

Abstract
Tumor necrosis factor receptor 1 (TNFR1) activation in hepatocytes can trigger apoptotic or inflammatory signaling. The factors that determine which signaling pathway dominates are not clear and are thought to relate to the efficiency of death-inducing signaling complex (DISC) formation. However, the steps involved in DISC formation in hepatocytes are poorly understood. In characterizing DISC formation within cultured hepatocytes, we demonstrated that TNF-α exposure leads to the rapid formation of a DISC involving TNF-α, the TNFR-associated death domain adaptor molecule (TRADD), the Fas-associated death domain adaptor molecule (FADD), caspase-8, TNFR-associated factor 2 (TRAF2), and receptor-interacting protein (RIP). The inclusion of the sensitizing agent actinomycin D both accelerated and amplified the appearance of the DISC. Notably, TNFR1 along with some DISC components also appeared within mitochondria within 30 minutes. Whereas TNFR1 consistently co-localized with the TRADD, FADD, the caspase-8, and TRAF2 in the cytosolic fraction, TNFR1 in the mitochondria was associated only with caspase-8 after TNF-α exposure. Similar observations were made in vivo using TNF-α with D-galactosamine. Actinomycin D alone also enhanced the appearance of DISC components in both cytosol and the mitochondria. Thus the DISC that includes TNFR1 forms in the cytosol of hepatocytes under both survival and pro-apoptotic conditions. The observations also suggest that TNF-α–mediated signaling includes the translocation of TNFR1 to mitochondria.
Tumor necrosis factor (TNF)-α is a key cytokine that can induce apoptosis in many transformed and non-transformed cell types. In the liver, TNF-α has been implicated in the excessive hepatocyte apoptosis seen in many conditions associated with liver damage.1–3
Paradoxically, TNF-α signaling through the TNF-α receptor 1 (TNFR1) can activate concurrent signaling pathways leading either to cell survival or cell death in the liver and other tissues.4 However, the molecular mechanism that determines which of these two opposing signaling pathway is preferentially activated in hepatocytes is unclear.5,6 Based on studies in HT1080 and 293T cell lines, Micheau and Tschopp7 proposed distinct signaling complexes. The prosurvival pathway or complex I is thought to rapidly assemble on the cytosolic death domain of TNFR1 and to be composed of the TNFR-associated death domain (TRADD) adaptor molecule, protein kinase receptor-interacting protein 1 (RIP1), and the TNFR-associated factor 2 (TRAF2) signal transducer. This complex promotes cell survival through the activation of NF-κβ. In this paradigm, once the receptor complex is internalized, TRADD, RIP, and TRAF2 dissociate from TNFR1. According to Schneider et al, 8 TRADD and RIP then bind to the Fas-associated death domain (FADD) adaptor molecule, resulting in caspase-8 recruitment in the cytosol (complex II). This complex lacks TNFR1 but contains the proapoptotic molecules. The interaction between FADD and pro-caspase-8 leads to apoptosis through protein cleavage and activation of the downstream caspase cascade, as well as the cleavage and activation of pro-apoptotic targets. It is thought that the activation of NF-κβ through complex I can antagonize apoptotic signaling through the expression of genes that prevent the pro-death potential of complex II. In the absence of a signal from complex I, complex II formation can dominate and lead to death.9 It is important to stress that the paradigm detailed above was described mostly in transformed cell lines. Whether these events pertain to hepatocytes is uncertain.
TNF-α–induced apoptosis in cultured hepatocytes requires the addition of a sensitizing agent such as actinomycin D (Act D) or cycloheximide, suggesting that the NF-κβ survival pathway predominates in hepatocytes.10 However, the molecular mechanisms that account for the shift toward apoptosis following sensitization are not known. It is known that the induction of apoptosis by the TNF-α/TNFR1 system involves the mitochondrial amplification pathway in hepatocytes. This process is initiated by the cleavage of Bid by caspase-8, followed by the translocation of truncated Bid to the mitochondria. This process in turn induces the release of pro-apoptotic molecules to the cytoplasm, which results in effective induction of apoptosis. Although deletion of Bid leads to nearly complete blockage of Fas-mediated hepatocyte apoptosis and liver injury,11,12 deletion of Bid only delays and does not block TNF-α–mediated hepatocyte apoptosis and liver injury or the induction of mitochondrial changes.11–14
Traditionally, TNFR1 is regarded as a cell surface receptor, although studies in endothelial cells have shown that TNFR1 is predominantly expressed in the trans Golgi network, with only low levels being detected at the plasma membrane.15,16 However, the localization of TNFR1 based on studies in cell lines is a controversial subject in the literature. Cottin et al17 have reported that TNFR1 was predominantly localized to the lipid raft. Furthermore, there is no information in hepatocytes on the distribution of the TNFR1 either at baseline or under stress conditions. Given the importance of the mitochondria in TNF-α signaling of hepatocytes, we assessed the relationship between death-inducing signaling complex (DISC) components and the mitochondria in hepatocytes in the resting state and following either TNF-α or TNF-α in combination with Act D. Several unexpected findings were observed. First, the sensitizing agent alone [Act D and D-galactosamine (D-gal)] induced DISC formation in hepatocytes. Second, TNF-α with or without sensitizing agent resulted in the translocation of TNFR1 to mitochondria. Finally, TNFR1 within mitochondria associated only with caspase-8. Other DISC components, including TRADD, FADD, TRAF2, and RIP1, were not part of the mitochondrial TNFR1-associated complex. Thus the hepatocyte response to TNF-α exposure or stress includes the translocation of TNFR1 to the mitochondria.
Materials and Methods
Williams E medium, penicillin, streptomycin, l-glutamine, and HEPES were purchased from Invitrogen (Carlsbad, CA). Insulin (Humulin) was purchased from Eli Lilly (Indianapolis, IN), and calf serum was obtained from HyClone Laboratories (Green Bay, WI). TNF-α was obtained from R&D Systems (Minneapolis, MN). Unless indicated otherwise, all other chemicals and proteins were purchased from Sigma-Aldrich (St. Louis, MO).
