

Abstract
Obliterative bronchiolitis is a frequent, morbid, and usually refractory complication of lung transplantation. Mechanistic study of obliterative bronchiolitis would be aided by development of a relevant model that uses human immune effector cells and airway targets. Our objective was to develop a murine chimera model that mimics obliterative bronchiolitis of lung allograft recipients in human airways in vivo. Human peripheral blood mononuclear cells were adoptively transferred to immunodeficient mice lacking activity of T, B, and NK cells, with and without concurrent transplantations of human small airways dissected from allogeneic cadaveric lungs. Chimerism with human T cells occurred in the majority of recipient animals. The chimeric T cells became highly activated, rapidly infiltrated into the small human airway grafts, and caused obliterative bronchiolitis. In contrast, airways implanted into control mice that did not also receive human peripheral blood mononuclear cell transfers remained intact. In vitro proliferation assays indicated that the chimeric T cells had enhanced specific proliferative responses to donor airway alloantigens. This model confirms the critical role of T cells in development of obliterative bronchiolitis among human lung allograft recipients and provides a novel and easily implemented mechanism for detailed, reductionist in vivo studies of human T-cell responses to allogeneic human small airways.
Chronic lung allograft rejection typically manifests with obliterative bronchiolitis (OB), a small airway fibroproliferative disease process characterized by varying degrees of airway luminal obliteration with fibrinous granulation tissue.1 Lung transplant recipients with OB develop expiratory airflow obstruction, which is often inexorably progressive, and have increased predilection for infection.2 Despite extensive investigation, as well as incremental advances in donor organ preservation, surgical techniques, immunosuppressive regimens, and infection prophylaxis, OB remains the single most frequent cause of late graft dysfunction and death after lung transplantation.2
Current limitations in prevention and treatment of OB likely reflect incomplete understanding of the responsible immunopathogenic mechanisms. Accordingly, having a model to generate novel insights regarding the disease paradigm, and a ready means to test these hypotheses, could be a boon for the development of more effective modalities to prevent or counter this frequent complication of lung transplantation.
Animal models have been immeasurably important in advancing our understanding of many disease processes, including allograft rejection mechanisms.3 With particular respect to pulmonary transplantation, heterotopic tracheal allografts in mice undergo histological abnormalities that reproduce the progression of OB in humans, including an initial lymphocytic infiltration, followed by successive stages of epithelial metaplasia and denudation and, ultimately, intraluminal fibroproliferation.3–7 Although this model successfully recapitulates the histological abnormalities of OB, the system is limited to study of nonhuman immune effector cells and airways.
Mice and humans diverged approximately 70 million years ago, however, and have since evolved striking differences of size, reproductive capability, longevity, diet, and habitat, as well as having varied exposures to particular microbial and viral pathogens.8 All of these factors, and doubtless others, have exerted idiosyncratic selection pressures on the evolution of the respective host defense mechanisms. Even though the immune systems of mice and humans share many common elements and general functions, they also differ in myriad important details.8,9 Not surprisingly, some immunological therapies that appeared promising in murine subjects have not translated into comparably effective human treatments.10,11 The availability of a facile model of human immune effector cell and target organ interactions that mimics the phenotype of clinical disease in situ could be very useful for fundamental studies, and a unique, potentially more valid tool for early trials of immunosuppressive agents and biological response modifiers.
We have developed a novel human-murine chimeric model, in which both the immune effector cells and allograft tissues are of human derivation. Small airways, the primary target structure involved in OB, are harvested from human cadaveric lung explants and are heterotopically implanted into the dorsal subcutaneous tissue of NOD/SCID mice, followed by adoptive transfer of human T cells from a donor allogeneic to the airway donor. In conjunction with anti-murine NK-cell treatments, the adoptively transferred human T cells engraft, proliferate, and mediate airway injuries that mimic OB among lung transplant recipients.1,2 This model provides a ready means to study the immunopathogenesis of lung allograft rejection, and may also be especially useful for early-phase pharmacotherapeutic trials. Minor modifications of this model may also have utility for in situ generation of antigen-specific human T cells in large numbers for further study or other applications.
Materials and Methods
Animals
NOD.CB17-Prkdcscid/J mice (NOD/SCID mice) were acquired from the Jackson Laboratory (Bar Harbor, ME) and maintained under pathogen-free conditions. In some cases, a genetically modified mouse strain that is also deficient in inherent NK-cell activity (NOD/SCID IL-2Rγnull) was similarly maintained and used. This protocol was approved by the Institutional Animal Care and Use Committee and the Institutional Review Board of the University of Pittsburgh.
Human Peripheral Blood Mononuclear Cells for Adoptive Transfers
Blood was obtained from a single healthy volunteer human, allogeneic to the airway donors, and peripheral blood mononuclear cells (PBMNCs) were isolated by Ficoll density gradient centrifugation. The cells were washed in complete RPMI 1640 medium (supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mmol/L l-glutamine, 50 μmol/L 2-mercaptoethanol, and 25 mmol/L HEPES) and then resuspended at 2 × 107 cells/mL in Hank's buffered saline solution (HBSS). A 0.5-mL volume of this suspension was adoptively transferred into each recipient animal by intraperitoneal injection.
In some cases, adoptive transfers consisted of either CD4 or CD8 T-cell populations that had been individually isolated from the human PBMNCs by negative selection with immunomagnetic beads (Miltenyi Biotec, Auburn, CA). Because the numbers of CD8 T cells in the PBMNC donor preparations were often limiting, these adoptive transfers of isolated CD4 or CD8 T cells consisted of 7 × 106 cells/recipient mouse, to maintain comparable inocula.
Human Airway Grafts
Normal human lung explants, the source for small airway grafts, were obtained during cadaveric multiorgan harvests; these explants had been deemed not suitable for therapeutic pulmonary transplantations (eg, due to size or ABO incompatibility). The explants were kept on ice and were processed within 10 hours of harvest. Specimens were excluded if they had evidence of infection or other underlying lung disease by clinical history, radiography, gross examination, or histology. Small airways (2 to 5 mm in diameter, ∼6 mm in length) were harvested from these explants by blunt dissection of peripheral parenchyma. After isolation, the human airways were flushed with complete RPMI 1640 medium, irradiated at 9 Gy to eliminate residual lymphocytes, suspended in freezing medium (10% glycerol:90% fetal bovine sera), and stored at −80°C.
