrane glycoprotein 2 (LAMP2) mAb (rat IgG2a, cell culture concentrate) (ABL-93-c) was purchased from Developmental Studies Hybridoma Bank at the University of Iowa (Iowa City, IA). The following antibodies were also purchased for use in IHC: mouse IgM (M 2521) and mouse IgG (I 5381) from Sigma Immunochemicals (St. Louis, MO), biotinylated goat anti-mouse IgM (BA-2020), biotinylated goat anti-mouse IgG (BA-9200), and biotinylated goat anti-rabbit IgG (BA-1000) from Vector Laboratories (Burlingame, CA). The following secondary and tertiary antibodies were purchased for use in immunofluorescence: Texas Red-conjugated goat anti-mouse IgM (μ chain specific, JGN075075) from AxCell Biosciences (Newtown, PA); fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (γ specific-Fc, 55517) from MP Biomedicals (Solon, OH); Alexa Fluor 488-conjugated goat anti-rat IgG (A11006), Alexa Fluor 546-conjugated goat anti-mouse IgM (A21045), Alexa Fluor 488-conjugated streptavidin (S32354), and Alexa Fluor 488 Signal-Amplification Kit for FITC probes (A11053) from Molecular Probes/Invitrogen (Carlsbad, CA). Filipin complex from Streptomyces filipinensis (F9765) was purchased from Sigma-Aldrich (St. Louis, MO). BCθ20 was provided by Dr. Yoshiko Ohno-Iwashita (Cellular Signaling Group, Tokyo Metropolitan Institute of Gerontology, Tokyo, Japan). High performance silica gel 60 thin layer chromatographic plates were from Merck (Darmstadt, Germany).
Animals and Genotyping Procedures
Mice with NPC1 and NPC2 diseases, GM1 gangliosidosis, and knockouts for GalNAcT (GM2/GD2 synthase) (B4galnt1) and GD3 synthase (St8sia1) were obtained from breeding colonies maintained at the Albert Einstein College of Medicine. The NPC1 mouse model (BALBc/NPCnih) (referred to here as Npc1−/−) was originally obtained from Dr. Peter Pentchev; the Npc2 model (Npc2−/−) mice on a BALB/c background were provided by Drs. Peter Lobel and David Sleat; the knockouts for individual GSL synthetic enzymes (B4galnt1−/− and St8sia1−/−) were from Drs. Richard Proia and Ronald Schnaar; and the GM1 gangliosidosis mouse model (β-gal−/−) was from Dr. Alessandra d'Azzo. Individual colonies were maintained according to published methods with animals identified by routine PCR genotyping.13,18,21,22 Double-heterozygous animals were generated and subsequently bred for production of double mutants, with the genotype of each animal established and analyzed in littermate cohorts. These studies evaluated 7 Npc2−/−;B4galnt1−/− double mutants (with 3 Npc2−/−, 3 B4galnt1−/−, and 4 wild types as littermates), 4 Npc1−/−;St8sia1−/− double mutants (with 3 Npc1−/− mutants, 3 St8sia1−/− knockouts, 3 wild types as littermates), and 4 Npc1−/−;β-gal−/− double mutants (with 3 Npc1−/− mutants; 3 β-gal−/− knockouts and 3 wild types as littermates). Both male and female mice were analyzed in these studies. Littermates from each were cross identified as homozygous for one gene defect and heterozygous for the other generally were not included in this analysis. Mutant mice with single lysosomal diseases (Npc1 and Npc2 and GM1 gangliosidosis) from other litters were also available for comparison in terms of longevity data versus the shorter lived double mutants. At termination, mice were deeply anesthetized and insensate after a dose of pentobarbital (Nembutal) (40 mg/kg) and perfused with 0.9% saline solution. This was followed by a craniotomy and removal of the right cerebral hemisphere, which was immediately frozen. Perfusion was then reinitiated using 4% paraformaldehyde in 0.1 M phosphate buffer. The brains were removed; immersion was fixed overnight in additional fixative, which was then transferred into 0.1 M phosphate buffer and stored cold. All procedures using animals were approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine.
Immunoperoxidase and Histochemical Procedures
Procedures used were as previously published.15,23 Antibodies to GM2 and GM3 gangliosides have been extensively documented to label punctate cytoplasmic structures in the neurons of a variety of lysosomal diseases known to exhibit accumulation of these two gangliosides. Hemi-coronal sections (30 μm) were cut with a Leica VT-1000S Vibratome (Leica Microsystems, Buffalo Grove, IL). Sections were washed 2 × 10 minutes in 10 mmol/L pH 7.5 PBS and were then incubated at room temperature for 1 hour in blocking buffer [PBS with 1% bovine serum albumin (BSA) (Sigma A7906), 1.5% normal goat serum (NGS) (Vector Labs S-1000), and 0.02% saponin (Sigma S7900)]. Sections were then incubated overnight at 4°C with primary antibodies [anti-GM2 (1:15), anti-GM3 (1:100), anti-GM1 (1:5000), or anti-GD3 (1:150)] in diluent (PBS with 1% bovine serum albumin, 1% NGS, and 0.02% saponin). Then sections were washed 4 × 10 minutes in wash solution (0.02% saponin in PBS) and incubated for 1 hour at room temperature with biotinylated secondary antibodies, goat anti-rabbit IgG (1:200), goat anti-mouse IgM (1:200), or goat anti-mouse IgG (1:200) in diluent. The sections were washed 5 × 10 minutes in wash solution and then incubated in Vectastain ABC kit (Vector Labs PK-4000) for 1 hour at room temperature followed by 4- to 10-minute washes in PBS. The sections were then incubated with Peroxidase Substrate Kit DAB (Vector Labs SK-4100) for 10 minutes, washed 2 × 5 minutes with PBS, and mounted with Permount (Fisher SP15-500).
Filipin has been widely documented to label unesterified cholesterol in cells.24 A newer probe for unesterified cholesterol, BCθ, shows a similar staining pattern.25 For filipin labeling, sections were washed 2 × 10 minutes in PBS and 2 × 10 minutes in wash solution (0.02% saponin in PBS). Sections were then incubated for 20 minutes with PBS containing 0.005% Filipin Complex (Sigma F9765) from a 25 mg/mL solubilized stock in dimethyl sulfoxide (Sigma D128-500), or with an equivalent concentration of dimethyl sulfoxide only to serve as a control. The sections were washed 2 × 10 minutes in wash solution, 2 × 10 minutes in PBS, and finally mounted on Gold Seal fluorescent antibody slides (Fisher 12-568-20) with Prolong Antifade Kit (Molecular Probes P7481) and circular #1½ coverslips (Fisher 12-545-81).
For BCθ labeling, sections were washed 2 × 10 minutes in PBS then incubated in blocking buffer [PBS with 1% reduced fatty acid bovine serum albumin (BSA) (Sigma A3803), 10% NGS, and 0.02% saponin] for 90 minutes at room temperature. After the blocking step, sections were washed 5 × 10 minutes in PBS and then incubated overnight at 4°C with BCθ (12 μg/mL) in diluent (PBS with 1% reduced fatty acid BSA and 0.02% saponin). Sections were washed 5 × 10 minutes in wash solution and incubated for 1 hour at room temperature with Alexa Fluor 488-conjugated streptavidin in diluent, followed by 4- to 10-minute washes in PBS. Finally, sections were mounted on Gold Seal fluorescent antibody slides with Prolong Antifade Kit and circular #1½ coverslips.