Hepatocyte Isolation and Culture
All procedures involving animals were approved by the Animal Care and Use Committee of the University of Pittsburgh. Primary hepatocytes were harvested from male Sprague-Dawley rats (Harlan, Indianapolis, IN) as previously described in detail.18 The cells were washed and further cultured in fresh medium containing 5% calf serum in various treatments of reagents as specifically indicated in the figure legends.
Mitochondria Isolation
Cells were collected and subcellular fractions were obtained as previously described.19 Cells were collected in buffer A (250 mmol/L mannitol, 70 mmol/L sucrose, 0.5 mmol/L EGTA, and 5 mmol/L HEPES-NaOH, pH 7.2) with freshly added protease inhibitors spun at 400 × g for 10 minutes at 4°C, and the supernatant was discarded. The cells were then resuspended in 1 mL buffer A with protease inhibitors and placed on ice for 10 minutes and homogenized with a Teflon glass Dounce homogenizer (25 strokes). Homogenates were centrifuged at 700 × g for 10 minutes and the supernatant was separated into two tubes. One of the tubes was used as whole cell lysates. The other tube was spun (1000 × g for 30 minutes at 4°C) to obtain the heavy membrane pellet. The supernatant from this tube was removed and spun in an ultracentrifuge at 100,000 × g for 30 minutes and represented the cytosolic fraction. The mitochondria were purified from the heavy membrane pellet by stepwise sucrose-density-gradient centrifugation. The heavy membrane pellet was resuspended in 1 mL buffer A with protease inhibitors and placed on the top of a 1.0, 12, and 1.6 M sucrose buffer gradient before being centrifuged. Gradient samples were placed in a SW 50.1 rotor (Beckman-Coulter, Brea, CA) and spun at 39,000 × g for 30 minutes with slow brake at 4°C. The brownish band containing the mitochondria at the 1.2 M sucrose and 1.6 M sucrose interface was carefully removed (<200 μL) and diluted 5× with buffer A. Samples were then spun at 10,000 × g for 10 minutes and the supernatant was removed and resuspended in buffer B (250 mmol/L sucrose, 10 mmol/L HEPES-NaOH, pH 7.5, 2 mmol/L KH2PO4, 5 mmol/L sodium succinate, 25 μmol/L EGTA, and 0.1 mmol/L phenylmethylsulfonyl fluoride). Contamination assays were performed on fractions using an equal number of cells. β-Actin, COX IV, calreticulin, TGN-38, and lamin B antibodies were used for immunoblot analysis.
Co-Immunoprecipitation and Immunoblotting
Whole cell lysates, cytosol, or mitochondrial fractions were incubated overnight with rabbit anti-TNFR1 antibody. The immune complexes were then precipitated with protein A/G-agarose beads for 6 hours and washed extensively with radioimmunoprecipitation assay buffer. Immunoprecipitated proteins were eluted with 2× SDS loading buffer, separated by SDS gels, and transferred onto a nitrocellulose membrane, and immunoblot [Western blot (WB)] was processed as described previously.18
Immunostaining and Confocal Microscopy
Primary hepatocytes were plated on collagen-1 coated 22 mm coverslips (BD Pharmingen, San Diego, CA) overnight and treated the next day. Cells were fixed with 2% (w/v) paraformaldehyde for 15 minutes and processed as previously described20 with the exception of labeling cells with rabbit anti-TNFR1 and mouse anti-ATP synthase at 4°C for 1 hour. Cells were viewed on an Olympus Fluoview 1000 (Olympus America Inc., Center Valley, PA).
Immunogold Labeling and Immunoelectron Microscopy
Cells were fixed with 2% paraformaldehyde and in 0.1 mol/L phosphate buffer (pH 7.4) and processed as previously described20 with the exception of labeling sections with rabbit anti-TNFR1 for 1 hour. Sections were observed on a JEM 1210 electron microscope (JEOL Ltd, Akishima, Tokyo) at 80 kV.20
In Vivo Experiments
All procedures were performed according to the guidelines of the Council on Animal Care at the University of Pittsburgh and the National Research Council's Guide for the Care and Use of Laboratory Animals.
Statistical Analysis
Hepatocyte viability is presented as the mean ± SEM. One-way analysis of variance followed by the Student–Neuman–Keuls test was used to determine the significance of the differences between the experimental groups. A P value <0.05 was considered significant.
Results
Effects of TNF-α, TNF-α/Act D, and Act D on Hepatocyte Cell Death
We first assessed the timing and extent of TNF-α–induced hepatocyte death in the absence and presence of Act D. Cell viability was measured by crystal violet staining at 3, 6, 9, and 18 hours following treatment. As shown in Figure 1, no significant cell death occurred in hepatocytes treated with TNF-α alone at any time point. Act D alone caused some degree of cell death that was first seen at 6 hours. However, the co-treatment of TNF-α with Act D led to significant loss of viability by 6 hours after treatment. Viability continued to decline in a time-dependent manner (percentage of live cells at 18 hours, TNF-α alone: 95.9 ± 1.8%; TNF-α + Act D: 39.9 ± 1.9%; and Act D alone: 78.4 ± 2.5%). The loss of cell viability was associated with an increase in the percentage of TUNEL-positive cells (data not shown).
Figure 1.
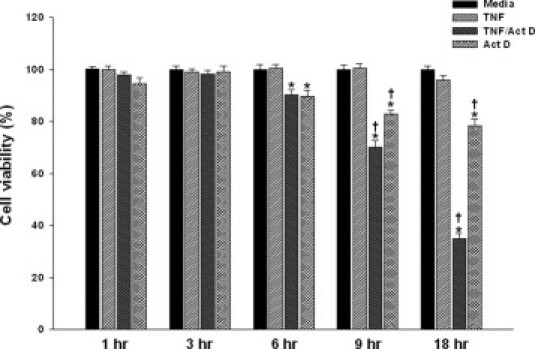
Effects of TNF-α, TNF-α/Act D, and Act D on hepatocyte cell death. Primary rat hepatocytes were treated with TNF-α (2000 units/mL) ± Act D (200 ng/mL) for 3-, 6-, 9-, and 18-hour treatment(s). *P < 0.01, media group; †P < 0.01, group treated with TNF-α alone.