The cryopreserved airways were later thawed in a 37°C water bath and were successively flushed with complete RPMI 1640 medium and then sterile HBSS immediately before subcutaneous dorsal implantation into recipient animals, using sterile technique.12
Hilar Lymph Node Cells
Hilar lymph nodes were dissected from the cadaveric human lung explants at the time of small airway harvest. Single-cell suspensions were made by forcing the nodes through sterile wire mesh. The cells were washed in complete RPMI 1640, resuspended at a concentration of 5 × 107 cells/mL in lymphocyte freezing medium (10% dimethyl sulfoxide, 90% fetal calf serum), and stored at −80°C.
Human-Murine Chimeras
Three treatments were performed in each series of studies: i) airway control mice had heterotopic airway graft (HAG) implantations but did not receive human PBMNCs; ii) PBMNC control mice received adoptive transfers of human PBMNCs, but did not have HAG implantations; and iii) experimental mice had concurrent HAG implantations and human PBMNC adoptive transfers. Within a given experimental series, the HAGs of both airway control and experimental recipients were procured from a single donor lung.
NOD/SCID animals were treated by injections of anti-murine CD122 antibody (0.5 mg i.p.), purified by protein G from supernatants of hybridoma TMB1 and 25 μL of anti-mouse asialo-GM1 antibody (Wako Chemicals, Richmond, VA) twice weekly, beginning on the same day as the adoptive transfers of human PBMNCs or T cells.
Harvests of Tissues and Cells from Recipient Animals
Most treated mice were maintained for 4.5 ± 0.5 weeks, at which time they were euthanized and their spleens and HAGs were harvested. Single-cell suspensions of mouse splenocytes were made by forcing these organs through sterile wire mesh, followed by erythrocyte lysis with ACK buffer (0.15 mol/L NH4Cl, 10 mmol/L KHCO3, and 0.1 mmol/L Na2EDTA, pH 7.3). Splenocytes were repeatedly washed and resuspended in complete RPMI 1640 medium supplemented with 10% fetal calf serum (RPMI-10) for functional studies, or in staining buffer (HBSS supplemented with 0.1% BSA, pH 7.5) for flow cytometry analyses. Circulating leukocytes from recipient mice were also obtained by tail-vein phlebotomy and density gradient centrifugation of blood using Ficoll specific gravity 1.079.
Human airway grafts were harvested and fixed in 10% neutral buffered formalin, paraffin-embedded, sectioned, and mounted on glass slides.
Flow Cytometry
General aspects of these methods and materials have been described previously.13–15 In brief, cell-surface molecule expressions of freshly isolated cells were ascertained using anti-human fluorochrome-conjugated monoclonal antibodies (mAb) against the following cell-surface determinants: CD3, CD4, CD8, CD14, CD19, CD28, CD45, and HLA class II (DR) from BD Biosciences (San Jose, CA).13
In some cases, cells were stained with anti-human T-cell antigen receptor variable β chain (TCRBV) mAb with specificity for 2S1, 3S1, and 4S1 (Immunotech, Marseilles, France), as well as 5S1 and 8S1 (BD Biosciences). These particular TCRBVs correspond to T-cell subpopulations in high frequencies among healthy humans.16 Each of the cell-surface anti-human monoclonal antibodies was tested in pilot trials to confirm that it did not cross-react with murine splenocytes (data not shown).
Intracellular cytokine elaborations were determined in PBMNCs stained with anti-human CD4 and were stimulated for 5 hours with phorbol 12-myristate 13-acetate (PMA) (5 ng/mL) and calcium ionomycin (500 ng/mL) in the presence of GolgiStop protein transport inhibitor (BD Biosciences). Cells were then fixed and permeabilized, and stained with fluorochrome-conjugated mAb against selected human cytokines (or CD68). Reagents for these determinations were purchased from BD Biosciences and were used according to the manufacturer's instructions, as described previously.15
Quantitations were performed on ≥10,000 live cells and analyzed using a FACSCalibur system (BD Biosciences).13–15
Proliferation Assays
Proliferation assays were established with 2 × 105 splenocytes from recipient animals in 200 μL RPMI-10 medium, supplemented with 10 U/mL IL-2, in individual wells of 96-well plates. Splenocytes from all chimeric populations (PBMNC-only controls, HAG only, and experimental mice that had received concurrent HAG and adoptive transfers of PBMNCs) were individually cultured in triplicate with equal numbers of irradiated (20 Gy) stimulator cells from each of three sources: i) syngeneic control stimulator cells consisting of splenocytes from unmanipulated NOD/SCID mice; ii) allogeneic stimulator cells consisting of cryopreserved pulmonary hilar lymph node cells derived from the same human donor as the HAG; and iii) third-party pulmonary hilar lymph node cells that had been harvested from a human lung explant unrelated to either the HAG or human PBMNC donors. These cocultures were incubated in a 7% CO2 environment at 37°C for 3 days before addition of [3H]thymidine (1 μCi), and were harvested 16 hours later; counts per minute were determined with a beta counter.
Rejection Scoring
HAG sections recovered from recipient animals were stained with H&E and were histologically evaluated using a scoring system that has been described previously12 (Table 1). Investigators (M.P.G. and S.R.D.) blinded to treatment assigned the scores based on the most severe histological abnormality present in several sections of each individual HAG.
Table 1.
Histological Scoring of Airway Grafts
| Score | Histological features |
|---|---|
| 0 | Normal |
| 1 | Focal squamous metaplasia over ≤10% of luminal circumference |
| 2 | Squamous metaplasia over >10% of luminal circumference |
| 3 | Focal epithelial denudation over ≤10% of luminal circumference |
| 4 | Denudation comprising >10% of luminal circumference |
| 5 | Intraluminal granulation tissue (obliterative bronchiolitis) |
Source: Duncan et al.12
Immunohistochemistry
Paraffin-embedded HAG sections were deparaffinized in three washes of xylene and rehydrated in successively dilute ethanol baths (100%, 95%, 80%, 70%, 50%). Endogenous peroxidase activity was quenched with 0.05% hydrogen peroxide for 10 minutes. Antigen retrieval was performed for 15 minutes in a pressure cooker using citrate buffer (10 mmol/L sodium citrate, 0.05% Tween 20, pH 6). After three washes in PBS, the sections were blocked with 5% goat serum in PBS for 45 minutes. These sections were then incubated overnight at 4°C with murine anti-human CD3 (Dako, Glostrup, Denmark) at 1:200 dilution. Biotinylated anti-mouse secondary antibody (Vector Laboratories, Burlingame, CA), also at a dilution of 1:200, was then incubated for 30 minutes before immunoperoxidase staining with Vectastain ABC (Vector Laboratories) according to the manufacturer's instructions. After five rinses in PBS, the sections were counterstained for 5 seconds with hematoxylin, and then were mounted with coverslips and mounting medium.
