

Abstract
Type 2 granular corneal dystrophy (GCD2) is caused by point mutation R124H in the transforming growth factor-β–induced gene (TGFBI) and is characterized by age-dependent progression of corneal deposits. Mitochondrial features in heterozygous GCD2 and normal corneal tissues was evaluated using electron microscopy. Primary corneal fibroblasts of homozygous and normal corneas were cultured to passage 4 or 8. Keratocytes of normal corneal tissue are narrow, and details of their intracellular organelles are difficult to distinguish. Keratocytes of heterozygous GCD2 tissues exhibited many degenerative mitochondria. MitoTracker and cytochrome c staining demonstrated increased mitochondrial activity in mutated cells at early passages. Decreases in depolarized mitochondria, cellular proliferation, and expression of complexes I to V and increases in apoptotic change were observed in late-passage mutant fibroblasts. PGC-1α, ANT-1, p-Akt, and p-mTOR but not NF-κB expression demonstrated a passage-dependent decrease in all cells. Increased passage- or mutation-related intracellular reactive oxygen species and delayed proliferation of methanethiosulfonate (MTS) were recovered using application of antioxidant butylated hydroxyanisole. Mitochondrial features and function were altered in mutated GCD2 keratocytes, in particular in older cells. Alteration of mitochondrial function is critical for understanding the pathogenesis of GCD2.
The corneal manifestation of type 2 granular corneal dystrophy (GCD2) involves age-dependent progression of opacity, with clinically severe vision impairment.1–3 The size and number of deposits change slowly but worsen progressively. A mutation of codon 124 that changes arginine to histidine in the transforming growth factor β-induced gene (TGFBI, formerly known as BIGH3) on human chromosome 5 (5q31) causes the condition.4,5 This mutation is also responsible for abnormal protein deposits in the corneal stroma of GCD2. The major component of these deposits is TGFBI protein (TGFBIp, also known as BIGH3).6,7 TGFBIp contains an N-terminal signal peptide that targets the protein to the lumen of the endoplasmic reticulum and the trans-Golgi network for eventual secretion.8–10 The corneal dystrophy–associated R124H mutation causes aberrant redistribution of the mutant TGFBI into lysosomes.8 Excessive accumulation of mutated protein are observed in the cytoplasm, and this abnormal distribution may influence cellular metabolism.
Mitochondria are essential to cellular metabolism and cell survival, and any deviation from their normal operation is certainly fatal. Aging is frequently characterized by accumulation of altered proteins and dysfunctional mitochondria. The effect of energy metabolism on proteostasis, in particular the formation and elimination of altered proteins, is also vital to cell survival and degeneration.11 In other diseases associated with abnormal protein accumulation such as Alzheimer disease, increasing oxidative stress and altered mitochondrial dynamics contribute to the course of the disease and functional decline.12 The accumulation of mutated protein in GCD2, which becomes diverted from the ordinary protein secretion pathway, may induce intracellular damage and an altered intracellular metabolic state by affecting mitochondrial function. Moreover, extracellular deposits of mutated protein could lead to cellular toxicity.
In the present study, we compared the structure and function of mitochondria in wild-type and GCD2 mutated corneal tissues and fibroblasts, and observed that the GCD2 mutation alters mitochondrial function, potentially explaining the age-dependent progression of the disease.
Materials and Methods
Corneal Tissues
Corneal tissues were obtained during keratoplasty from patients with GCD2, in whom DNA analysis confirmed heterozygosity for the R124H TGFBI gene mutation. Healthy corneal tissues were supplied by the Yonsei University Eye Bank. The study was performed in accordance with the Helsinki agreement, and informed consent was obtained from all patients. Corneas were stored in 2.5% glutaraldehyde in PBS solution overnight at 4°C.
Primary Culture of Homozygote and Wild-Type Corneal Fibroblasts
Primary corneal fibroblast cultures were prepared from healthy corneas from 20-year-old donors obtained from the eye bank or from mutant corneal fibroblasts from 8-year-old patients homozygous for GCD2 after penetrating keratoplasty (cornea tissue study approved by Severance Hospital Institutional Review Board, IRB-2006-0139). Homozygous GCD2 diagnoses were based on the results of DNA analysis for the R124H TGFBIp gene mutation using peripheral blood samples. Stromal explants were prepared by removing the epithelium and endothelium and culturing in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum at 37°C in 5% CO2 in six-well tissue culture plates. Corneal fibroblasts migrated from the explants along the bottom of the plates. Cells were confluent within 15 to 21 days, after which they were enzymatically detached using 0.05% trypsin at 37°C for 3 minutes and centrifuged at 1400 rpm for 5 minutes, and the supernatant was removed. The cell pellet was resuspended in 20 mL medium and cultured in 75-mL flasks at 37°C in 5% CO2 until confluent. Cells were then serially trypsinized and passaged three or seven times before use in experiments. In preparation for experiments, cells were plated at concentrations of 3 to 5 × 103 per well in 96-well tissue culture plates and incubated in 1 mL 10% FBS in DMEM at 37°C in 5% CO2 for 24 to 48 hours.