TNFR1 Accumulates in the Mitochondria Following TNF-α, TNF-α/Act D, or Act D Stimulation in Hepatocytes
TNFR1 has been shown to localize to different subcellular compartments but not to mitochondria in cell lines.15–17 Because TNF-α signaling includes mitochondria-dependent steps, we sought to determine whether TNFR1 also translocated to mitochondria following TNF-α stimulation. At 0, 15, 30, and 60 minutes after stimulation, mitochondrial and cytosolic fractions were isolated using differential and density gradient centrifugation. To verify purity of cell fractions, marker enzymes were evaluated by immunoblot analysis using antibodies that recognize β-actin, COX IV, calreticulin, TGN-38, and lamin B (Figure 2A). Immunoblotting confirmed the presence of TNFR1 in whole cell lysates and in the cytosolic fraction over the first 60 minutes following TNF-α, TNF-α/Act D, or Act D exposure (Figure 2B). TNFR1 was absent at baseline in mitochondria but became detectable by 30 minutes following TNF-α alone and TNF-α/Act D. Act D alone resulted in the appearance of TNFR1 in the mitochondrial fraction by 60 minutes. To further confirm the localization of TNFR1 in the mitochondria, confocal microscopy was performed following double-staining with anti-TNFR1 antibody and anti-ATP synthase antibody as a mitochondrial marker. In untreated hepatocytes, an even distribution of TNFR1 in the cytoplasm was observed (Figure 3A). At 60 minutes following TNF-α or TNF-α/Act D, a punctate pattern appeared. Furthermore, co-localization of TNFR1 with mitochondria, as demonstrated by a yellow color, could be easily detected following TNF-α or TNF-α/Act D treatment and faintly detected with Act D alone. We then performed electron microscopy with immune gold labeling of TNFR1 in hepatocytes at baseline and at 60 minutes following stimulation (Figure 3B). A few beads could be seen in mitochondria at baseline (culture media with no additive treatments). Stimulation with TNF-α, TNF-α/Act D, or Act D alone led to a dramatic increase in the number of gold beads associated with mitochondria (Figure 3B). Interestingly, changes in mitochondrial membrane potential, as measured with use of the fluorescent probe JC-1, decreased by 15 minutes in cells treated with TNF-α and TNF-α + ActD (see Supplemental Figure S1 at http://ajp.amjpathol.org). Furthermore, the reactive oxygen species scavenger (N-acetylcysteine) but not the JNK inhibitor (SP600125) prevented the TNF-α + ActD–induced translocation of TNFR1 to the mitochondria (see Supplemental Figure S2 at http://ajp.amjpathol.org). Taken together, these findings establish that TNFR1 translocates to mitochondria by 30 minutes and following changes in mitochondrial membrane potential in cultured cells exposed to TNF-α.
Figure 2.
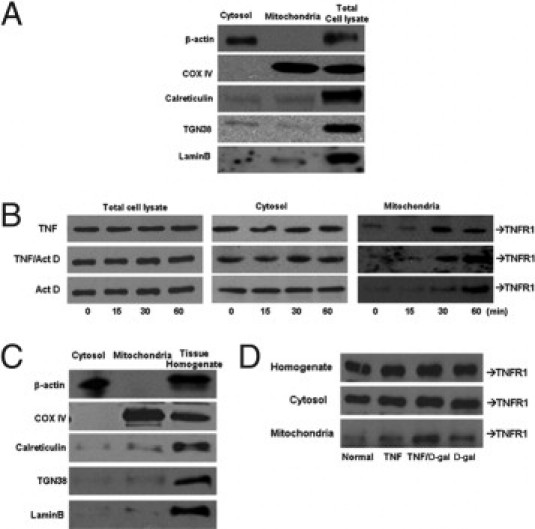
TNFR1 accumulates in the mitochondria following TNF-α, TNF-α/Act D, or Act D treatment in vitro and in vivo. Primary hepatocytes were treated with TNF-α (2000 units/mL) ± Act D (200 ng/mL). A: Fraction purity was assessed using antibodies to β-actin, COX IV, calreticulin, TGN-38, and lamin B. B: The level of TNFR1 protein in cytosolic or mitochondrial was determined by immunoblot (N = 3). Livers were harvested and separated into mitochondrial and cytosolic fractions from rats injected with sterile TNF-α (10 μg/kg) ± D-gal (750 mg/mL). C: Contamination assays using equal amount of proteins measured β-actin, COX IV, calreticulin, TGN-38, and lamin B by immunoblot analysis. D: TNFR1 protein in cytosolic or mitochondrial fractions were determined by immunoblot analysis.
Figure 3.

Immunofluorescent confocal microscopy and immunoelectron microscopy confirmed localization of TNFR1 in TNF-α–stimulated primary hepatocytes and isolated mitochondria. Primary rat hepatocytes were treated with TNF-α (2000 units/mL) ± Act D (200 ng/mL) for 1 hour. A: Fluorescent labeling: TNFR1, red; ATP synthase, green; nucleus, gray. B: Sections were labeled with rabbit anti-TNFR1 and then labeled with 10 nmol/L gold-conjugated second antibodies. Note the labeling of TNFR1 in basal level (media) of TNFR1 in mitochondria and increased TNFR1 when treated with TNF-α, TNF-α/Act D, or Act D. Arrows point to TNFR1 positive 10-nm gold beads.
TNFR1 Localizes to Liver Mitochondria Following TNF-α, TNF-α/D-gal, or D-gal Stimulation in Vivo
To determine whether TNFR1 associated with the mitochondria in intact liver, rats were treated with TNF-α alone or with D-gal. Whole liver homogenates, as well as the cytosolic and mitochondrial fractions, were subjected to immunoblotting for TNFR1 for 60 minutes. As shown in Figure 2C, TNFR1 levels did not change in liver homogenates or the cytosolic fractions. Low baseline levels were seen in the mitochondrial fraction, which markedly increased with TNF-α alone, TNF-α/D-gal, and D-gal alone. The purity of the fractions was confirmed using the appropriate markers (Figure 2D).