Immunofluorescence and Confocal Imaging Procedures
Procedures used were as previously published.15,23 Vibratome sections cut at 35-μm were washed twice with PBS, incubated in blocking buffer (PBS with 1% BSA, 10% NGS, and 0.02% saponin) for 90 minutes at room temperature and incubated overnight at 4°C in diluent (PBS with 1% BSA, 10% NGS, and 0.02% saponin) with desired combinations of primary antibodies to GM2 (1:5), GM3 (1:10), LAMP2 (1:500), BCθ (12 μg/mL), or with nonspecific mouse IgM (1:1250), mouse IgG (1:500), or rat IgG (1:625) as controls. On day 2, the sections were washed five times with wash solution for 10 minutes and incubated for 1 hour at room temperature with the secondary antibodies Texas Red-conjugated goat anti-mouse IgM (1:150), FITC goat anti-mouse IgG (1:200), Alexa Fluor 488 goat anti-rat IgG (1:150), and/or Alexa Fluor 488-conjugated streptavidin (1:750). Sections were then washed 3 × 10 minutes in PBS and mounted as previously described for filipin labeling, using Prolong Antifade with or without DAPI (Molecular Probes P36935 with DAPI). A three-step indirect detection method was used when using anti-GM3 mAb. Sections were washed 4 × 10 minutes in wash solution after secondary incubation and were subsequently incubated with tertiary antibody [Alexa Fluor 488-conjugated rabbit anti-FITC (1:80)] for 1 hour at room temperature. Sections were washed 4 × 10 minutes in wash solution and incubated with 0.005% Filipin complex or dimethyl sulfoxide for 20 minutes at room temperature. Again, sections were washed 3 × 10 minutes in PBS and mounted as previously described. Double-labeling experiments using BCθ in combination with anti-GM1, anti-GM2, or anti-GM3 were performed as previously described, except all solutions were made with reduced fatty acid BSA. Confocal images were obtained with either an Olympus Fluoview 500 (Olympus America, Center Valley, PA) or a Zeiss 510 Duo V2 system laser scanning microscope using a 40× (1.1 NA) or 63× (1.4 NA) objective (Carl Zeiss AG, Oberkochen, Germany). A zoom setting of 3 was used for higher magnification images. Metamorph software (Molecular Devices, Sunnyvale, CA) was used to process images and assess degree of co-localization.
Electron Microscopy Procedures
Analyses were performed using published procedures.15,23 Tissues were postfixed in 2% glutaraldehyde, dehydrated, and embedded in Epon (Electron Microscopy Sciences, Hatfield, PA). Thin sections were cut on a Sorvall ultramicrotome (Dupont Instruments, Cincinnati, OH) and examined on a Philips CM10 electron microscope (FEI, Hillsboro, OR).
Biochemical Analysis of Gangliosides and Neutral Glycolipids
Analyses were performed on frozen cerebral hemispheres using procedures similar to those used in previous studies.13,18 Briefly, total lipids were extracted from a 20% tissue homogenate in water with chloroform:methanol 1:2 (v/v).26 Part of the extract was desalted and separated into two fractions using 100 mg reverse-phase Bond Elut C18 (Agilent Technologies, Massy, France) columns.27 Neutral glycosphingolipids were studied after saponification of the chloroform-methanol 1:2 (v/v) fraction.26 The acidic lipid fraction, eluted with methanol-water 12:1 (v/v), contained all of the gangliosides and was used without further purification. Total sialic acid was measured by the Svennerholm's resorcinol-HCl method as described.26 A suitable aliquot of the extract (1.5 to 3 mg tissue equivalent) was spotted using a CAMAG Linomat 4 device on high-performance thin layer chromatography silica gel 60 plates developed with chloroform-methanol-0.2% CaCl2 55:45:10 (v/v/v) and sprayed with resorcinol-HCl reagent to visualize the sialic acid moiety of individual gangliosides. Densitometric quantification at 580 nm was done using a CAMAG TLCII scanner equipped with the Cats software (CAMAG, Muttenz, Switzerland). After normalization to the total sialic acid content, individual concentrations were calculated by taking into account the number of sialic acids for each ganglioside.
Results
Gangliosides Govern the Extent of Sequestration of Unesterified Cholesterol in Most Neurons Lacking NPC2
Previous studies established that major storage products in neurons in NPC disease include several glycosphingolipids (GM2 and GM3 gangliosides; and possibly GM1; and also lactosylceramide and glucosylceramide), as well as unesterified cholesterol.1–3,16,28 Although most studies support a primary role for NPC1 and NPC2 proteins in cholesterol transport from LEs/LYs, two independent reports18,19 have shown that cholesterol storage within neurons of Npc1−/− mice could be dramatically reduced by limiting synthesis of complex gangliosides. This finding suggested a pivotal role for sphingolipid processing and storage in determining the extent of cholesterol accumulation in NPC disease.19 In the present work, we have extended these studies by investigating whether NPC disease caused by deficiency of NPC2, a well-documented cholesterol-binding protein, would exhibit the same ganglioside-dependence in terms of cholesterol sequestration.
Analogous to previous experiments with NPC1, we crossed mice with NPC2 disease to others lacking GalNAcT (B4galnt1), a critical enzyme for the synthesis of all complex gangliosides (Figure 1). The Npc2−/−;B4galnt1−/− double mutants allowed to survive to end-stage disease revealed a pattern of clinical disease onset, progression, and duration similar to Npc2−/− mutants, reaching end-stage disease at 14 to 15 weeks of age. As previously reported,18,29 B4galnt1−/− mice at this age resembled WT and only later developed tremors related to peripheral nervous system disease.
Ganglioside patterns in brain tissue from end-stage Npc2−/−;B4galnt1−/− mice were compared to those in age-matched single mutants and WT mice. The biochemical data (ganglioside profiles) are depicted in Table 1 and Figure 2A. In single Npc2−/− mutants, gangliosides GM3 and GM2, normally very minor components, were markedly increased, as reported previously.13 As expected, in the B4galnt1−/−, but also the double mutant (Npc2−/−;B4galnt1−/−) mice, only GM3 and GD3 and trace amounts of GT3 gangliosides were present, with GD3 and GT3 appearing in both O-Ac and N-Ac forms.18,29 When quantified from the total and relative lipidic sialic acid values, the concentrations of GM3 ganglioside reached 13 times the normal (WT) value in Npc2−/− mutants, and as much as 21 times the normal value in the B4galnt1−/− mice. Double-mutant mice exhibited GM3 levels that were close to those in the B4galnt1 single mutants, with a nonsignificant trend toward a slight further increase (to 23 times the normal value). Total ganglioside levels in the double mutants, interestingly, resembled those of WT and B4galnt1−/− mice, which were essentially equivalent, rather than those of Npc2−/− mutants, indicating that the lack of complex ganglioside production led to an overall reduction in ganglioside accumulation.
Table 1.
Concentrations of Main Individual Gangliosides in Brain Tissue from Normal Mice
| Wild type | Npc1−/− | Npc2−/− | B4galnt−/− | Npc2−/−; B4galnt−/− | St8sia1−/− | Npc1; St8sia1−/− | β-Gal−/− | Npc1−/−; β-Gal−/− | |
|---|---|---|---|---|---|---|---|---|---|
| (n = 7) | (n = 5) | (n = 3) | (n = 3) | (n = 7) | (n = 3) | (n = 3) | (n = 2) | (n = 3) | |
| Age range (days) | 54 to 108 | 54 to 77 | 93 to 108 | 93 to 108 | 93 to 108 | 54 to 62 | 54 to 62 | 60 to 68 | 60 to 68 |
| GM3 | 32 ± 4 | 448 ± 67 | 414 ± 42 | 688 ± 50 | 749 ± 72 | 60 ± 4 | 701 ± 41 | 49 | 57 ± 8 |
| GM2 | 18 ± 2 | 484 ± 60 | 429 ± 35 | — | — | 25 ± 5 | 709 ± 31 | 20 | 30 ± 11 |
| GD3-O-Ac | — | — | — | 317 ± 12 | 337 ± 18 | — | — | — | — |
| GM1 | 324 ± 77 | 589 ± 107 | 457 ± 19 | — | — | 645 ± 55 | 961 ± 41 | 2162 | 4100 ± 257 |
| GD3 | 79 ± 13 | 107 ± 12 | 120 ± 120 | 969 ± 17 | 1066 ± 37 | — | — | 103 | 171 ± 6 |
| GD1a | 562 ± 44 | 708 ± 62 | 560 ± 28 | — | — | 1201 ± 33 | 1293 ± 98 | 697 | 901 ± 93 |
| GD2 | 75 ± 9 | 96 ± 19 | 107 ± 16 | — | — | — | — | 106 | 168 ± 19 |
| GD1b + unidentified | 266 ± 45 | 303 ± 52 | 280 ± 18 | — | — | — | — | 361 | 506 ± 35 |
| GT1b+GQ | 354 ± 19 | 384 ± 82 | 360 ± 7 | 464 | 578 ± 43 | ||||
| Other | — | — | — | 60 ± 8 | 65 ± 10 | 126 ± 16 | 177 ± 25 | — | — |
Animals included NPC mice (Npc1−/−and Npc2−/− single mutants), mutant mice devoid of a-series (B4galnt−/−) or b-series (St8sia1−/−) gangliosides, β-galactosidase-deficient mice (β-gal−/−), and NPC double mutants (Npc2−/−; B4galnt−/−) (Npc1−/−; St8sia1−/−) (Npc1−/−; β-gal−/−). The results are expressed as nmol of ganglioside per g wet weight (mean ± SD when n > 2).