Statistical Analyses
Two-group and three-group comparisons of continuous or ordered data were made by Mann-Whitney U-test or Kruskal-Wallis test, respectively. Replicate measures over time were evaluated by Wilcoxon paired-sample test. Dichotomous variables were analyzed by χ2 test. Data were analyzed using a StatView software package version 5.0 (SAS Institute, Cary, NC). Data are reported as means ± SEM, and P values of <0.05 were considered significant.
Results
Engraftment of Human T Cells in Immunocompromised Mice
Initial attempts to engraft human PBMNCs in B6;129S7-Rag11tm1Mom or NOD/SCID animals without anti-NK-cell treatments were only sporadically and partially successful. However, use of NOD/SCID recipients, in conjunction with twice weekly anti-murine NK-cell mAb (anti-CD122 and anti-AGM1), resulted in far more frequent engraftment and expansion of adoptively transferred human T cells.
Human T cells were evident in the circulation of most recipient animals by 2 weeks and usually increased further during the next 2 weeks (Figure 1A). More importantly, human T cells comprised a significant proportion of cells in the spleens of recipient animals at 4.5 ± 0.5 weeks after adoptive transfers (Figure 1B). There were no consistent differences in the proportions of chimeric human T cells in the spleens between recipient mice treated only with adoptive PBMNC transfers and those that received both PBMNC and HAG implantations (Figure 1B).
Figure 1.
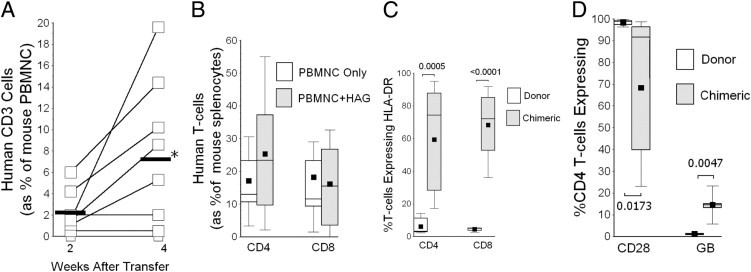
A: Human T cells (as indicated by CD3 surface marker) were evident in the peripheral blood of most recipient mice by 2 weeks after adoptive transfers, and typically increased over the next 2 weeks. *P = 0.04. Heavy horizontal line indicates mean. B: Human T cells comprised significant proportions of cells within spleens of recipient mice after adoptive transfer. There were no significant differences in the extent of splenic chimerism between recipient animals that received only adoptive transfers of peripheral blood mononuclear cells (PBMNC) (n = 15) and those that received concomitant PBMNCs and heterotopic airway grafts (HAG) (n = 14). C: The activation marker HLA class II DR was significantly up-regulated on chimeric T cells (n = 17), compared with expression on the original, donor lymphocytes (n = 6). There were no apparent differences in extent of chimeric T-cell activation between PBMNC only and PBMNC + HAG, so both groups were combined for this analysis. P values are indicated next to brackets. D: A large proportion of chimeric CD4 T cells did not coexpress CD28, but these cells more frequently produced granzyme B, relative to the original donor lymphocytes, which is analogous to findings in human lung allograft recipients with chronic allograft rejection.14P values are indicated next to brackets. In box plots (B–D), the lower whisker denotes the 10th percentile; the lower edge of the box, the 25th percentile; the middle line, the median; the square, the mean; the upper edge of box, the 75th percentile; and the upper whisker, 90th percentile.
There was a significantly greater proliferation of CD8 T cells relative to the initial adoptive transfer inocula (17.4 ± 3.3 fold increase) than of CD4 T-cells (6.0 ± 1.2 fold increase) (P = 0.008). Nonetheless, in the absence of CD4 T cells, adoptive transfers of isolated CD8 T cells alone did not engraft well and/or proliferate (0.3 ± 0.3% of recipient spleen cells, n = 5), whereas pure inocula of CD4 T cells underwent substantial (and perhaps enhanced) proliferation after adoptive transfers, constituting 51 ± 13% of recipient spleen cells at harvest (n = 5).
Human PBMNC leukocyte subpopulations other than T cells did not undergo comparable engraftments or expansions after adoptive transfer, in that ≥98% of human CD45+ cells within recipient spleens coexpressed the specific T-cell marker CD3, whereas the percentage of CD3+/CD45+ among the initial donor human PBMNC inocula, before adoptive transfers into these mice, was 46.4 ± 5.6%.
Characterization of Chimeric T Cells
We hypothesized a priori that adoptively transferred human T cells bearing antigen receptors (TCR) with appropriate avidities would become activated by engagement with recipient xenoantigens, and perhaps also by engagement with human alloantigens among those mice that also received HAG.
Chimeric human T cells within the spleens of murine recipients were highly activated, as evidenced by their increased expressions of HLA class II (DR) compared with the initial donor lymphocytes (Figure 1C). Although a large proportion of circulating CD8 T cells in healthy humans do not express CD28, and many produce granzyme B, analogous findings of CD4 T lymphocytes that lack CD28 expression or make granzyme B are distinctly abnormal findings that show these cells have undergone multiple prior antigen-driven divisions.13,14,17–20 Considerable CD28 down-regulation and granzyme B up-regulation were evident among the chimeric CD4 T cells harvested from these animals (Figure 1D), findings that are highly analogous to those seen in human lung transplant recipients with chronic allograft rejection.14
The cytokine production profile of chimeric T cells was altered, relative to that of the original donor cells, as illustrated here by CD4 lymphocyte elaborations (Figure 2A). Although there was no clear TH1 versus TH2 bias among these chimeric T cells, and, with the exception of IL-2 and tumor necrosis factor-α, their production of other disease-relevant mediators was increased, and in some cases severalfold (eg, interferon-γ, IL-1β, IL-4) (Figure 2A).
Figure 2.