Different DISC Components Localize with TNFR1 in the Cytosolic and Mitochondrial Fractions
Immunoblotting confirmed the presence of DISC components in hepatocytes including TRADD, FADD, RIP1, TRAF2, and caspase-8 (Figures 4, 5, 6, 7, and 8). Levels of these proteins changed minimally in the first 60 minutes. To define the composition of the DISC in hepatocytes, immunoprecipitation and immunoblot was performed on whole cell lysates at 15-minute intervals following stimulation. TNF-α alone or with Act D led to a similar level of interaction of TNFR1 with TRADD (Figure 4), FADD (Figure 5), and TRAF2 (Figure 6). Both TNF-α alone and TNF-α with Act D brought caspase-8 (Figure 7) and RIP1 (Figure 8) into the DISC; however, the association was enhanced with Act D or D-gal. Interestingly, exposure to the sensitizing agents alone led to an interaction of all five DISC components with TNFR1, although it was somewhat delayed and to a lesser extent than that seen with TNF-α.
Figure 4.
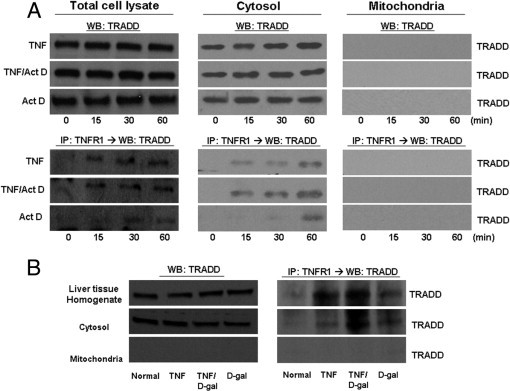
TNFR1 complexes isolated from hepatocytes treated with TNF-α ± sensitizing agents do not contain TRADD in the mitochondria. A: Rat primary hepatocytes were collected and subcellular populations were isolated following treatment with TNF-α (2000 units/mL) ± Act D (200 ng/mL) for the indicated times. Each fraction was analyzed by WB for TRADD. TNFR1 complex was isolated by immunoprecipitation with TNFR1 antibody and then analyzed for the presence of TRADD interaction with TNFR1 by WB. B: TNF-α (10 μg/kg) or D-gal (750 mg/mL) was injected into rats, and the livers were harvested 1 hour later. Cytosolic and mitochondrial fractions were isolated from these livers, followed by WB for TRADD. TNFR1 complex was isolated by immunoprecipitation with TNFR1 antibody and then analyzed for the presence of TRADD interaction with TNFR1 by WB. IP, immunoprecipitated protein.
Figure 5.
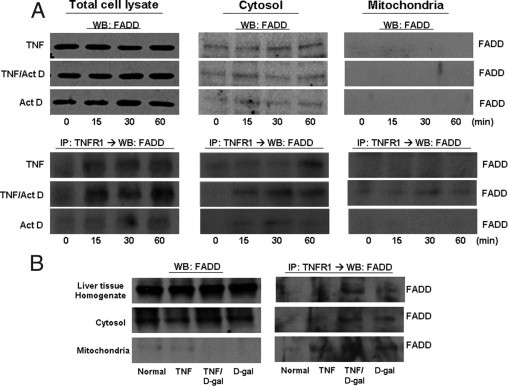
TNFR1 complexes isolated from hepatocytes treated with TNF-α ± Act D do not contain FADD in the mitochondria. Rat primary hepatocytes were treated with TNF-α (2000 units/mL) for the indicated times ± Act D (200 ng/mL). A: Hepatocytes were then fractionated into cytosolic and mitochondrial lysates and analyzed by WB for FADD. TNFR1 complex was isolated by immunoprecipitation with TNFR1 antibody and then analyzed for the interaction with TRADD with TNFR1 by WB. B: Livers were harvested 1 hour after injections with TNF-α (10 μg/kg) or D-gal (750 mg/mL); mitochondrial and cytosolic fractions were obtained and analyzed by WB for FADD. TNFR1 complex was isolated by immunoprecipitation with TNFR1 antibody and then analyzed for presence of FADD interaction with TNFR1 by WB. IP, immunoprecipitate.
Figure 6.
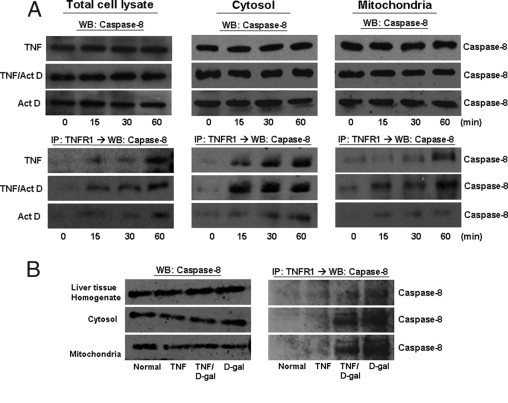
TNFR1 complexes isolated from hepatocytes treated with TNF-α ± D-gal or Act D contain caspase-8 in the mitochondria. Rat primary hepatocytes were collected following treatment with TNF-α (2000 units/mL) for the indicated times ± Act D (200 ng/mL). A: Cytosolic and mitochondrial hepatocyte fractions were analyzed by WB for caspase-8. TNFR1 complex was isolated by immunoprecipitation with TNFR1 antibody and then analyzed by immunoblot for presence of caspase-8 interaction with TNFR1. Livers from rats injected with TNF-α (10 μg/kg) or D-gal (750 mg/mL) were harvested after 1 hour and fractionated into cytosol and mitochondria. B: Each fraction was analyzed by WB for caspase-8. TNFR1 complex was isolated by immunoprecipitation with TNFR1 antibody and then analyzed for presence of caspase-8 interaction with TNFR1 by WB. IP, immunoprecipitated protein.
Figure 7.