Figure 2.
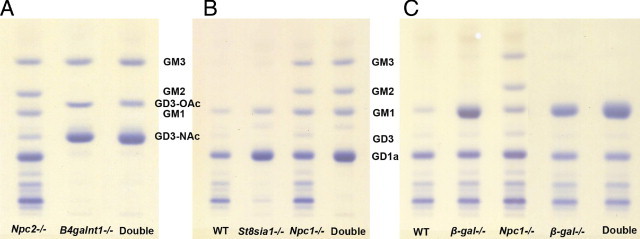
Chromatographic profiles of gangliosides in cerebral hemispheres of the 3 sets of double mutants used in this study. A: Results of the Npc2;B4galnt− double mutant study with lane 1 showing Npc2−/− mice with typical elevations of GM2 and GM3 gangliosides,13 lane 2 showing B4galnt1−/− mice, and lane 3 showing Npc2−/−;B4galnt1−/− double mutant mice. Both of the latter show an absence of GM2 and higher a-series gangliosides with abundance of GD3 in O-Ac and N-Ac forms. These findings are essentially the same as those reported for Npc1−/−;B4galnt1−/− double mutant mice.18B: Results of the Npc1;St8sia1 double mutant study with lane 1 showing WT, lane 2 showing St8sia1−/− mice, lane 3 showing Npc1−/− mice, and lane 4 showing Npc1−/−;St8sia1−/− double mutant mice. Note the persistence of only ‘a’ series gangliosides in the double mutant mice. C: Results of the Npc1−/−;β-gal−/− double mutant study with lane 1 showing WT, lanes 2 and 4 showing two different β-gal−/− mice, lane 3 showing a single Npc1−/− mutant, and lane 5 showing the Npc1−/−;β-gal−/− double mutant mice. Note the absence of GM2 and GM3 in the double mutants and the relative increase in GM1 in the double mutant compared to its adjacent Npc1 mutant. An equivalent of 3 mg wet weight tissue was spotted for all samples except for lanes 4 and 5 in C for which 1.5 mg was spotted.
Brain tissue from the Npc2−/−;B4galnt1−/− double-mutant mice was also analyzed morphologically (Figure 3). As expected, filipin staining for unesterified cholesterol revealed extensive labeling within cells of the cerebral cortex of Npc2−/− mice and essentially no labeling in WT (Figure 3A) and B4galnt1−/− mice (not shown) as expected.13,19 Similarly, immunoperoxidase labeling revealed that GM3 ganglioside storage was readily detectable in vesicular structures in cortical neurons of Npc2−/− mutants, but not in WT (Figure 3A) or B4galnt1−/− mice (not shown). Immunoperoxidase labeling for GM2 ganglioside in cerebral cortex, as previously reported,13 revealed no staining in B4galnt1−/− mice, little to no staining in WT mice, and abundant vesicular GM2 immunoreactivity in Npc2−/− mutants. In Npc2−/− mice, consistent with an earlier study,13 analysis of GM2, GM3, and cholesterol distribution across different neuronal populations showed evidence of differential distribution of these storage compounds. In cerebral cortex, cholesterol (Figure 3A) and GM2 ganglioside was elevated in neurons in both supra- and infra-granular layers, whereas GM3 predominated in infra-granular neurons (Figure 3A). In hippocampus, GM2 and cholesterol were again found to predominate in vesicular structures within neurons in all areas (fascia dentata and areas CA1 to CA4) (Figure 3B), whereas detectable GM3 accumulation was limited to cells in CA3 and CA4 (not shown). Similarly, although GM2 and cholesterol accumulation was evident in Purkinje cells of Npc2−/− mice, GM3 storage was not detectable in these cells (Figure 3C).
Figure 3.

Immunoperoxidase and filipin labeling of different brain regions in the Npc2;B4galnt1 double mutant study. A: cerebral cortex. Filipin staining (white labeling in cells) for cholesterol (left column) and immunostaining (brown labeling in cells) for GM3 ganglioside (right column) in Wt, Npc2−/−, and Npc2−/−;B4galnt1−/− (DKO) mice. WT brain showed no cholesterol or GM3 staining, as expected. NPC2 brain showed an abundance of both cholesterol and GM3, both of which were significantly lower in Npc2−/−;B4galnt−/− (DKO) DKO brain. Note that the GM3 immunostaining in panels for NPC2 and DKO mice includes minor background microglial and blood vessel labeling likely due to the presence of residual endogenous peroxidases. B: CA1 region of the hippocampus. Filipin and immunoperoxidase staining for GM2, GM3, and GD3 gangliosides in WT, Npc2−/−, and Npc2−/−;B4galnt1−/− (DKO) mice. Note the abundance of cholesterol and GM2 storage in CA1 pyramidal neurons of the Npc2−/− mouse (double arrows) and the relative absence of cholesterol storage in the DKO mouse coincident with the absence of GM2. Occasional filipin-positive cells in CA1 of this mouse correlate with rare GM3-positive neurons in this same area (single arrows); no GD3 storage was evident in this region in any of the mice. C: Purkinje cells of the cerebellum. Filipin and immunoperoxidase staining for GM2, GM3, and GD3 gangliosides in WT, Npc2−/−, and Npc2−/−;B4galnt1−/− DKO mice. Both cholesterol and GM2 storage is abundant in Purkinje cells of the Npc2−/− mouse and is, as expected, absent in WT (arrows); the DKO mouse still shows abundant filipin labeling but an absence of GM2, GM3 or GD3 storage (arrows). Note the presence of some minor background astroglial fiber staining with the GM2 antibody23 used in these studies. All immunoperoxidase images are Nissl counterstained; Calibration bar for all panels equals 75 μm.
In Npc2−/−;B4galnt1−/− double mutants, the absence of GM2 and complex gangliosides was accompanied by significant reduction of filipin labeling for unesterified cholesterol in most brain regions, including cerebral cortex and hippocampus (Figure 3, A and B). The loss of GM2 storage also did not lead to compensatory increases in vesicular GM3 or GD3 staining. For example, neurons in the CA1 region of the hippocampus, which exhibited GM2, but not GM3 accumulation in single Npc2−/− mice, did not reveal evidence for GM3 or GD3 storage in the double mutants (Figure 3B). Reductions in cholesterol sequestration in neurons of the double mutants also varied by brain region, with neurons storing GM3 ganglioside in all areas consistently possessing filipin-positivity. Thus, although cholesterol staining remained evident in the infra-granular cerebral cortex and in the CA3 and CA4 regions of the hippocampus (coincident with persisting GM3 storage), it was now minimal to absent in the supragranular cerebral cortex and CA1 region of the hippocampus where GM3 storage was also barely detectable (Figures 3, A and B). Thus, in neurons of both the cerebral cortex and hippocampus, the reduction of cholesterol sequestration in double mutants consistently coincided with the knockout-induced absence of GM2 and a concomitant lack of GM3 (and GD3) storage. On the other hand, neurons continuing to exhibit GM3, such as those in the lower cerebral cortex (Figure 3A), also continued to exhibit cholesterol storage. Interestingly, although GD3 (the only other abundantly expressed ganglioside in the Npc2−/−;B4galnt1−/− double mutants) could be found as vesicular staining in many neurons of these mice, it was not evident in neurons of the CA1 region of the hippocampus (Figure 3B) or in the upper cerebral cortex (not shown). These findings as a whole are fully equivalent to those reported for Npc1−/−; B4galnt1−/− double mutants.19 Together, they provide compelling support to the view that the limiting ganglioside storage concomitantly limits that of cholesterol in most neurons in NPC disease, and that overall the cholesterol sequestration in this disorder is tightly coupled to vesicular ganglioside accumulation.
A single, notable exception to the link between cholesterol and GM2 and GM3 ganglioside sequestration occurred in the cerebellum. Here, Purkinje cells, which in Npc2−/− mutants accumulate cholesterol and GM2, but not GM3 (Figure 3C), showed a persistent cholesterol accumulation in the double mutants, despite essentially no detectable GM3. We also examined Purkinje cells in the Npc2−/−;B4galnt1−/− double mutants for evidence of GD3 accumulation, but little or no vesicular labeling for this ganglioside was detected (Figure 3C). These studies suggest that Purkinje cells, unlike other CNS neurons examined, undergo cholesterol sequestration in the absence of comparable ganglioside accumulation.