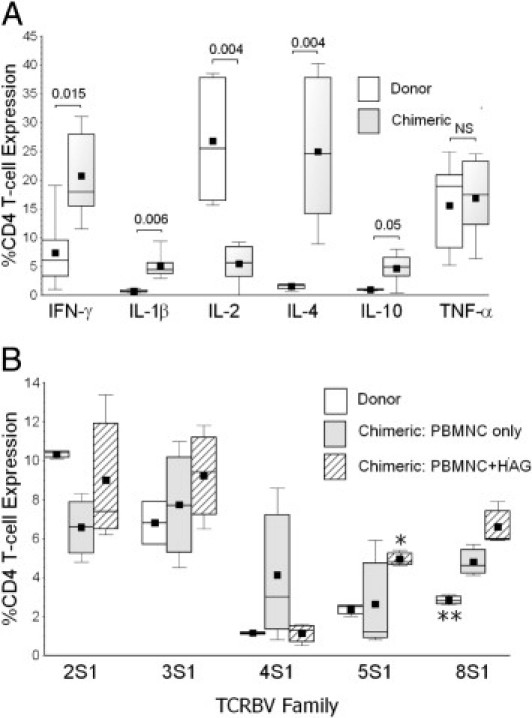
A: Chimeric CD4 T cells in the spleens of recipient animals (n = 12) had altered cytokine production profiles, compared with the original donor T cells before adoptive transfers (n = 5). There were no evident differences in chimeric T-cell cytokine productions among the two subpopulations of recipients (PBMNC versus PBMNC + HAG), and both groups are combined here. P values for intergroup comparisons of each parameter measured are indicated next to brackets. B: T-cell antigen receptor variable β-chain (TCRBV) expression repertoires of chimeric T cells were highly altered, compared with the original donor lymphocytes, as illustrated here by CD4 T-cell TCRBV expression profiles. *P = 0.036, HAG + PBMNC (n = 6) versus PBMNC only (n = 10); **P = 0.0096 , donor T cells (n = 5) versus both chimeric populations. In box plots, the lower whisker denotes the 10th percentile; the lower edge of the box, the 25th percentile; the middle line, the median; the square, the mean; the upper edge of box, the 75th percentile; and the upper whisker, 90th percentile.
T-cell antigen receptor variable β chain repertoires were also biased among chimeric T lymphocytes, indicating preferential expansions and contractions of T cells bearing distinct TCRBV families after adoptive transfers by engagement with xenoantigens and/or alloantigens (Figure 2B). There was a prominent increase of TCRBV 8S1 frequency among both populations of chimeric T cells, relative to the original donor lymphocytes, possibly reflecting preferential activation and expansion of one or more founder T cells bearing this TCRBV that have xenoantigen specificity. TCRBV 5S1 frequency was increased among chimeric T cells of recipients with concurrent HAG transplantation, perhaps reflecting particular expansions of T cells in this subpopulation that have alloantigen specificity.
Alloantigen Avidities of Chimeric Human T Cells
Based on the preceding results, which we interpreted as indicative of cognate in situ T-cell activation and proliferation, we hypothesized that some of the lymphocytes among the recipients with concurrent HAG implantation might represent expansion of daughter progeny with specific avidity for the distinct human alloantigens of the airway allografts.
Splenocytes from all recipient populations had similar baseline thymidine incorporation and de novo responses to irrelevant human antigens of third-party stimulator cells (Figure 3A). However, proliferative responses of the chimeric T cells from recipients with concurrent HAG implantation were significantly increased in cocultures with lymph node cells from the original HAG donor, consistent with a second-set response (Figure 3A).
Figure 3.
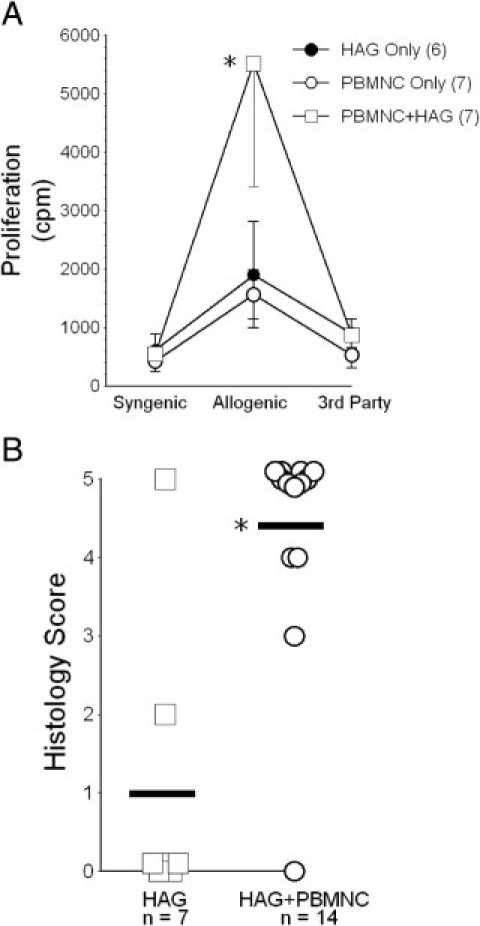
A: Proliferative responses of recipient T cells to allogeneic donor-strain target cells were greatest among those animals that had been previously exposed to the particular alloantigens of the heterotopic airway grafts (HAG). Numbers within parentheses denote number of subjects. *P = 0.02 for the intergroup comparison between HAG + PBMNC versus both other groups. cpm, counts per minute. B: Most heterotopic airway grafts (HAG) from recipient animals that did not also receive peripheral blood mononuclear cells (PBMNC) maintained normal histological appearance. In contrast, HAG among animals with concurrent adoptive transfers of PBMNC allogeneic to the HAG developed severe histological abnormalities. The latter were scored irrespective of whether or not successful T-cell engraftment occurred. Heavy horizontal line indicates mean. *P = 0.003.
Human Airway Heterotopic Grafts as a Model of Obliterative Bronchiolitis
The ultimate aim of these experiments was to develop a model that would enable study of human T-cell responses to allogeneic human airways in vivo.
Pilot studies had shown that human HAGs became heavily enveloped with a richly vascularized fascia originating from the recipient panniculus carnosus21 within a few days of implantation into these mice, as described previously with murine tracheal transplantations.12 Moreover, with only two exceptions (Figure 3B), human HAGs, in animals not reconstituted with human T cells, maintained normal histological appearance and apparent preservation of airway epithelial mucous production for at least 2 months after implantation (Figure 4A).
Figure 4.