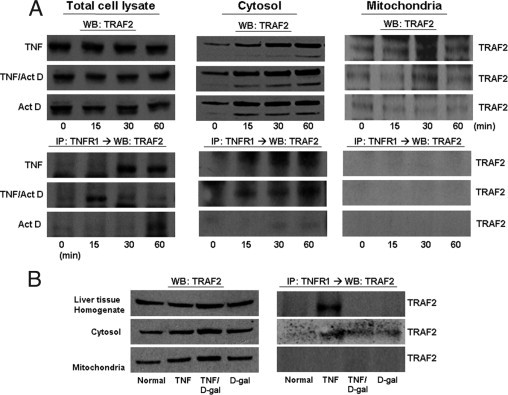
TNFR1 complexes isolated from TNF-α with or without hepatocytes treated with a sensitizing agent do not contain TRAF2 in the mitochondria. Rat primary hepatocytes were harvested following treatment with TNF-α (2000 units/mL) for the indicated times ± Act D (200 ng/mL). A: Cytosolic and mitochondrial fractions were analyzed by immunoblot for TRAF2. TNFR1 complex was isolated by immunoprecipitation with TNFR1 antibody and then analyzed for presence of TRAF2 interaction with TNFR1 by WB. B: Livers harvested from rats 1 hour after injection with sterile TNF-α (10 μg/kg) or D-gal (750 mg/mL) were separated into cytosolic and mitochondrial fractions and assayed by immunoblot for TRAF2. TNFR1 complex was isolated by immunoprecipitation with TNFR1 antibody and then analyzed for the presence of TRAF2 interaction with TNFR1 by WB. IP, immunoprecipitated protein.
Figure 8.
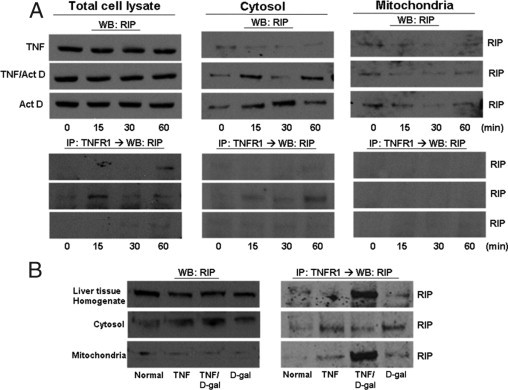
TNFR1 complexes isolated from hepatocytes treated with TNF-α ± a sensitizing agent contain RIP in the mitochondria. A: Rat primary hepatocytes treated with TNF-α (2000 units/mL) for the indicated times ± Act D (200 ng/mL) were fractionated into cytosolic and mitochondrial fractions to be analyzed for RIP content by WB. TNFR1 complex was isolated by immunoprecipitation with TNFR1 antibody and then inmmunoblotted to measure TRAF2 interaction with TNFR1. Livers were harvested from rats 1 hour after injection with sterile TNF-α (10 μg/kg) or D-gal (750 mg/mL). B: Cytosolic and mitochondrial fractions were analyzed by WB for RIP. TNFR1 complex was isolated by immunoprecipitation with TNFR1 antibody and then analyzed for presence of RIP interaction with TNFR1 by WB. IP, immunoprecipitated protein.
To determine whether these same DISC components are present in the mitochondria before or after TNF-α exposure, the mitochondrial and cytosolic fractions were subjected to immunoblotting. Furthermore, to determine whether these DISC members interaction with TNFR1 in the mitochondria, the mitochondrial and cytosolic fractions were subjected to immunoprecipitation. This analysis was performed on both cultured hepatocytes and liver isolated from TNF-α–treated rats. TRADD was present in the cytosolic fraction at baseline, and levels of TRADD in the cytosol did not change after any treatment (Figure 4). As expected, TNFR1 was found to interact with TRADD in the cytosol following TNF-α treatment with or without sensitization. However, TRADD could not be detected in the mitochondria under any condition tested (Figure 4). Hence no interaction between TNFR1 and TRADD was seen in the mitochondria.
FADD association with TNFR1 could be detected in the cytosolic fraction after TNF-α, TNF-α/Act D, or D-gal treatment. Again, the sensitizing agent alone induced an interaction between TNFR1 and FADD in the cytosol (Figure 5). FADD was found to co-immunoprecipitate with TNFR1 in the mitochondria only with TNF-α + Act D or D-gal. Otherwise, FADD could not be detected in the mitochondrial fraction.
Procaspase-8 was easily detectable in the cytosol and mitochondria at both baseline and following the various treatments both in vitro and in vivo (Figure 6). As would be expected, caspase-8 strongly associated with TNFR1 in the cytosol fraction following exposure to TNF-α with sensitizing agent, but also to some extent with TNF-α alone and with either Act D or D-gal alone. Caspase-8 was also found to co-immunoprecipitate with TNFR1 in mitochondria. This effect was most pronounced with TNF-α combined with a sensitizing agent but also was seen with TNF-α and the sensitizing agents alone (Figure 6). This finding is consistent with previously published findings.21–24
TRAF2 was detected at baseline in both the cytosol and mitochondria in vitro and in vivo (Figure 7). As expected, TNF-α induced an association between TNFR1 and TRAF2 in the cytosol. Despite the presence of TRAF2 in the mitochondrial fraction, under no condition could we detect an interaction between TNFR1 and TRAF2 in the mitochondria. RIP1 also was detected in both the cytosol and the mitochondria. A strong interaction between RIP1 and TNFR1 in the cytosolic fraction was seen with TNF-α and the sensitizing agent. Interestingly, RIP co-immunoprecipitated with TNFR1 in the mitochondria in the whole liver in vivo with TNF-α/D-gal but not in vitro with TNF-α/Act D (Figure 8).
Discussion
This study was undertaken to gain a better understanding of the initial signaling steps following TNF-α/TNFR1 interaction in hepatocytes under conditions leading to cell survival or cell death. We found that a typical DISC rapidly forms in the cytoplasm of cells that undergo apoptosis (TNF-α with Act D or D-gal) but also in cells exposed to TNF-α or the sensitizing agents alone. It also appears that TNFR1 remains part of the signaling complex within the cytoplasm of hepatocytes. Somewhat unexpected was the observation that TNFR1 also appears within the mitochondria where TNFR1 associates with caspase-8 but not other DISC components. Thus cytoplasmic DISC formation involving TNFR1 forms under both cell survival and death signaling and even after cell stress in the absence of exogenous TNF-α. Translocation of TNFR1 to the mitochondria also may contribute to cellular signaling responses under the conditions studied. Our observations provide the basis for further studies to define the pathways involved in TNFR1 translocation and function within the mitochondria.