Importantly, all of the previously mentioned changes for the Npc2−/−;B4galnt1−/− double mutants essentially replicate those observed in the same cross involving Npc1−/− mutants,18,19 consistent with the close similarity between metabolic abnormalities caused by defects in either the NPC1 or NPC2 protein.13
Gangliosides Synthesized in the a-Series Pathway Exhibit Greatest Influence over Cholesterol Sequestration in Neurons Lacking NPC1
Absence of GD3 synthase in mice results in an inability to synthesize complex gangliosides of the b- and c- series, but retention of synthesis of all gangliosides in the a-series (Figure 1A), including GD1a and GM1, two of the most abundant brain gangliosides, as well as GM2 and GM3, the two major storage gangliosides in NPC disease. A key question in this study was whether limiting expression to a-series gangliosides alone would change the pattern or extent of cholesterol sequestration in neurons.
Npc1−/−;St8sia1−/− double-mutant mice allowed to survive to end-stage disease revealed clinical disease progression that appeared slightly more severe than the single Npc1−/− mutants. The four double mutants available in this study reached end-stage disease by 54 to 62 days of age compared with 70 to 77 days for Npc1−/− mice. St8sia1−/− mice, as reported previously,22 did not show evidence of clinical disease.
The brain ganglioside profiles of mice with the four genotypes are shown in Figure 2B and Table 1. The pattern observed in the Npc1−/− mice was essentially identical to that previously described for the Npc2−/− mice, qualitatively and quantitatively. In the St8sia1−/− mice, GD1a was the predominant ganglioside, followed by GM1 and absence of gangliosides from the b-series, in good accordance with published data.22 GM2 and GM3 gangliosides were present in minute, normal amounts. The brain ganglioside profiles of the Npc1−/−;St8sia1−/− double mutant mice resembled those seen in the St8sia1−/− mice, however with a major difference, namely that the concentrations of GM2, GM3, and GM1 gangliosides were very elevated, reaching even higher levels than observed in the Npc1−/− single mutant mice (Table 1). Thus, unlike the Npc2−/−;B4galnt1−/− double mutants, which showed no evidence of ganglioside elevations beyond that of WT or B4galnt1−/− mice, the Npc1−/−;St8sia1−/− double mutants did exhibit ganglioside storage that could be attributed principally to the increases in monosialogangliosides.
Immunocytochemical staining for GM2 and GM3 gangliosides in the cerebral cortex of 8- to 10-week-old St8sia1−/− mutant mice used in this study (Figure 4A) resembled that seen in WT. The Npc1−/−;St8sia1−/− double-mutant mice revealed an abundance of storage of both GM2 and GM3 gangliosides and filipin labeling showed accumulation of unesterified cholesterol in cells throughout the cerebral cortex (Figure 4A) and elsewhere (including hippocampus and cerebellum; not shown) that was indistinguishable from Npc1−/− mice. These findings indicate that a-series gangliosides alone are sufficient to generate the NPC-cellular phenotype, including widespread intraneuronal cholesterol accumulation.
Figure 4.
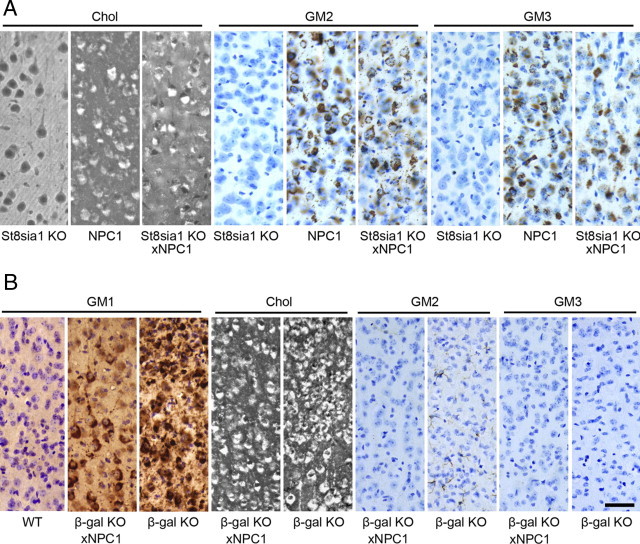
Immunoperoxidase and filipin labeling of cerebral cortex in the Npc1;St8sia1 and Npc1;βgal double mutant studies. A: Filipin staining (white labeling in cells) for cholesterol and immunostaining (brown labeling cells) for GM2 and GM3 gangliosides in St8sia1−/−, Npc1−/−, and the Npc1−/−;St8sia1−/− double mutant. Note the St8sia1−/−appeared similar to WT in Figure 3, whereas the Npc1−/− and Npc1−/−;St8sia1−/− double mutants showed abundant staining for filipin and GM2 and GM3 gangliosides. B: Filipin staining for cholesterol and immunostaining for GM1, GM2 and GM3 gangliosides in WT, Npc1−/−;βgal−/− double mutant and βgal−/− single mutant mice. Note the abundance of labeling for cholesterol and GM1 in both the double and single mutants, and the absence of GM2 and GM3 in these mice. All immunoperoxidase images are Nissl counterstained. Calibration bar in B equals 50 μm and applies to all images.
GM2 and GM3 Gangliosides and Cholesterol Are Not Sequestered in the Same Storage Vesicles in Neurons of Npc1−/− or Npc2−/− Mice
The previous studies are consistent with gangliosides having significant influence over the sequestration of cholesterol in NPC disease and suggested that the relationship between the various storage compounds needed closer examination. Confocal microscopy was used to examine the location of each storage compound. Combined immunofluorescence for GM2 and GM3 gangliosides in mice lacking either the NPC1 or NPC2 protein revealed a similar distribution of individually-labeled punctate throughout neuronal cell bodies, but with the two gangliosides exhibiting almost no overlap (Figure 5, A and B). NPC1-deficient neurons also exhibited occasional punctate vesicular labeling for GM1 ganglioside and also here these appeared independent of those labeling for GM2 ganglioside (Figure 5C). We also analyzed the intraneuronal accumulation of gangliosides in NPC1-deficient neurons relative to that of cholesterol (labeled with either filipin or BCθ). No significant overlap was found between cholesterol and GM2 (Figure 5D) or GM3 (Figure 5E), whereas GM1 and cholesterol were occasionally co-localized (Figure 5F). However, even for the latter significant areas of cholesterol sequestration showed no evidence of accompanying GM1 accumulation.
Figure 5.
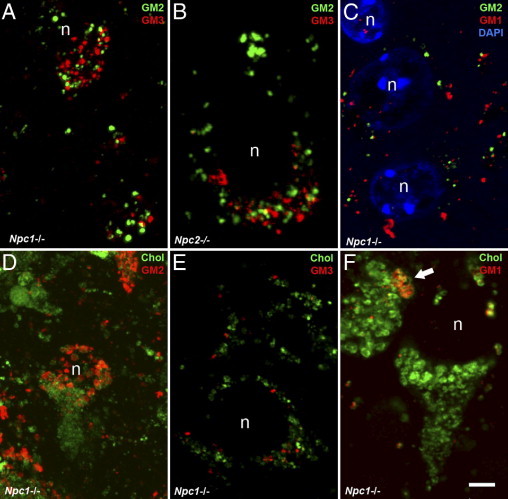
Confocal/immunofluorescence analysis of the localization of GM2, GM3, and GM1 gangliosides and cholesterol within neurons of Npc1−/− and Npc2−/− mice. A-B: Vesicular accumulation of GM2 (green label) and GM3 (red label) is abundant in cortical pyramidal neurons of Npc1−/− (A) and Npc2−/− (B) mutant mice but only very rare sites of co-localization are apparent (yellow label). C: Vesicular accumulation of GM2 (green label) and GM1 (red label) in cortical pyramidal neurons of an Npc1−/− mouse also shows little to no visible overlap; blue label for DAPI indicate cell nuclei. D-F: In NPC1−/− neurons, vesicular accumulation of gangliosides (red label) and cholesterol using BCθ (green label) reveals no major sites of co-localization between sequestered cholesterol and GM2 (D) or GM3 (E), and at most partial overlap (yellow label) for GM1 (arrow). Nucleus, n. Calibration bar in F equals 5 μm and applies to all images.