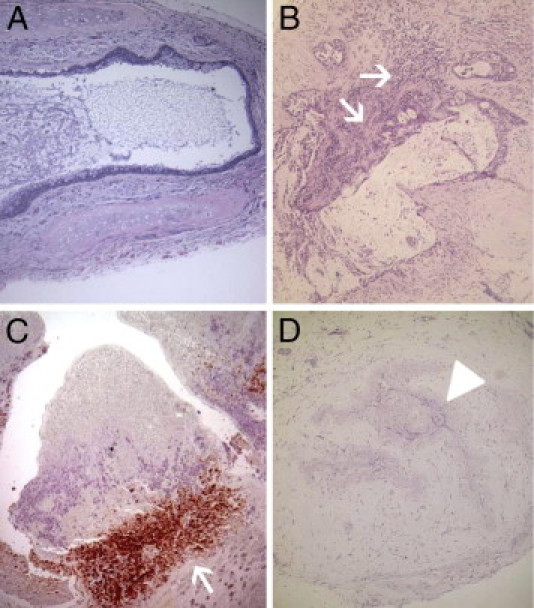
A: Heterotopic airway grafts (HAG) from recipient animals that did not also receive PBMNC transfers typically showed normal appearance. Mucus within the airway lumen indicates preserved secretory function of the airway epithelial cells. B: HAG harvested 3 weeks after implantation with concurrent PBMNC transfers typically showed inflammatory infiltrates (arrows) at the base of fibroproliferative granulation extending into the airway lumen. C: Immunohistochemistry images confirmed these inflammatory infiltrations as human CD3 T cells (arrow). D: Most HAG harvested at 4.5 weeks or later after transplantation into animals that also received PBMNC transfers showed extensive injuries, including loss of airway epithelium and obliteration of airway lumens (arrowhead) with fibroproliferative granulation tissue. Note that the extent of inflammatory infiltration in these later lesions had typically diminished, which is also a common feature of advanced obliterative bronchiolitis in human lung allografts.1 Original magnification: ×10 (A, B, and D); ×20 (C).
However, HAGs implanted into mice that also received adoptive transfers of human PBMNCs showed inflammatory infiltrations within 1 to 2 weeks, along with progressive airway epithelial metaplasia and denudation. Granulation tissue encroaching into the airway lumen was evident in 3 weeks (Figure 4, B and C), and complete luminal obliteration (along with disappearance of the inflammatory cellular infiltrate) occurred soon thereafter in nearly all HAG transplantations (Figures 3B and 4D). HAGs in the animals that received adoptive transfers of pure human CD4 T cells allogeneic to the graft underwent identical histological abnormalities (data not shown).
Studies in NOD/SCID IL-2Rγnull Mice
Analogous experiments were performed using NOD/SCID IL-2Rγnull animals, except that these animals were not treated with the NK-cell-depleting mAb regimen (ie, anti-CD122 and anti-AGM1). This recently developed strain is reportedly deficient in inherent NK-cell activity, as well as being devoid of T and B lymphocytes.22 Aside from obviating the need for frequent injections of the NK mAb, these animals also support engraftment of other human cell lineages, in addition to T cells.23
Identical adoptive transfers of PBMNCs (from the same healthy human donor as for the previous studies) into the NOD/SCID IL-2Rγnull resulted in enhanced engraftment and propagation of activated human T cells (Figure 5, A and B), compared with the original NOD/SCID animals (Figure 1B). Donor human B cells (CD19+), monocytes (CD14+), and some macrophages (CD68+) were also evident in the spleens of the NOD/SCID IL-2Rγnull recipients.
Figure 5.
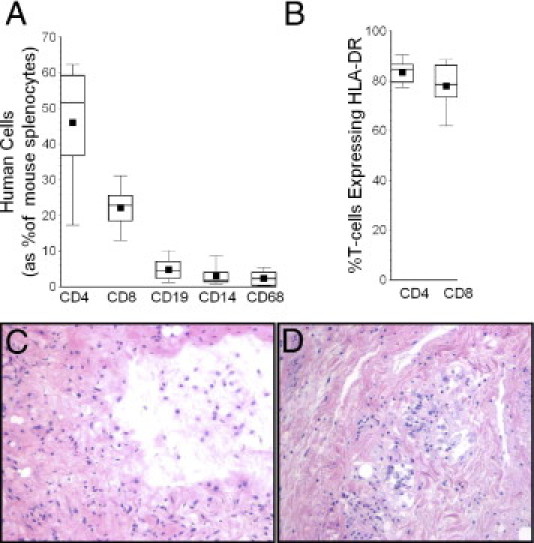
A: Engraftments of T cells and other mononuclear leukocytes in NOD/SCID IL-2Rγnull mice, 4 weeks after adoptive transfers of human PBMNCs from the same donor used for the earlier NOD/SCID experiments (Figures 1–4). Total splenocyte counts in these animals (including chimeric human cells) were 65 ± 14 × 106. B: The chimeric human T cells were highly activated, as evidenced by HLA class II DR expression. C: HAG harvested at 4 weeks from control animals (which had not received adoptive transfers of human PBMNCs) invariably showed loss of small airway epithelium. D: HAG of mice that were also reconstituted with human PBMNCs showed more extensive changes, including focal inflammatory infiltrates and intraluminal granulation tissue. Original magnification, ×10. In box plots, the lower whisker denotes the 10th percentile; the lower edge of the box, the 25th percentile; the middle line, the median; the square, the mean; the upper edge of box, the 75th percentile; and the upper whisker, 90th percentile.
Human small airway grafts did not maintain normal histological appearance after heterotopic transplantation into control NOD/SCID IL-2Rγnull mice (those not receiving PBMNCs) (Figure 5C). No epithelial layer was present in HAGs harvested from these animals 4 weeks after transplantation. In many cases, the airways appeared to be partially collapsed, seemingly due to extrinsic compression and/or loss of structural integrity, but in the absence of dense intraluminal granulation tissue. HAGs harvested from the NOD/SCID IL-2Rγnull that had also been reconstituted with human PBMNCs more closely recapitulated findings of human OB, having dense intraluminal granulation tissue, as well as patchy inflammatory infiltrates (Figure 5D). These experiments were repeated in two experimental trials separated by several weeks, with n ≥ 6 in each experimental arm on both occasions, using airways harvested from two different human cadavers.