In cell lines sensitive to TNF-α–induced apoptosis, it has been shown that two types of signaling complexes form following TNF-α exposure.7,8 One (complex I) forms at the cell surface and involves TNFR1 interaction with TRADD, RIP1, and TRAF2, and a second (complex II) is devoid of TNFR1 but engages FADD and caspase 8. It is well known that the TNFR1-TRADD-RIP-TRAF2 complex initiates a pathway leading to survival through NF-κB.25,26 Our results indicate that TNFR1 remains engaged with TRADD, FADD, RIP1, and TRAF-2 in a cytoplasmic signaling complex in hepatocytes. That this complex forms in both cells that survive (TNF-α alone) and in cells that go on to die (TNF-α + Act D) suggests that pathways that determine cell survival are downstream of the initial complex.
Act D amplified TNF-α–induced DISC formation and even stimulated DISC formation when added alone. Our work does not illuminate how this transcription inhibitor induces DISC formation, but we also have observed this action with other sensitizing agents, including protein synthesis inhibitors (data not shown). Therefore cell stress resulting from the suppression gene expression could trigger DISC formation as a stress signaling response. The inclusion of TNFR1 in the complex raises the possibility that sensitizing agents trigger TNF-α production. However, we have not detected TNF-α production by hepatocytes, and anti-TNF-α antibodies do not prevent Act D–induced DISC formation in cultured cells (data not shown).
The appearance of TNFR1 in the mitochondria under the same conditions associated with DISC formation suggests that the receptor participates in mitochondrial changes induced by TNF-α exposure. Changes in mitochondrial membrane potential occurred as early as 15 minutes following TNF-α exposure, whereas TNFR1 accumulation becomes prominent at 30 minutes. Whether these two events are interrelated was not established by our work. Whereas translocation of cytoplasmic proteins to the mitochondria is well described in apoptotic signaling as a mechanism for signal amplification,27 the translocation of a receptor to the mitochondria during cytokine signaling has not been reported. TNFR1 has been reported on the cell surface28 and Golgi.29 The source of the mitochondrial TNFR1 is uncertain and could include TNFR1 from the cell surface or intracellular sources such as the Golgi. The selective presence of DISC components in the mitochondria and limited interactions between TNFR1 and DISC components suggest that the DISC does not simply transfer to the mitochondria. The suppression of TNFR1 translocation by a reactive oxygen species scavenger suggests that mitochondria changes may be required for the translocation to occur. Mitochondrial reactive oxygen species production is known to occur following TNF-α exposure.30,31
Some DISC components have been shown to be localized in the mitochondria following Fas stimulation.31 TRADD and TRAF2 have been detected in the nucleus and cytoplasm32,33; however, these proteins have not been reported to interact with the mitochondria. Although both TRADD and TNFR1 are found in the Golgi in endothelial cells, they do not appear to have an interaction.15 In this study we found that TRAF2 is present in mitochondria under all conditions studied. In contrast, TRADD could not be detected in the mitochondria under any condition.
The presence of caspase-8 in the mitochondria has been controversial, with conflicting reports in the literature.15,34,35 Temkin et al36 have determined that RIP1 protein is present in the mitochondria at baseline and that RIP1 accumulates in mitochondria following TNF-α exposure in THP-1 cells. We also found that RIP1 and caspase-8 are present in the mitochondria both in cultured hepatocytes and liver tissue in the resting state and following either exposure to TNF-α or TNF-α in combination with sensitizing agents. Furthermore, our data show the interaction of RIP and caspase-8 with TNFR1 in mitochondria with either TNF-α or D-gal alone; however, the association was much stronger in the liver treated with TNF-α/D-gal. The differences of DISC composition between cytosolic fraction and mitochondrial fraction suggest that there may be a distinct complex that forms involving TNFR1 in mitochondria. The function of the TNFR1-associated complex in the mitochondria is uncertain and is the subject of ongoing research.
Footnotes
Supported by NIH grant R37-GM044100 (T.R.B.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.05.046.
Supplementary data
Effects on ratiometric change in JC-1red/JC-1green in hepatocytes treated with TNF−α ± a sensitizing agent as a measure of mitochondrial depolarization were examined using a mitochondria-specific probe, JC-1. Primary rat hepatocytes were seeded to 70% confluence on MatTek glass bottom culture dishes (MatTek, Ashland, MA) overnight. The mitochondrial membrane potential indicator JC-1 (1 mg/mL) was incubated for 30 minutes at 37°C (Invitrogen, Carlsbad, CA); the nuclei indicator Draq 5 was incubated for 5 minutes (1:1000; Biostatus Limited, Leicestershire, UK) and then removed. Cells were maintained at 37°C in a heated micro-incubator (Tokai Hit Co., Shizuoka-ken, Japan) in Williams E media–buffered media plus treatment additives during imaging. A: Images were acquired at 10 different positions using a Nikon Sweptfield (TSI inverted, Nikon Instruments, Melville, NY) 40× objective (0.70 NA) every 5 minutes for 60 minutes. In calculating the red/green fluorescence ratio, background correction was used for each field and time point. B: Inclusion of TNF−α and to a lesser extent H2O2 resulted in a rapid depolarization of the rat hepatocytes mitochondria. Cells treated with Act D alone or media exhibited a gradual mitochondrial depolarization. DIC, differential interference contrast.
TNFR1 accumulates in the mitochondria on treatment(s) with TNF−α + Act D ± JNK II inhibitor (SP600125; 20 μmol/L) ± reactive oxygen species scavenger (NAC; 5 mmol/L). Primary rat hepatocytes were treated with TNF−α (2000 units/mL) and Act D (200 ng/ml) for 1 hour. TNFR1 accumulation in the nucleus occurs with media treatment ± inhibitors and to a lesser extent TA + inhibitors. None of the cells were TUNEL positive. Fluorescent labeling: TNFR1, red; ATP synthase, blue; nuclei, white.