Accumulation of GM2 and GM3 Gangliosides, and Lactosylceramide, in NPC Disease Results Exclusively from the Degradation of Complex Gangliosides in the E/L Pathway
That GM2 and GM3 gangliosides in Npc1−/− and Npc2−/− mice showed little or no co-localization (Figure 5, A and B) suggested that independent mechanisms may be responsible for their sequestration. This possibility was reinforced by studies showing that lack of GM2 in the Npc2−/−;B4galnt1−/− mice failed to cause any compensatory increase in storage of GM3 (Table 1; Figures 2B and 3A). A possible explanation for this apparent independence is that one of the two gangliosides (eg, GM2) could be elevated within the glycosphingolipid synthetic pathway and detected in the Trans Golgi Network (TGN) and/or exocytic vesicles destined for the plasmalemma, with the other (eg, GM3) being generated in LEs and LYs by the action of GSL degradative enzymes. To test this possibility, mice lacking acidic β-galactosidase (β-gal−/−) and therefore unable to degrade GM1 ganglioside in the endosomal/lysosomal (E/L) system were crossed with mice lacking NPC1. If either GM2 or GM3 ganglioside was being generated through synthesis and vesicular packaging at the Golgi-TGN, these ganglioside-laden vesicles would not be expected to be affected by a degradative block in the E/L system. If, however, either ganglioside was being generated specifically in the E/L system, this staining would be eliminated because the degradation of GM1 ganglioside to GM2 requires the presence of β-gal (Figure 1B).
Npc1−/−;β-gal−/− double mutants allowed to survive to end-stage disease revealed a pattern of disease onset and progression that was more severe than the NPC1 mutation alone (9 to 10 weeks versus 11 weeks for Npc1−/−). The β-gal−/− mice, as reported previously21, generally develop a neurological phenotype at 4 months of age followed by death by 6 to 7 months.
Biochemical analysis of ganglioside profiles in brain tissue from the β-gal−/− mice, as expected, showed an overwhelming storage of GM1, whereas the concentrations of GM2 and GM3 were essentially normal (Table 1 and Figure 2C). In the end-stage Npc1−/−;β-gal−/− mice, the profile was very similar to that in the single β-gal−/− mutant. As shown from both the chromatographic separation (Figure 2C) and the quantitative data (Table 1), and at variance with the findings in the Npc1−/−;St8sia1−/− mice, there was no increase of GM2 or GM3 ganglioside [or of lactosylceramide (data not shown)], in this double mutant. The biochemical findings were confirmed with immunocytochemical labeling for GM2 and GM3 gangliosides, as neither was detectable in the double-mutant animals (Figure 4B). These data indicated that the presence of both GM2 and GM3 gangliosides in vesicles in NPC1 disease is the result of degradation of GM1 ganglioside in the E/L pathway and that synthesis and packaging in exocytic vacuoles at the Golgi/TGN does not contribute to the presence of GM2- or GM3-labeled vesicle populations in Npc1−/− mice. Cellular GM1 and cholesterol accumulation revealed by IHC, as expected, was abundant in all of the disease models in this experiment (Figure 4B). Indeed, as revealed by both the immunoperoxidase labeling and biochemical analysis (Table 1; Figure 4B), storage of GM1 ganglioside was significantly higher in Npc1−/−;β-gal−/− mice than in either single mutant. Asialo-GM1 was elevated in the GM1 mutants and in the double mutants to the same degree [ie, there was no further increase in the latter (not shown)]. As discussed below, an increase in GM1 in the double-mutant mice may be revealing an important role for NPC1 in salvaging simple gangliosides from the E/L system.
We also performed ultrastructural studies to characterize the morphology of storage bodies in the Npc1−/−;β-gal−/− double mutants compared with single mutants. As expected, neurons in the β-gal−/− mutant mice exhibited the presence of membranous cytoplasmic bodies and Npc1−/− mice polymorphous cytoplasmic bodies (Figure 6A–B), as previously reported.13,21 These structural differences appear to reflect the underlying molecular defects in that lack of NPC1, which is believed to lead to failure of egress of materials from LEs and LYs, resulting in storage bodies resembling defective multivesicular bodies, whereas the β-gal defect leads to failure to degrade GM1 ganglioside and the presence of ganglioside-enriched vesicles. Ultrastructural analysis of the double-mutant mice revealed hybrid storage bodies that most closely resembled those in NPC disease (Figure 6C) (ie, the multivesicular-like morphology of the inclusion bodies persisted).
Figure 6.
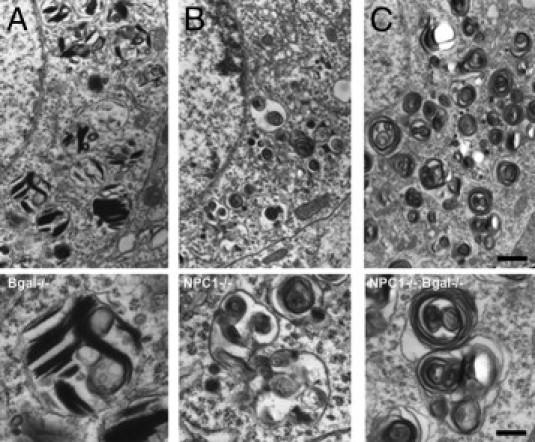
Electron microscopic analysis of the ultrastructure of storage body inclusions in cortical neurons of βgal−/−, Npc1−/−, and Npc1−/−;βgal−/− double mutant mice. A: Inclusion bodies from a βgal−/− mouse showing the presence of vesicular structures containing membranous profiles consistent with GM1 storage; Higher magnification image below. B: Inclusion bodies from a Npc1−/− mouse showing the presence of polymorphous cytoplasmic bodies13 characteristic of this disease; Higher magnification image below. C: Inclusion bodies from a Npc1−/−;βgal−/− double mutant showing predominance of the abnormal multivesicular body morphology closely resembling that at the Npc1−/− mutant; Higher magnification image below. Calibration bar in C equals 0.82 μm and also applies to A-B; bar in micrograph below C equals 0.25 μm and also applies to the other two higher magnification images.
Cholesterol and Ganglioside Storage in NPC Disease Occurs Predominately but Not Exclusively within LAMP2-Positive Vesicles
To further characterize the location of sequestered gangliosides and cholesterol, and to confirm the results of the Npc1−/−;β-gal−/− double-mutant study, we next analyzed the subcellular location of three gangliosides (GM2, GM3, and GM1) and cholesterol using antibodies to LAMP2, a known marker of LEs/LYs. Consistent with the double-mutant study, most individual sites of ganglioside labeling were surrounded by LAMP2-positve profiles (Figure 7, A–C) indicating that the ganglioside was sequestered within LEs or LYs. Similarly, filipin labeling with LAMP2 revealed the presence of unesterified cholesterol within LAMP2-positive vesicles (Figure 7D). However, in each case a small proportion of ganglioside- and cholesterol-positive punctae in neurons showed no associated LAMP2, suggesting involvement of vesicles other than LEs and LYs. Given that cholesterol and GM1 are believed to be endocytosed from the neuronal plasmalemma their presence in LAMP2-negative vesicles, such as early endosomes or recycling endosomes would be anticipated. However, because GM2 and GM3 gangliosides are products of GM1 and GM2 degradation, respectively, a process generally believed to occur exclusiveld in LEs/LYs, the presence of these two simple gangliosides in occasional LAMP2-negative vesicles was unexpected and implicates involvement of early endosomal compartments in the disease process.
Figure 7.
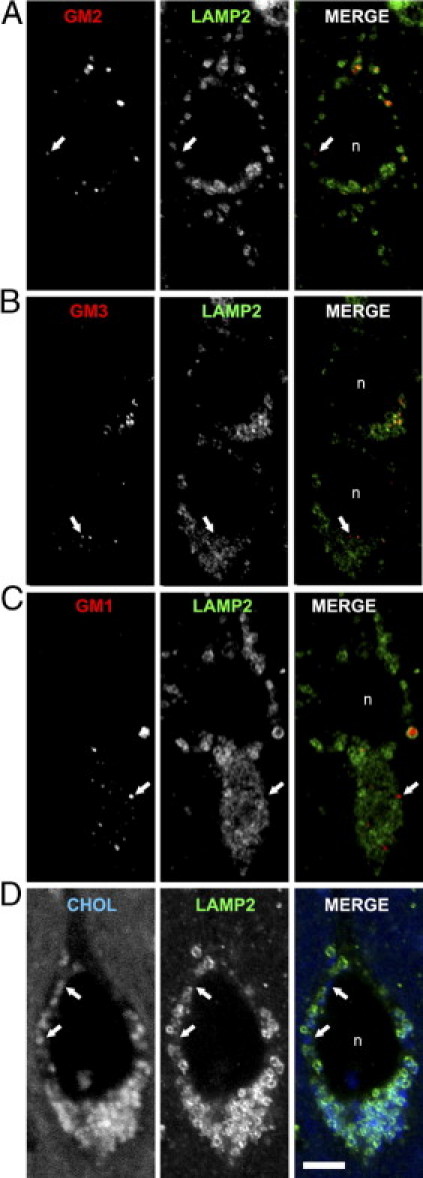
Confocal/immunofluorescence analysis of the localization of GM2, GM3, and GM1 gangliosides (red) and cholesterol (blue) relative to LAMP2 (green) within individual neurons of Npc1−/− mice. A: GM2 and LAMP2. B: GM3 and LAMP2. C: GM1 and LAMP2. D: Cholesterol (filipin) and LAMP2. Note that while there is extensive overlap between LAMP2 and these storage materials, there are also examples of ganglioside- and cholesterol-positive vesicles that are LAMP2 negative (indicated by arrows in each panel). Nucleus, n. Calibration bar in D equals 5 μm and applies to all images.