Transmission Electron Microscopic Examination of Wild-Type and Mutant Corneal Tissue and Fibroblasts
Corneal tissue or cultured cells were fixed in 1% OsO4 in phosphate buffer for approximately 90 minutes at room temperature, washed twice in phosphate-buffered fixative vehicle, and dehydrated in a graded ethanol series. The transition from 100% ethanol to epoxy was mediated by two changes of propylene oxide, and a pure epoxy medium was used for infiltration and embedding. The fragments were mounted in flat molds and hardened at 80°C overnight before sectioning. Both 600- to 1000-nm sections and 60- to 80-nm sections for transmission electron microscopy were cut onto polyvinyl butyral–coated grids (Pioloform; Sigma-Aldrich Corp., St. Louis, MO). One-micrometer to 2-μm sections were stained using alkaline toluidine blue and previewed using light microscopy to select areas for electron microscopic analysis. Selected sections were stained using saturated aqueous uranyl acetate and lead citrate. Evaluation was performed using a transmission electron microscope (JEM1200 EX2; JEOL Ltd., Tokyo, Japan).
MitoTracker and Cytochrome c Staining
Mitochondria were detected using MitoTracker stock solution (Invitrogen Corp., Carlsbad, CA). Cells were plated in two-well chambers, 1 × 104 cells per well. After 24 hours, cells were incubated with 10% fetal bovine serum in DMEM, washed with fresh prewarmed serum-free DMEM, and fixed in DMEM containing 4% formaldehyde for 1 hour. After incubating with PBS containing 0.2% Triton X-100 for 10 minutes, the cells were washed with PBS containing 0.02% Tween-20 and treated with 1% bovine serum albumin for 5 minutes. Then the cells were stained with 1 mmol/L MitoTracker Red 580 (Molecular Probes, Inc., Eugene, OR) for 30 minutes. After staining, cells were washed three times with serum-free DMEM. For cytochrome c staining, cells were incubated with goat anti–cytochrome c antibody (Abcam, Inc., Cambridge, MA) to a final working concentration of 1:650 in PBS containing 3% bovine serum albumin. After several washes in buffer, fluorescein isothiocyanate (green)–labeled anti-mouse IgG (1:200 dilution; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) was applied as a secondary antibody. Images of stained cells were acquired using a confocal microscope (TCS-SP5; Leica Microsystems Inc., Bannockburn, IL), and were analyzed using Scion Image software (Scion Corp., Frederick, MD).
Annexin V Staining
Cells were stained with annexin V (BioVision, Inc., Mountain View, CA) and incubated with avidin–horseradish peroxidase complex (1:300), and binding was visualized using 0.05% diaminobenzidine or 0.01% H2O2 solution. Cells were photographed using a fluorescence microscope equipped with an annexin V filter.
JC-1 Assay for Mitochondrial Membrane Potential
The mitochondrial membrane potential was assayed using the MitoProbe JC-1 Assay Kit for Flow Cytometry (Molecular Probes, Inc.) according to the manufacturer's directions. In brief, cells were washed and then incubated with 2 μmol/L JC-1 at 37°C in 5% CO2 for 30 minutes. Cells were washed with 2 mL PBS and pelleted using centrifugation, then resuspended in 500 μL PBS. Cells were analyzed using a flow cytometer with 488-nm excitation using emission filters appropriate for Alexa Fluor 488 dye (Molecular Probes, Inc.) and R-phycoerythrin. The dye, which exists as a green fluorescent monomer (excitation, 485 nm; emission, 535 nm) in the cytosol, is absorbed by healthy mitochondria and forms a red fluorescent aggregate (excitation, 550 nm; emission, 600 nm).
MTS and CCK-8 Assay
Cell proliferation was measured using the methanethiosulfonate [MTS; 3-(4,5-dimethylthiazol-2-yl)−5-(3-carboxymethoxyphenyl)−2-(4-sulfophenyl)-2H-tetrazolium] mix–based cell titer assay and Cell Counting Kit-8 (CCK-8) assay after 6, 24, and 48 hours of culture. Cells were plated in 96-well plates at 1 × 104 cells per well overnight. Cell proliferation was determined using the Cell Titer 96 AQueous One Solution Reagent Cell Proliferation Assay Kit with the tetrazolium compound (inner salt; MTS. (Promega Corp., Madison, WI). In brief, the culture medium was removed, and 20 μL MTS solution was added to each well in 100 μL culture medium. The reaction was terminated by adding 10% SDS, and optical density was measured using a plate reader with a filter setting at 450 nm.
For the CCK-8 assay, 10 μL CCK-8 solution (Dojindo Laboratories, Kumamoto, Japan) was added to each well in 100 μL culture medium. The cultures were incubated at 37°C for 2 to 4 hours under 95% humidity and 5% CO2. Optical density was measured using a plate reader with a filter setting of 490 nm.