References
- 1.Rudiger H.A., Clavien P.A. Tumor necrosis factor alpha, but not Fas, mediates hepatocellular apoptosis in the murine ischemic liver. Gastroenterology. 2002;122:202–210. doi: 10.1053/gast.2002.30304. [DOI] [PubMed] [Google Scholar]
- 2.Simeonova P.P., Gallucci R.M., Hulderman T., Wilson R., Kommineni C., Rao M., Luster M.I. The role of tumor necrosis factor-alpha in liver toxicity, inflammation, and fibrosis induced by carbon tetrachloride. Toxicol Appl Pharmacol. 2001;177:112–120. doi: 10.1006/taap.2001.9304. [DOI] [PubMed] [Google Scholar]
- 3.Yin M., Wheeler M.D., Kono H., Bradford B.U., Gallucci R.M., Luster M.I., Thurman R.G. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- 4.Leist M., Gantner F., Jilg S., Wendel A. Activation of the 55 kDa TNF-α receptor is necessary and sufficient for TNF-α-induced liver failure, hepatocyte apoptosis, and nitrite release. J Immunol. 1995;154:1307–1316. [PubMed] [Google Scholar]
- 5.Garcia-Ruiz C., Fernandez-Checa J.C. Redox regulation of hepatocyte apoptosis. J Gastroenterol Hepatol. 2007;22(Suppl 1):S38–S42. doi: 10.1111/j.1440-1746.2006.04644.x. [DOI] [PubMed] [Google Scholar]
- 6.Kimura T., Sakaida I., Terai S., Matsumura Y., Uchida K., Okita K. Inhibition of tumor necrosis factor-alpha production retards liver regeneration after partial hepatectomy in rats. Biochem Biophys Res Commun. 1997;231:557–560. doi: 10.1006/bbrc.1997.6135. [DOI] [PubMed] [Google Scholar]
- 7.Micheau O., Tschopp J. Induction of TNF-α receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 8.Muppidi J.R., Tschopp J., Siegel R.M. Life and death decisions: secondary complexes and lipid rafts in TNF-α receptor family signal transduction. Immunity. 2004;21:461–465. doi: 10.1016/j.immuni.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Schneider-Brachert W., Tchikov V., Neumeyer J., Jakob M., Winoto-Morbach S., Held-Feindt J., Heinrich M., Merkel O., Ehrenschwender M., Adam D., Mentlein R., Kabelitz D., Schutze S. Compartmentalization of TNF-α receptor 1 signaling: internalized TNF-α receptosomes as death signaling vesicles. Immunity. 2004;21:415–428. doi: 10.1016/j.immuni.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 10.Nagaki M., Moriwaki H. How are hepatocytes sensitized to apoptosis? J Gastroenterol. 2002;37:1094–1096. doi: 10.1007/s005350200187. [DOI] [PubMed] [Google Scholar]
- 11.Yin X.M., Wang K., Gross A., Zhao Y., Zinkel S., Klocke B., Roth K.A., Korsmeyer S.J. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y., Li S., Childs E.E., Kuharsky D.K., Yin X.M. Activation of pro-death Bcl-2 family proteins and mitochondria apoptosis pathway in tumor necrosis factor-alpha-induced liver injury. J Biol Chem. 2001;276:27432–27440. doi: 10.1074/jbc.M102465200. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Y., Ding W.X., Qian T., Watkins S., Lemasters J.J., Yin X.M. Bid activates multiple mitochondrial apoptotic mechanisms in primary hepatocytes after death receptor engagement. Gastroenterology. 2003;125:854–867. doi: 10.1016/s0016-5085(03)01066-7. [DOI] [PubMed] [Google Scholar]
- 14.Ding W.X., Ni H.M., DiFrancesca D., Stolz D.B., Yin X.M. Bid-dependent generation of oxygen radicals promotes death receptor activation-induced apoptosis in murine hepatocytes. Hepatology. 2004;40:403–413. doi: 10.1002/hep.20310. [DOI] [PubMed] [Google Scholar]
- 15.Jones S.J., Ledgerwood E.C., Prins J.B., Galbraith J., Johnson D.R., Pober J.S., Bradley J.R. TNF-α recruits TRADD to the plasma membrane but not the trans-Golgi network, the principal subcellular location of TNF-α-R1. J Immunol. 1999;162:1042–1048. [PubMed] [Google Scholar]
- 16.Bradley J.R., Thiru S., Pober J.S. Disparate localization of 55-kd and 75-kd tumor necrosis factor receptors in human endothelial cells. Am J Pathol. 1995;146:27–32. [PMC free article] [PubMed] [Google Scholar]
- 17.Cottin V., Doan J.E., Riches D.W. Restricted localization of the TNF-α receptor CD120a to lipid rafts: a novel role for the death domain. J Immunol. 2002;168:4095–4102. doi: 10.4049/jimmunol.168.8.4095. [DOI] [PubMed] [Google Scholar]
- 18.Metukuri M.R., Beer-Stolz D., Namas R.A., Dhupar R., Torres A., Loughran P.A., Jefferson B.S., Tsung A., Billiar T.R., Vodovotz Y., Zamora R. Expression and subcellular localization of BNIP3 in hypoxic hepatocytes and liver stress. Am J Physiol Gastrointest Liver Physiol. 2009;296:G499–G509. doi: 10.1152/ajpgi.90526.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim T.H., Zhao Y., Ding W.X., Shin J.N., He X., Seo Y.W., Chen J., Rabinowich H., Amoscato A.A., Yin X.M. Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome C release. Mol Biol Cell. 2004;15:3061–3072. doi: 10.1091/mbc.E03-12-0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stolz D.B., Zamora R., Vodovotz Y., Loughran P.A., Billiar T.R., Kim Y.M., Simmons R.L., Watkins S.C. Peroxisomal localization of inducible nitric oxide synthase in hepatocytes. Hepatology. 2002;36:81–93. doi: 10.1053/jhep.2002.33716. [DOI] [PubMed] [Google Scholar]
- 21.Kim P.K., Wang Y., Gambotto A., Kim Y.M., Weller R., Zuckerbraun B.S., Hua Y., Watkins S.C., Billiar T.R. Hepatocyte Fas-associating death domain protein/mediator of receptor-induced toxicity (FADD/MORT1) levels increase in response to pro-apoptotic stimuli. J Biol Chem. 2002;277:38855–38862. doi: 10.1074/jbc.M203484200. [DOI] [PubMed] [Google Scholar]