Discussion
NPC disease, whether caused by defects in NPC1 or NPC2 proteins, is characterized by chronic accumulation of unesterified cholesterol and sphingolipids, progressive neurological dysfunction, and premature death. Although elevations of cholesterol seen in liver and spleen do not occur in cerebral gray matter,1 numerous studies using filipin and BCθ as probes have documented its sequestration within neurons.18,19,23,25 The lack of total brain cholesterol increase detected biochemically, coupled with its widespread intraneuronal storage, suggests that cholesterol distribution is abnormal in NPC disease. Consistent with this are studies showing that while cholesterol is abundant in NPC neuronal cell bodies, it is diminished in axons.30 Sequestration of unesterified cholesterol in late endosomes and lysosomes in NPC disease is consistent with the discovery that NPC1 and NPC2 are cholesterol-binding proteins acting in a common processing pathway responsible for cholesterol egress.5–7,11–13 Failure to traffic cholesterol out of late endosomes/lysosomes not only leads to its sequestration within these organelles, but also results in significant increases in cholesterol biosynthesis in some cell types,31 presumably further exacerbating cholesterol storage.
In addition to cholesterol, GM2 and GM3 gangliosides and two neutral glycosphingolipids (lactosylceramide and glucosylceramide) have long been known to be prominent storage compounds in NPC brain.1,2,13,23 Although studies in yeast have suggested a role for NPC1 homologues in glycosphingolipid efflux,32 studies directly linking NPC1 or NPC2 function to glycosphingolipid trafficking in mammalian cells are lacking. Glycosphingolipids are synthesized in the Golgi/TGN and trafficked to the plasmalemma.17,33 After endocytosis from the plasmalemma and degradation in the endosomal/lysosomal system, simple glycosphingolipids (such as GM2 and GM3) are salvaged by poorly understood mechanisms and transported to the Golgi where they are used for synthesis of higher order gangliosides.34 In mature neurons, this process is believed to be the predominate source of precursors for the synthesis of complex glycosphingolipids rather than complete de novo synthesis. Thus, as for cholesterol, there is the possibility that ganglioside storage in NPC disease triggers increases in glycosphingolipid synthesis, followed by a further exacerbation of the storage process.23,35
The purpose of the present study was to expand our understanding of the relationship between the sequestration of cholesterol and gangliosides in neurons in NPC disease relative to the functions of NPC1 and NPC2 proteins. Our studies show that mice lacking the NPC2 protein and GalNAcT, and therefore unable to generate complex gangliosides, exhibit dramatically reduced cholesterol storage in most neurons, just as it was found in Npc1−/− mice lacking this enzyme.8,19 Cholesterol sequestration remaining in neurons of Npc2−/−;B4galnt1−/− mice (with the notable exception of Purkinje cells, see as follows) consistently coincided with the presence of GM3 storage, again similar to Npc1−/−;B4galnt1 −/− mice.19 However, when glycosphingolipid synthesis in Npc1−/− mice was restricted to a-series (and asialo) gangliosides by generating Npc1−/−;St8sia1−/− double mutants, cholesterol sequestration in neurons resembled that of single NPC mutants, suggesting links between the storage of cholesterol and a-series gangliosides (Figure 4).
Biochemical studies of double mutants similar to the ones used in the current study, including Npc1−/−;B4galnt1−/− mice, were recently shown to exhibit no cholesterol reductions in visceral organs, such as liver despite the absence of complex gangliosides.36 Similarly, brain tissue from the double mutants also failed to show cholesterol reductions, a finding believed to indicate lack of linkage between cholesterol and ganglioside storage. However, as is well documented, total brain cholesterol is not elevated in NPC disease.1 Indeed, in NPC mouse brain, interpretation of cholesterol levels is difficult because these mice also show a loss of myelin lipids, and analyses are typically made on mixtures of unmyelinated and myelinated tissue. However, above all, whole brain biochemical analyses cannot disclose abnormalities in distribution within cells. Using filipin and BCθ to label unesterified cholesterol in neuronal cell bodies, we show that reduction in complex gangliosides in brain significantly lowers the amount of cholesterol labeling in most neurons. Although consistent with cholesterol sequestration being dependent on storage of simple gangliosides, such as GM2 or GM3, this finding does not necessarily argue against a primary role for NPC1 to NPC2 proteins in intracellular cholesterol trafficking. Reductions in cholesterol sequestration reported here could occur if the trafficking of complex gangliosides in neurons influences the delivery of cholesterol to LEs/LYs for processing. Such a mechanism is plausible because both complex gangliosides and cholesterol are constituents of membrane microdomains (rafts)37, and therefore presumably share common pathways for endocytosis. Alternatively, gangliosides in LEs/LYs may influence cholesterol egress from these compartments. Regardless of the mechanism, the current studies clearly show that in most CNS neurons the degree of cholesterol sequestration in NPC disease is significantly influenced by the ability of the neuron to synthesize complex gangliosides.
There was one notable exception to the ability of gangliosides to influence cholesterol sequestration in NPC disease (ie., Purkinje cells). Here, cell bodies exhibited conspicuous cholesterol sequestration, even in the absence of any detectable ganglioside in the double-mutant mice (Figure 3C). This finding suggests that Purkinje cells may differ from most neurons and share characteristics of cholesterol-ganglioside homeostasis with non-neuronal cells (eg, hepatocytes) in terms of and NPC1-2 function. Interestingly, Purkinje cells are also one of the most vulnerable cell types in NPC disease, showing earlier death than most other types of neurons.23
In the current study, we also show that accumulation of GM2 and GM3 gangliosides in both the Npc1−/− and Npc2−/− mice largely occurs in separate vesicle populations within the same neurons (Figure 5). Furthermore, GM2 and cholesterol were not co-localized in the Npc1−/− mice. Both of these findings replicate the situation described in several types of mucopolysaccharidose disease, conditions in which ganglioside storage is believed secondary, and cholesterol tertiary, to glycosaminoglycan accumulation.15,38 The lack of GM2 to GM3 co-localization in Npc1−/− and Npc2−/− mice suggested to us that the two gangliosides might be generated independently of one another (eg, one in exocytic vesicles after synthesis in the Golgi/TGN, and the other in lysosomes after degradation of complex gangliosides). By making Npc1−/−;β-gal−/− double mutants, we were able to answer this question with the absence of GM2 and GM3 storage in vesicles in the double mutants, revealing that accumulation of both was dependent on normal processing of GM1 ganglioside within the endosomal/lysosomal system (Figure 4). Interestingly, ultrastructural examination of storage vacuoles in these double mutants revealed a persistence of NPC-like polymorphous cytoplasmic bodies rather than membranous cytoplasmic bodies, which are characteristic of GM1 gangliosidosis (Figure 6). This suggests that factors leading to the generation of abnormal multivesicular bodies in NPC disease have an overriding effect relative to major stored substrates (cholesterol and GM1 ganglioside).