Discussion
Our present data show that human airway implantations into NK-cell-depleted NOD/SCID mice that also received adoptive transfers of allogeneic human PBMNCs develop progressive histological changes that recapitulate lesions of chronic allograft rejection in human lung transplant recipients. The chimeric human T cells in recipient animals are highly activated (Figure 1C), and have both skewed TCRBV repertoires (Figure 2B) and enhanced proliferative responses to the alloantigens of the airway grafts (Figure 3A). These findings indicate that the chimeric T-cell expansions within these mice were preferentially biased for those lymphocytes with avidity for mouse xeno- and/or airway allo-antigens, analogous to homeostatic repopulation of lymphopenic hosts.24 In addition, chimeric T cells within recipient animals develop the unusual and potentially highly pathogenic phenotypic and functional changes that occur in human patients with chronic immunological diseases, including lung transplant recipients with OB13,14,17–20 (Figure 1D). Thus, the reconstituted human T-cell population in these chimeric animals appears to successfully mimic key features of the activated, oligoclonal, and end-differentiated cellular immune response of lung transplant recipients with chronic allograft rejection.14,16
This model may have considerable utility for studies of obliterative bronchiolitis (OB) in lung transplant recipients. The methodology is facile per se, and requires only limited surgical skills. A novel and potentially important feature of this model is that both the immune effector cells and the allograft target are of human derivation, and thus are singularly relevant to studies of pathogenic processes that occur in human lung transplant patients. Although useful information has been and will continue to be gleaned from traditional studies of airway and other transplantations among allogeneic mouse strains,3–7 there are a considerable number of differences between mouse and human immune systems.8,9
As one of many examples, oligoclonal daughter progeny of human T cells that have undergone repetitive, antigen-driven proliferations develop markedly dysregulated and pathogenic characteristics and functions, including increased expression of NK-cell receptors, absence of cell-surface CD28, discordant expression of many activation and maturation markers, telomere shortening, and diminished production of FoxP3.13,14,17–20 Unlike normal human lymphocytes, these CD4 T-cell progeny of repetitive prior cell divisions also produce cytolytic mediators, have enhanced facultative and constitutive production of diverse proinflammatory and profibrotic cytokines, and are resistant to antiproliferative effects of calcineurin inhibitors14 and glucocorticoids (unpublished data). The importance of this T-cell differentiation is highlighted by studies showing that the extent of these lymphocytic changes is associated with the manifestations and outcomes of patients affected by diverse chronic immunological diseases,13,18–20 including lung allograft rejection.14 Murine T cells do not undergo comparable phenotypic and functional differentiation after repetitive cycles of proliferation.9,17
Multiple other species-specific differences in structure and functions of adaptive immune mechanisms have been described.8,9 Unlike the T cells of mice, activated human T lymphocytes express MHC class II, which may in turn may play a role (or multiple roles) in amplification of immune responses and/or in homeostatic regulation of these processes.25,26 Calcium influxes during activation of human T cells can occur through Kv1.3 K+ channels, currently under study as a potential target for therapeutic modulation,27 whereas murine T cells do not express this ion-flux apparatus. interferon-α production by macrophages is an important innate mediator of the response to viral infections in humans, with actions that include induction of T-cell STAT4 activation and TH1 polarization. In mice, however, this cytokine neither activates T-cell STAT4 nor promotes TH1 differentiation.28 Moreover, TH polarization of murine T cells generally appears to be relatively more dichotomous and immutable, whereas T cells that cause human diseases may be more plastic or, in any event, pathogenesis can less readily be attributed to distinct TH subpopulations.29
Numerous other important elements of target organs or cells that can influence the nature and extent of adaptive immune responses are also species-specific. Chemokine receptors and their ligands mediate important functions, notably including chemotaxis of immune effectors into inflammatory foci, and numerous elements of these processes in humans are not replicated by murine orthologs.30 Species-specific effects of proinflammatory cytokines on expressions of selectins seem likely to have important differential influences on leukocyte adhesion and ingress into target organs.31 Mice and humans differ broadly in the expression, cellular distribution, and presumably effector functions of their varied Toll-like receptors (TLR),32 among many other potentially relevant innate immune elements.
The capability of allogeneic human T cells to directly initiate and/or mediate chronic allograft injuries is supported by findings in the present study that show a paucity of other chimeric donor cells among the recipient animals, aside from the expanded T cells, and the proximate localization of T cells at the base of the OB granulation lesions (Figure 4, B and C). Moreover, the generation of OB among recipients of pure CD4 T-cell inocula here, as well as analogous findings in other murine model systems6,7,33 illustrates the pathogenic potential of isolated T-cell subpopulations. Thus, the present studies corroborate and extend previous experimental findings that implicate the critical dependence of T-cell alloresponses in the development of OB.16,33–35
Although this human-murine chimeric system appears to have singular relevance for study of OB in lung transplantation recipients, the methodology has some potential limitations (as do most other animal models of human disease). Among other considerations, the apparent inability of this particular mouse strain to support engraftment and expansions of other relevant human immune effector cells (eg, B cells) seems to preclude study of contributions by these other, potentially important elements.
This initial series of studies was focused on development and validation of the model. The operating characteristics of this system were therefore defined in the absence of immunosuppressive treatments, to minimize the number and complexity of experimental variables, with the net result that differences of HAG appearance between the control and immune-reconstituted recipients occurred rapidly and were striking. A similar approach was adopted in development and description of the murine heterotopic airway model,4 which has since seen wide use,5–7,33 as well as in the development of other animal models used to study the immunobiology of transplantation.3 Although not tested here, we hypothesize that treatment of experimental animals with effective immunosuppressants, to recreate conditions in human allograft recipients, could have a measurable effect on the development of OB (for example, it might slow the progression or extent of airway pathology). Moreover, depending on the agent or agents used, addition of immunosuppressants might also alter specific characteristics of the allogeneic T-cell responses (eg, activation, TCRBV repertoire biases, or antigen-specific proliferation). Indeed, one of the rationales for development of this chimeric model was to provide a unique system that will enable subsequent in situ trials of experimental pharmaceuticals to modify human allograft responses. In this context, the use of humanized mice could add biological credence to initial, murine-based preclinical studies, given the many known species-specific differences between immune systems of mice and humans.8,9,25–32
Other potential limitations of heterotopic airway implantations in general have been described,3 although we think that none of these objections obviate the utility of the model described herein. Heterotopic airway implants are not surgically vascularized at the time of implantation and, indeed the lack of having to perform what would be very intricate microsurgical procedures enhances the feasibility of this model for a wider range of investigators. Moreover, the rapid envelopment of the HAG by a highly vascularized fascia, persistence of normal-appearing allografts in control recipient animals (Figure 4A), and rapid cellular influxes into the implanted airways (Figure 4B), speak, in the absence of compelling contrary data, to the adequacy of graft revascularization and inflammatory cell accessibility. It can also be argued that, unlike human lung allografts, heterotopic airway grafts in mice are somehow deficient because they are not exposed to the environment.3 A contrary opinion could be advanced, however, that this lack of environmental exposure precludes experimental confounding by idiosyncratic immunological responses to inhaled antigens that likely differ between, and perhaps even temporally within, individual vivaria. Furthermore, the more generally relevant alloantigen-specific responses that lead to fibroproliferative lesions of chronic rejection that are common to all transplanted organs (including those not exposed to the outside environment, such as hearts, kidneys, or livers), can be more readily parsed in the absence of highly variable inhaled dusts, microbes, and the like in a given environment. In any event, it may also be possible to orthotopically implant cadaveric small human airway grafts in humanized, immune-reconstituted mice to specifically and comparatively study effects of environmental exposure.