- 22.Kim Y.M., Kim T.H., Chung H.T., Talanian R.V., Yin X.M., Billiar T.R. Nitric oxide prevents tumor necrosis factor alpha-induced rat hepatocyte apoptosis by the interruption of mitochondrial apoptotic signaling through S-nitrosylation of caspase-8. Hepatology. 2000;32:770–778. doi: 10.1053/jhep.2000.18291. [DOI] [PubMed] [Google Scholar]
- 23.Li J., Yang S., Billiar T.R. Cyclic nucleotides suppress tumor necrosis factor alpha-mediated apoptosis by inhibiting caspase activation and cytochrome c release in primary hepatocytes via a mechanism independent of Akt activation. J Biol Chem. 2000;275:13026–13034. doi: 10.1074/jbc.275.17.13026. [DOI] [PubMed] [Google Scholar]
- 24.Kim H.S., Loughran P.A., Kim P.K., Billiar T.R., Zuckerbraun B.S. Carbon monoxide protects hepatocytes from TNF-α-alpha/Actinomycin D by inhibition of the caspase-8-mediated apoptotic pathway. Biochem Biophys Res Commun. 2006;344:1172–1178. doi: 10.1016/j.bbrc.2006.03.180. [DOI] [PubMed] [Google Scholar]
- 25.Hsu H., Shu H.B., Pan M.G., Goeddel D.V. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF-α receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 26.Wajant H., Pfizenmaier K., Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 27.Gross A., Yin X.M., Wang K., Wei M.C., Jockel J., Milliman C., Erdjument-Bromage H., Tempst P., Korsmeyer S.J. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- 28.Madge L.A., Pober J.S. TNF-α signaling in vascular endothelial cells. Exp Mol Pathol. 2001;70:317–325. doi: 10.1006/exmp.2001.2368. [DOI] [PubMed] [Google Scholar]
- 29.Al-Lamki R.S., Wang J., Skepper J.N., Thiru S., Pober J.S., Bradley J.R. Expression of tumor necrosis factor receptors in normal kidney and rejecting renal transplants. Lab Invest. 2001;81:1503–1515. doi: 10.1038/labinvest.3780364. [DOI] [PubMed] [Google Scholar]
- 30.Martindale J.L., Holbrook N.J. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 31.Wang X., Wang Y., Kim H.P., Nakahira K., Ryter S.W., Choi A.M. Carbon monoxide protects against hyperoxia-induced endothelial cell apoptosis by inhibiting reactive oxygen species formation. J Biol Chem. 2007;282:1718–1726. doi: 10.1074/jbc.M607610200. [DOI] [PubMed] [Google Scholar]
- 32.Lin Y., Ryan J., Lewis J., Wani M.A., Lingrel J.B., Liu Z.G. TRAF2 exerts its antiapoptotic effect by regulating the expression of Kruppel-like factor LKLF. Mol Cell Biol. 2003;23:5849–5856. doi: 10.1128/MCB.23.16.5849-5856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morgan M., Thorburn J., Pandolfi P.P., Thorburn A. Nuclear and cytoplasmic shuttling of TRADD induces apoptosis via different mechanisms. J Cell Biol. 2002;157:975–984. doi: 10.1083/jcb.200204039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van L.G., Saelens X., Matthijssens F., Schotte P., Beyaert R., Declercq W., Vandenabeele P. Caspases are not localized in mitochondria during life or death. Cell Death Differ. 2002;9:1207–1211. doi: 10.1038/sj.cdd.4401101. [DOI] [PubMed] [Google Scholar]
- 35.Zhivotovsky B., Samali A., Gahm A., Orrenius S. Caspases: their intracellular localization and translocation during apoptosis. Cell Death Differ. 1999;6:644–651. doi: 10.1038/sj.cdd.4400536. [DOI] [PubMed] [Google Scholar]
- 36.Temkin V., Huang Q., Liu H., Osada H., Pope R.M. Inhibition of ADP/ATP exchange in receptor-interacting protein-mediated necrosis. Mol Cell Biol. 2006;26:2215–2225. doi: 10.1128/MCB.26.6.2215-2225.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects on ratiometric change in JC-1red/JC-1green in hepatocytes treated with TNF−α ± a sensitizing agent as a measure of mitochondrial depolarization were examined using a mitochondria-specific probe, JC-1. Primary rat hepatocytes were seeded to 70% confluence on MatTek glass bottom culture dishes (MatTek, Ashland, MA) overnight. The mitochondrial membrane potential indicator JC-1 (1 mg/mL) was incubated for 30 minutes at 37°C (Invitrogen, Carlsbad, CA); the nuclei indicator Draq 5 was incubated for 5 minutes (1:1000; Biostatus Limited, Leicestershire, UK) and then removed. Cells were maintained at 37°C in a heated micro-incubator (Tokai Hit Co., Shizuoka-ken, Japan) in Williams E media–buffered media plus treatment additives during imaging. A: Images were acquired at 10 different positions using a Nikon Sweptfield (TSI inverted, Nikon Instruments, Melville, NY) 40× objective (0.70 NA) every 5 minutes for 60 minutes. In calculating the red/green fluorescence ratio, background correction was used for each field and time point. B: Inclusion of TNF−α and to a lesser extent H2O2 resulted in a rapid depolarization of the rat hepatocytes mitochondria. Cells treated with Act D alone or media exhibited a gradual mitochondrial depolarization. DIC, differential interference contrast.
TNFR1 accumulates in the mitochondria on treatment(s) with TNF−α + Act D ± JNK II inhibitor (SP600125; 20 μmol/L) ± reactive oxygen species scavenger (NAC; 5 mmol/L). Primary rat hepatocytes were treated with TNF−α (2000 units/mL) and Act D (200 ng/ml) for 1 hour. TNFR1 accumulation in the nucleus occurs with media treatment ± inhibitors and to a lesser extent TA + inhibitors. None of the cells were TUNEL positive. Fluorescent labeling: TNFR1, red; ATP synthase, blue; nuclei, white.