The presence of GM2 and GM3 (as well as GM1), and cholesterol in separate vesicles in the E/L system in the Npc1−/− and Npc2−/− mice, although unanticipated, is consistent with studies showing considerable heterogeneity of endosomal processing.39 Studies here also indicated that a small proportion of sequestered gangliosides and cholesterol were in LAMP2-negative vesicles (Figure 7), consistent with localization outside of LEs and LYs (possibly in EEs or REs), similar to that reported in mucolipidosis IV disease.40 Given the importance of these latter sites for a variety of signal transduction and processing events,41–44 better understanding of their possible involvement in storage diseases will be important and may provide insight into features, such as ectopic dendritogenesis, neuroaxonal dystrophy, and selective neuron death.30,45
One additional finding from the current studies was the significant increase in GM1 found in the brains of Npc1−/−;β-gal−/− mice compared to either single mutant mice. As illustrated in Figure 8, one simple interpretation of the increased GM1 storage is that these double mutants now lack two critical pathways (ie, lysosomal degradation and salvage) essential for glycosphingolipid processing and recycling; thus, neurons accumulate GM1 ganglioside in a manner equivalent to the sum (or greater) of GM1 stored in either single mutant alone. This finding suggests that the retroendocytic trafficking made possible by NPC1 and NPC2 proteins facilitates the salvage of simple gangliosides, and is a critical element in overall ganglioside homeostasis in neurons. It may also explain why so much GM2 and GM3, as well as lactosylceramide, accumulate in NPC disease, despite an absence of any known defects in ganglioside catabolic enzymes. Therefore, the recycling of simple ganglioside precursors may play a critical role in regulating overall glycosphingolipid synthesis, with failure of such feedback resulting in concomitant increases in synthesis not unlike that reported for cholesterol in liver and other tissues.31 Greater understanding of glycosphingolipid homeostatic regulation in NPC disease may provide insight into other lysosomal diseases that similarly show elevation of GM2 and GM3 gangliosides in the absence of known degradative defects.
Figure 8.
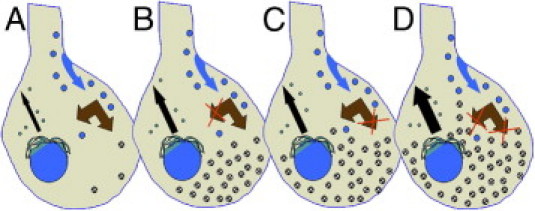
Summary schematic to explain unexpected increases in GM1 ganglioside in the Npc1−/−;βgal−/− double mutant mice. A: Normal neurons synthesize gangliosides in the Golgi/TGN followed by transport to the plasmalemma (black arrow) and subsequent endocytosis (blue arrow) and degradation in the E/L system. Simple gangliosides in the E/L system (double brown arrow) can be fully degraded or are salvaged intact and transported to the Golgi for re-utilization.34B: In NPC disease the absence of NPC1 or NPC2 proteins may cause a failure to efficiently salvage simple gangliosides (eg, GM2, GM3, lactosylceramide) leading to their storage. C: In neurons lacking lysosomal β-gal there is a failure to degrade GM1 followed by its subsequent accumulation. D: In mice lacking both NPC1 and β-gal these salvage and degradative pathways are each blocked leading to exacerbation of GM1 storage. A key question emerging from the current study is whether the failure to recycle simple ganglioside precursors leads to compensatory increases in ganglioside synthesis (black arrows).
Acknowledgments
We thank Drs. David Sleat and Peter Lobel for the mice with NPC2 disease; Drs. Ronald Schnaar and Richard Proia for the GalNAcT and GD3 synthase knockout mice; Dr. Alessandra d'Azzo (St. Jude Children's Research Hospital) for the mice with GM1 gangliosidosis; and Dr. Yoshiko Iwashita for the BCθ compound. We also thank Nafeeza Ali and Bin Cui for expert technical assistance.
Footnotes
Supported by grants from the National Institutes of Health (HD045561 to S.U.W.), the Ara Parseghian Medical Research Foundation (S.U.W.), and Vaincre les Maladies Lysosomales (M.T.V.).
A guest editor acted as editor-in-chief for the manuscript. No person at Thomas Jefferson University or Albert Einstein College of Medicine was involved in the peer review process or final disposition of this article.
Current address of R.McG., New York Eye and Ear Infirmary, 321 E. 14th St., New York, NY 10003.
References
- 1.Vanier M.T. Lipid changes in Niemann-Pick disease type C brain: personal experience and review of the literature. Neurochem Res. 1999;24:481–489. doi: 10.1023/a:1022575511354. [DOI] [PubMed] [Google Scholar]
- 2.Patterson M.C., Vanier M.T., Suzuki K., Morris J.A., Carstea E., Neufeld E.B., Branchette-Mackie J.E., Pentchev P.G. Niemann-Pick Disease type C: A lipid trafficking disorder. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., Childs B., Kinzler K.W., Vogelstein B., editors. The Metabolic and Molecular Bases of Inherited Disease. ed 4. McGraw-Hill; New York: 2001. pp. 3611–3634. [Google Scholar]
- 3.Walkley S.U., Suzuki K. Consequences of NPC1 and NPC2 loss of function in neurons. Biochem Biophys Acta. 2004;1685:48–62. doi: 10.1016/j.bbalip.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 4.Carstea E.D., Morris J.A., Coleman K.G., Loftus S.K., Zhang D., Cummings C., Gu J., Rosenfeld M.A., Pavan W.J., Krizman D.B., Nagle J., Polymeropoulos M.H., Sturley S.L., Ioannou Y.A., Higgins M.E., Comly M., Cooney A., Brown A., Kaneski C.R., Blanchette-Mackie E.J., Dwyer N.K., Neufeld E.B., Chang T.Y., Liscum L., Strauss J.F., Ohno K., Zeigler M., Carmi R., Sokol J., Markie D., O'Neill R.R., van Diggelen O.P., Elleder M., Patterson M.C., Brady R.O., Vanier M.T., Pentchev P.G., Tagle D.A. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 5.Ohgami N., Ko D.C., Thomas M., Scott M.P., Chang C.C., Chang T.Y. Binding between the Niemann-Pick C1 protein and a photoactivatable cholesterol analog requires a functional sterol-sensing domain. Proc Natl Acad Sci USA. 2004;101:12473–12478. doi: 10.1073/pnas.0405255101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Infante R.E., Abi-Mosleh L., Radhakrishnan A., Dale J.D., Brown M.S., Goldstein J.L. Purified NPC1 protein: I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J Biol Chem. 2008;283:1052–1063. doi: 10.1074/jbc.M707943200. [DOI] [PubMed] [Google Scholar]
- 7.Infante R.E., Radhakrishnan A., Abi-Mosleh L., Kinch L.N., Wang M.L., Grishin N.V., Goldstein J.L., Brown M.S. Purified NPC1 protein: iI. Localization of sterol binding to a 240-amino acid soluble luminal loop. J Biol Chem. 2008;283:1064–1075. doi: 10.1074/jbc.M707944200. [DOI] [PubMed] [Google Scholar]
- 8.Naureckiene S., Sleat D.E., Lackland H., Fensom A., Vanier M.T., Wattiaux R., Jadot M., Lobel P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–2301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- 9.Friedland H.L., Liou P., Lobel Stock A.M. Structure of a cholesterol-binding protein deficient in Niemann-Pick type C2 disease. Proc Natl Acad Sci USA. 2003;100:2512–2517. doi: 10.1073/pnas.0437840100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ko D.C., Milenkovic L., Beier S.M., Manuel H., Buchanan J., Scott M.P. Cell-autonomous death of cerebellar Purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet. 2005;1:81–95. doi: 10.1371/journal.pgen.0010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Infante R.E., Wang M.L., Radhakrishnan A., Kwon H.J., Brown M.S., Goldstein J.L. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc Natl Acad Sci USA. 2008;105:15287–15292. doi: 10.1073/pnas.0807328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwon H.J., Abi-Mosleh L., Wang M.L., Deisenhofer J., Goldstein J.L., Brown M.S., Infante R.E. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137:1213–1224. doi: 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sleat D.E., Jennifer J.A., Wiseman A., El-Banna M., Price S.M., Verot L., Shen M.M., Tint G.S., Vanier M.T., Walkley S.U., Lobel P. Genetic evidence for non-redundant functional co-operativity between NPC1 and NPC2 in lipid transport. Proc Natl Acad Sci USA. 2004;101:5886–5891. doi: 10.1073/pnas.0308456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pentchev P.G., Blanchette-Mackie E., Dawidowicz E.A. The NP-C gene: a key to pathways of intracellular cholesterol transport. Trends in Cell Biol. 1994;4:365–369. doi: 10.1016/0962-8924(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 15.McGlynn R., Dobrenis K., Walkley S.U. Differential subcellular localization of cholesterol, gangliosides and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J Comp Neurol. 2004;480:415–426. doi: 10.1002/cne.20355. [DOI] [PubMed] [Google Scholar]