The finding that HAG in control (no PBMNC transfers) NOD/SCID IL-2Rγnull mice did not maintain a normal phenotypic appearance was unexpected. The choice of NOD/SCID IL-2Rγnull recipients for chimera generation was intended not only to preclude the necessity of repetitive anti-NK-cell mAb treatments, but also to enable concurrent engraftments of other human lineages, more comparable proliferation of both CD4 and CD8 T-cell subpopulations, and generally more consistent and uniform engraftment of the chimeric human T cells (Figure 5A). Although the HAGs implanted into control NOD/SCID IL-2Rγnull do not evidence the fibroproliferative luminal obliteration of human OB, we believe the lesser distinction between the HAGs harvested from control and experimental animals (Figure 5, C and D, respectively) currently limits the usefulness of this mouse strain for these applications. For now, we can only speculate that the loss of HAG epithelium in the control NOD/SCID IL-2Rγnull might be due to relative deficiency of some cryptic trophic factor, or to compensatory increased activity of another immune element (eg, macrophages).
In summary, the present data show that small human airways can be harvested from cadaveric normal lungs, cryopreserved until convenient for use, and implanted into a particular strain of immunodeficient mice. In the absence of immune reconstitution, these airway xenografts maintain normal histological appearance and epithelial secretory function for prolonged periods. However, concomitant adoptive transfers of human T cells, allogeneic to the airway donor, will engraft and propagate in these animals, and mediate injuries that mimic OB of human lung transplant recipients.
Current medical practices have not been very effective at prevention or treatment of OB in human lung recipients,2 and there is an obvious need to develop better understanding of the causal immunological processes. Humanized mice models offer an inexpensive, ready means for study of relevant human immune responses to human airway allografts in highly reduced detail, and may be particularly useful for the preclinical development and testing of new therapeutics or other approaches that will be necessary to counter this frequent and morbid complication of transplantation.
Footnotes
Supported in part by NIH grants 1R01HL073241 (S.R.D.) and T32HL07563.
J.X. and X.Z. contributed equally to the present work.
Current address of X.Z., Department of Thoracic and Cardiovascular Surgery, Tongji Medical College, Wuhan, China PR 430030.
References
- 1.Stewart S., Fishbein M.C., Snell G.I., Berry G.J., Boehler A., Burke M.M., Glanville A., Gould F.K., Magro C., Marboe C.C., McNeill K.D., Reed E.F., Reinsmoen N.L., Scott J.P., Studer S.M., Tazelaar H.D., Wallwork J.L., Westall G., Zamora M.R., Zeevi A., Yousem S.A. Revision of the 1996 working formulation for the standardization of nomenclature in the diagnosis of lung rejection. J Heart Lung Transplant. 2007;26:1229–1242. doi: 10.1016/j.healun.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 2.Boehler A., Estenne M. Post-transplant bronchiolitis obliterans. Eur Respir J. 2003;22:1007–1018. doi: 10.1183/09031936.03.00039103. [DOI] [PubMed] [Google Scholar]
- 3.Sato M., Keshavjee S., Liu M. Translational research: animal models of obliterative bronchiolitis after lung transplantation. Am J Transplant. 2009;9:1981–1987. doi: 10.1111/j.1600-6143.2009.02770.x. [DOI] [PubMed] [Google Scholar]
- 4.Hertz M.I., Jessurun J., King M.B., Savik S.K., Murray J.J. Reproduction of the obliterative bronchiolitis lesion after heterotopic transplantation of mouse airways. Am J Pathol. 1993;142:1945–1951. [PMC free article] [PubMed] [Google Scholar]
- 5.Neuringer I.P., Aris R.M., Burns K.A., Bartolotta T.L., Chalermskulrat W., Randell S.H. Epithelial kinetics in mouse heterotopic tracheal allografts. Am J Transplant. 2002;2:410–419. doi: 10.1034/j.1600-6143.2002.20503.x. [DOI] [PubMed] [Google Scholar]
- 6.Chalermskulrat W., Neuringer I.P., Brickey W.J., Felix N.J., Randell S.H., Ting J.P., Aris R.M. Hierarchical contributions of allorecognition pathways in chronic lung rejection. Am J Respir Crit Care Med. 2003;167:999–1007. doi: 10.1164/rccm.200209-1099OC. [DOI] [PubMed] [Google Scholar]
- 7.Richards D.M., Dalheimer S.L., Ehst B.D., Vanasek T.L., Jenkins M.K., Hertz M.I., Mueller D.L. Indirect minor histocompatibility antigen presentation by allograft recipient cells in the draining lymph node leads to the activation and clonal expansion of CD4+ T cells that cause obliterative airways disease. J Immunol. 2004;172:3469–3479. doi: 10.4049/jimmunol.172.6.3469. [DOI] [PubMed] [Google Scholar]
- 8.Mestas J., Hughes C.C.W. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 9.Haley P.J. Species differences in the structure and function of the immune system. Toxicology. 2003;188:49–71. doi: 10.1016/s0300-483x(03)00043-x. [DOI] [PubMed] [Google Scholar]
- 10.Panitch H.S., Hirsch R.L., Haley A.S., Johnson K.P. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet. 1987;1(8538):893–895. doi: 10.1016/s0140-6736(87)92863-7. [DOI] [PubMed] [Google Scholar]
- 11.Monaco A.P. Chimerism in organ transplantation: conflicting experiments and clinical observations. Transplantation. 2003;75(9 Suppl):13S–16S. doi: 10.1097/01.TP.0000067945.90241.F4. [DOI] [PubMed] [Google Scholar]
- 12.Duncan S.R., Capetanakis N.G., Lawson B.R. Theofilopoulos AN: Thymic dendritic cells traffic to thymuses of allogeneic recipients and prolong graft survival. J Clin Invest. 2002;109:755–764. doi: 10.1172/JCI12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilani S.R., Vuga L.J., Lindell K.O., Gibson K.F., Xue J., Lindsay E.K., Kaminski N., Valentine V.G., George M.P., Steele C., Duncan S.R. D28 down-regulation on circulating CD4 T-cells is associated with poor prognoses of patients with idiopathic pulmonary fibrosis. PLoS One. 2010;5:e8959. doi: 10.1371/journal.pone.0008959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Studer S.M., George M.P., Zhu X., Song Y., Valentine V.G., Stoner M.W., Sethi J.M., Steele C., Duncan S.R. CD28 down-regulation on CD4 T cells is a marker for graft dysfunction in lung transplant recipients. Am J Respir Crit Care Med. 2008;178:765–773. doi: 10.1164/rccm.200701-013OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu X., Gadgil A.S., Givelber R., George M.P., Stoner M.W., Sciurba F.C., Duncan S.R. Peripheral T-cell functions correlate with the severity of chronic obstructive pulmonary disease. J Immunol. 2009;182:3270–3277. doi: 10.4049/jimmunol.0802622. [DOI] [PubMed] [Google Scholar]
- 16.Duncan S.R., Leonard C., Theodore J., Lega M., Girgis R.E., Rosen G.D., Theofilopoulos A.N. Oligoclonal CD4(+) T-cell expansions in lung transplant recipients with obliterative bronchiolitis. Am J Respir Crit Care Med. 2002;165:1439–1446. doi: 10.1164/rccm.2107009. [DOI] [PubMed] [Google Scholar]
- 17.Vallejo A.N., Weyand C.M., Goronzy J.J. T-cell senescence: a culprit of immune abnormalities in chronic inflammation and persistent infection. Trends Mol Med. 2004;10:119–124. doi: 10.1016/j.molmed.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 18.Martens P.B., Goronzy J.J., Schaid D., Weyand C.M. Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum. 1997;40:1106–1114. doi: 10.1002/art.1780400615. [DOI] [PubMed] [Google Scholar]
- 19.Liuzzo G., Goronzy J.J., Yang H., Kopecky S.L., Holmes D.R., Frey R.L., Weyand C.M. Monoclonal T-cell proliferation and plaque instability in acute coronary syndromes. Circulation. 2000;101:2883–2888. doi: 10.1161/01.cir.101.25.2883. [DOI] [PubMed] [Google Scholar]
- 20.Goronzy J.J., Matteson E.L., Fulbright J.W., Warrington K.J., Chang-Miller A., Hunder G.G., Mason T.G., Nelson A.M., Valente R.M., Crowson C.S., Erlich H.A., Reynolds R.L., Swee R.G., O'Fallon W.M., Weyand C.M. Prognostic markers of radiographic progression in early rheumatoid arthritis. Arthritis Rheum. 2004;50:43–54. doi: 10.1002/art.11445. [DOI] [PubMed] [Google Scholar]
- 21.McFarland H.L., Rosenberg A.S. Skin allograft rejection: Current Protocols in Immunology. In: Coligan J.E., Bierer B.E., Margulies D.H., Shevach E.M., Strober W., editors. John Wiley & Sons; Hoboken, NJ: 2006. pp. 4.4:1–4.4.13. [Google Scholar]
- 22.King M., Pearson T., Shultz L.D., Leif J., Bottino R., Truco M., Atkinson M.A., Wasserfall C., Herold K.C., Woodland R.T., Schmidt M.R., Woda B.A., Thompson M.J., Rossini A.A., Greiner D.L. A new Hu-PBL model for the study of human islet alloreactivity based on NOD-scid mice bearing a targeted mutation in the IL-2 receptor gamma chain gene. Clin Immunol. 2008;126:303–314. doi: 10.1016/j.clim.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Shultz L.D., Lyons B.L., Burzenski L.M., Gott B., Chen X., Chaleff S., Kotb M., Gillies S.D., King M., Mangada J., Greiner D.L., Handgretinger R. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2Rgnull mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 24.Datta S., Sarvetnick N. Lymphocyte proliferation in immune-mediated disease. Trends Immunol. 2009;9:430–438. doi: 10.1016/j.it.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Barnaba V., Watts C., de Boer M., Lane P., Lanzavecchia A. Professional presentation of antigen by activated human T cells. Eur J Immunol. 1994;24:71–75. doi: 10.1002/eji.1830240112. [DOI] [PubMed] [Google Scholar]
- 26.Taams L.S., van Eden W., Wauben M.H. Antigen presentation by T cells versus professional antigen-presenting cells (APC): differential consequences for T cell activation and subsequent T cell-APC interactions. Eur J Immunol. 1999;5:1543–1550. doi: 10.1002/(SICI)1521-4141(199905)29:05<1543::AID-IMMU1543>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 27.Wulff H., Calabresi P.A., Allie R., Yun S., Pennington M., Beeton C., Chandy K.G. The voltage-gated Kv1.3 K(+) channel in effector memory T cells as new target for MS. J Clin Invest. 2003;111:1703–1713. doi: 10.1172/JCI16921. [Erratum appeared in J Clin Invest 2003;112:298] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farrar J.D., Smith J.D., Murphy T.L., Leung S., Stark G.R., Murphy K.M. IL-12, but not IFN-alpha, promotes STAT4 activation and Th1 development in murine CD4+ T cells expressing a chimeric murine/human Stat2 gene. J Immunol. 2005;174:294–301. doi: 10.4049/jimmunol.174.1.294. [DOI] [PubMed] [Google Scholar]
- 29.Gor D.O., Rose N.R., Greenspan N.S. TH1-TH2: a procrustean paradigm. Nat Immunol. 2003;4:503–505. doi: 10.1038/ni0603-503. [DOI] [PubMed] [Google Scholar]
- 30.Zlotnik A., Yoshie O., Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 2006;7:243. doi: 10.1186/gb-2006-7-12-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pan J., Xia L., McEver R.P. Comparison of promoters for the murine and human P-selectin genes suggests species-specific and conserved mechanisms for transcriptional regulation in endothelial cells. J Biol Chem. 1998;273:10058–10067. doi: 10.1074/jbc.273.16.10058. [DOI] [PubMed] [Google Scholar]
- 32.Rehli M. Of mice and men: species variations of Toll-like receptor expression. Trends Immunol. 2002;23:375–378. doi: 10.1016/s1471-4906(02)02259-7. [DOI] [PubMed] [Google Scholar]
- 33.Higuchi T., Jaramillo A., Kaleem Z., Patterson G.A., Mohanakumar T. Different kinetics of obliterative airway disease development in heterotopic murine tracheal allografts induced by CD4+ and CD8+ T cells. Transplantation. 2002;74:646–651. doi: 10.1097/00007890-200209150-00010. [DOI] [PubMed] [Google Scholar]
- 34.Kitamura H., Tsubakihara M., Inayama Y., Ito T., Kanisawa M. Long-term maintenance of human distal airway epithelial cells in nude mice: a potentially useful model for the study of pulmonary carcinogenesis and lung cell biology. Lab Invest. 1990;62:383–389. [PubMed] [Google Scholar]
- 35.Yonou H., Yokose T., Yoshikawa T., Kanomata N., Kamijo T., Hasebe T., Nagai K., Ito H., Yamasaki A., Hatano T., Ogawa Y., Emura M., Ochiai A. Engraftment of adult human lung tissue in nonobese diabetic/severe combined immunodeficient mice: a novel lung epithelial regeneration model. Pathobiology. 2004;71:93–102. doi: 10.1159/000074422. [DOI] [PubMed] [Google Scholar]