- 16.Walkley S.U., Vanier M.T. Secondary lipid accumulation in lysosomal disease. Biochem Biophys Acta. 2009;1793:726–736. doi: 10.1016/j.bbamcr.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu R.K. Developmental regulation of ganglioside metabolism. Prog Brain Res. 1994;101:31–44. doi: 10.1016/s0079-6123(08)61938-x. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y., Wu Y.P., Wada R., Neufeld E.B., Mullin K.A., Howard A.C., Pentchev P.G., Vanier M.T., Suzuki K., Proia R.L. Alleviation of neuronal ganglioside storage does not improve the clinical course of the Niemann-Pick C disease mouse. Hum Mol Genet. 2000;9:1087–1092. doi: 10.1093/hmg/9.7.1087. [DOI] [PubMed] [Google Scholar]
- 19.Gondré-Lewis M., McGlynn R., Walkley S.U. Cholesterol accumulation in neurons with NPC1 dysfunction is ganglioside-dependent. Current Biology. 2003;13:1324–1329. doi: 10.1016/s0960-9822(03)00531-1. [DOI] [PubMed] [Google Scholar]
- 20.Iwamoto M., Morita I., Fukuda M., Murota S., Ando S., Ohno-Iwashita Y. A biotinylated perfringolysin O derivative: a new probe for detection of cell surface cholesterol. Biochim Biophys Acta. 1997;1327:222–230. doi: 10.1016/s0005-2736(97)00061-8. [DOI] [PubMed] [Google Scholar]
- 21.Hahn C.N., del Pilar Martin M., Schröder M., Vanier M.T., Hara Y., Suzuki K., Suzuki K., d'Azzo A. Generalized CNS disease and massive GM1-ganglioside accumulation in mice defective in lysosomal acid beta-galactosidase. Hum Mol Genet. 1997;6:205–211. doi: 10.1093/hmg/6.2.205. [DOI] [PubMed] [Google Scholar]
- 22.Kawai H., Allende M.L., Wada R., Kono M., Sango K., Deng C., Miyakawa T., Crawley J.N., Werth N., Bierfreund U., Sandhoff K., Proia R.L. Mice expressing only monosialoganglioside GM3 exhibit lethal audiogenic seizures. J Biol Chem. 2001;276:6885–6888. doi: 10.1074/jbc.C000847200. [DOI] [PubMed] [Google Scholar]
- 23.Zervas M., Dobrenis K., Walkley S.U. Neurons in Niemann-Pick disease type C accumulate gangliosides and cholesterol and undergo dendritic and axonal alterations. J Neuropath Exper Neurol. 2001;60:49–64. doi: 10.1093/jnen/60.1.49. [DOI] [PubMed] [Google Scholar]
- 24.Gimpl G., Gehrig-Burger K. Cholesterol reporter molecules. Biosci Rep. 2007;27:335–358. doi: 10.1007/s10540-007-9060-1. [DOI] [PubMed] [Google Scholar]
- 25.Reid P.C., Sakashita N., Sugii S., Ohno-Iwashita Y., Shimada Y., Hickey W.F., Chang T.Y. A novel cholesterol stain reveals early neuronal cholesterol accumulation in the Niemann-Pick type C1 mouse brain. J Lipid Res. 2004;45:582–591. doi: 10.1194/jlr.D300032-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Fujita N., Suzuki K., Vanier M.T., Popko B., Maeda N., Klein A., Henseler M., Sandhoff K., Nakayasu H. Targeted disruption of the mouse sphingolipid activator protein gene: a complex phenotype, including severe leukodystrophy and wide-spread storage of multiple sphingolipids. Hum Mol Genet. 1996;5:711–725. doi: 10.1093/hmg/5.6.711. [DOI] [PubMed] [Google Scholar]
- 27.Kyrklund T. Two procedures to remove polar contaminants from a crude brain lipid extract by using prepacked reverse-phase columns. Lipids. 1987;22:274–277. doi: 10.1007/BF02533991. [DOI] [PubMed] [Google Scholar]
- 28.Taniguchi M., Shinoda Y., Ninomiya H., Vanier M.T., Ohno K. Sites and temporal changes of gangliosides GM1/GM2 storage in the Niemann-Pick disease type C mouse brain. Brain Dev. 2001;23:414–421. doi: 10.1016/s0387-7604(01)00252-2. [DOI] [PubMed] [Google Scholar]
- 29.Matsuda J., Vanier M.T., Popa, Portoukalian J., Suzuki K. GD3- and O-acetylated GD3-gangliosides in the GM2 synthase-deficient mouse brain and their immunohistochemical localization. Proc Japan Academy. 2006;82(series B):189–196. doi: 10.2183/pjab.82.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karten B., Vance D.E., Campenot R.B., Vance J.E. Cholesterol accumulates in cell bodies but it decreased in distal axons of Niemann-Pick C1 deficient neurons. J Neurochem. 2002;83:1154–1163. doi: 10.1046/j.1471-4159.2002.01220.x. [DOI] [PubMed] [Google Scholar]
- 31.Xie C., Turley S.D., Pentchev P.G., Dietschy J.M. Cholesterol balance and metabolism in mice with loss of function of Niemann-Pick C protein. Am J Physiol. 1999;276:E336–E344. doi: 10.1152/ajpendo.1999.276.2.E336. [DOI] [PubMed] [Google Scholar]
- 32.Malathi K., Higaki K., Tinkelenberg A.H., Balderes D.A., Almanzar-Paramio D., Wilcox L.J., Erdeniz N., Redican F., Padamsee M., Liu Y., Khan S., Alcantara F., Carstea E.D., Morris J.A., Sturley S.L. Mutagenesis of the putative sterol-sensing domain of yeast Niemann Pick C-related protein reveals a primordial role in subcellular sphingolipid distribution. J Cell Biol. 2004;164:547–556. doi: 10.1083/jcb.200310046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwarzmann G., Sandhoff K. Metabolism and intracellular transport of glycosphingolipids. Biochemistry. 1990;29:10865–10871. doi: 10.1021/bi00501a001. [DOI] [PubMed] [Google Scholar]
- 34.Tettamanti G., Bassi R., Viani P., Riboni L. Salvage pathways in glycosphingolipid metabolism. Biochemie. 2003;85:432–437. doi: 10.1016/s0300-9084(03)00047-6. [DOI] [PubMed] [Google Scholar]
- 35.Walkley S.U. Pathogenic mechanisms in lysosomal disease: A reappraisal of the role of the lysosome. Acta Paediatrica. 2007;96(s455):26–32. doi: 10.1111/j.1651-2227.2007.00202.x. [DOI] [PubMed] [Google Scholar]
- 36.Li H., Turley S.D., Liu B., Repa J.J., Dietschy J.M. GM2/GD2 and GM3 gangliosides have no effect on cellular cholesterol pools or turnover in normal or NPC1 mice. J Lipid Res. 2008;49:1816–1828. doi: 10.1194/jlr.M800180-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simons K., Vaz W.L.C. Model systems, lipid rafts and cell membranes. Ann Rev Biophys Biomol Struct. 2004;33:269–295. doi: 10.1146/annurev.biophys.32.110601.141803. [DOI] [PubMed] [Google Scholar]
- 38.Walkley S.U. Secondary accumulation of gangliosides in lysosomal storage disorders. Seminars in Cell Biology & Development. 2004;15:433–444. doi: 10.1016/j.semcdb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 39.Griffiths G. On phagosome individuality and membrane signaling networks. Trends Cell Biol. 2004;14:343–351. doi: 10.1016/j.tcb.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 40.Micsenyi M.C., Dobrenis K., Stephney G., Pickel J., Vanier M.T., Slaugenhaupt S.A., Walkley S.U. Neuropathology of the Mcoln1−/− knockout mouse model of mucolipidosis IV. J. Neuropath Exp Neurol. 2009;68:125–135. doi: 10.1097/NEN.0b013e3181942cf0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barry M.F., Ziff E.B. Receptor trafficking and the plasticity of excitatory synapses. Current Opinion Neurobiology. 2002;12:279–286. doi: 10.1016/s0959-4388(02)00329-x. [DOI] [PubMed] [Google Scholar]
- 42.Bredt D.S., Nicoll R.A. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- 43.Pol S., Di Fiore P.P. Endocytosis conducts the cell signaling orchestra. Cell. 2006;124:897–900. doi: 10.1016/j.cell.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 44.Bronfman F.C., Escudero C.A., Weis J., Kruttgen A. Endosomal transport of neurotrophins: roles in signaling and neurodegenerative disease. Develop Neurobiol. 2007;9:1183–1203. doi: 10.1002/dneu.20513. [DOI] [PubMed] [Google Scholar]
- 45.Walkley S.U. Neurobiology and cellular pathogenesis of glycolipid storage diseases. Trans Royal Soc. 2003;358:893–904. doi: 10.1098/rstb.2003.1276. B. [DOI] [PMC free article] [PubMed] [Google Scholar]