Western Blot Analysis
Cells were lysed by incubation in 50 mmol/L Tris HCl (pH 7.5), 120 mmol/L NaCl, 20 mmol/L NaF, 1 mmol/L EDTA, 5 mmol/L EGTA, 15 mmol/L sodium pyrophosphate, 30 mmol/L p-nitrophenyl phosphate, 1 mmol/L benzamidine, 0.1 mmol/L phenylmethylsulfonyl fluoride, and 1% Nonidet P-40 for 20 minutes at 4°C and centrifuged at 15,000 × g for 15 minutes at 4°C. Cell lysates were boiled in Laemmli's sample buffer for 3 minutes, and 30 μg protein was subjected to SDS-PAGE. Proteins were then transferred to polyvinylidene difluoride membranes. The membranes were blocked for 30 minutes in Tris-buffered saline solution containing 0.1% Tween-20 and 5% (w/v) dry skim milk powder, then incubated overnight with primary antibodies [Total OXPHOS Complexes Detection Kit (1: 1000); Mitosciences Inc., Eugene, OR], PGC-1α (Novus Biologicals, LLC, Littleton, CO), ANT-1 (Abcam Inc.), VDAC (Cell Signaling Technology, Inc., Beverly, MA), mTsR, p-mTor (Ser 2448, Ser2481) Akt, or p-Akt (Ser473, Thr 308) (Cell Signaling Technology, Inc.). Nuclear extraction was performed using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific Inc., Waltham, MA), and the membranes were incubated with primary antibody of NF-κB (1:1000) (Cell Signaling Technology, Inc.) or PGC-1α. The membranes were then washed with Tris-buffered saline solution containing 0.1% Tween-20, incubated for 1 hour with a secondary antibody, and visualized using an enhanced chemiluminescence detection kit (Amersham Life Science, Arlington Heights, IL).
Measurement of Reactive Oxygen Species Production
Intracellular reactive oxygen species (ROS) generation was measured using CM-H2DCFDA (5,6-chloromethyl- 2′,7′-dichlorodihydrofluorescein diacetate) (Molecular Probes, Inc.). Cells, 3 × 105, were plated on 35-mm dishes and cultured for 24 hours. The cells were co-cultured with or without 100 μmol/L butylated hydroxyanisole (BHA) in phenol red–free medium. After stimulation, the cells were rinsed once with 2 mL Kreb's-Ringer's solution and incubated for 5 minutes with 5 μmol/L CM-H2DCFDA. The plate was mounted, and the dichlorofluorescein (DCF) fluorescence images were immediately acquired using fluorescence microscopy (Axiovert 200; Carl Zeiss MicroImaging, Inc., Thornwood, NY). Finally, the fluorescence intensities of 80 to 100 cells were measured and averaged using a Scion imaging system.
Statistical Analysis
Analysis of variance was used to compare cell viability detected in MTS and CCK-8 assays. The results for intensity of staining, the ratio of red to green fluorescence, expression of protein complexes, and expression of mitochondria-related factors were analyzed using the Student's t-test. Statistical analysis was performed using commercially available software (SigmaStat 3.5; Systat Software, Inc., San Jose, CA).
Results
Altered Mitochondrial Number and Structure in Mutant Corneal Tissue and Fibroblasts
The keratocytes of normal cornea have a characteristic narrow spindle shape. The cytoplasm of the cells is densely colored, and the intracellular organelles are barely discernible. The keratocytes of heterozygous GCD2 corneal tissue, in particular near the stromal deposits, were enlarged, and the intracellular organelles were clearly visible. These cells contained many dilated or degenerative mitochondria and vesicles containing amorphous material. Some of the keratocytes encircled deposits, and extended cellular processes reached the abnormal deposits (Figure 1). Wild-type cultured fibroblasts contained small round mitochondria and other cellular components in passages 4 and 8, whereas the homozygous mutant cells demonstrated elongated and swollen mitochondria with increasing numbers throughout the cytoplasm and limited numbers of other intracellular organelles. In passage 8 cultures of corneal fibroblasts with the homozygous genotype, fewer disorganized and dilated mitochondria were observed (Figure 2).
Figure 1.
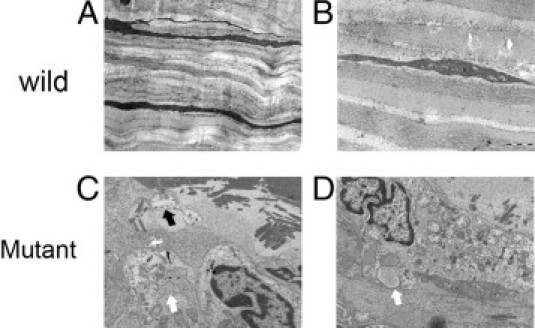
Electron microscopic images of wild-type and mutant corneal tissue and fibroblasts. A: Keratocytes of healthy cornea demonstrate the characteristic narrow spindle shape. B: Cytoplasm of the cell shows dense color, and details of the intracellular organelles are barely discernible. C: Enlarged keratocytes of heterozygous corneal tissue clearly demonstrate intracellular organelles. Some keratocytes encircled the deposits and extended cellular processes that reach the abnormal deposits (black arrow). D: Cells contained many dilated or degenerative mitochondria and vesicles containing amorphous material (white arrow). Original magnification: ×2000 (A); ×10,000 (B–D).
Figure 2.
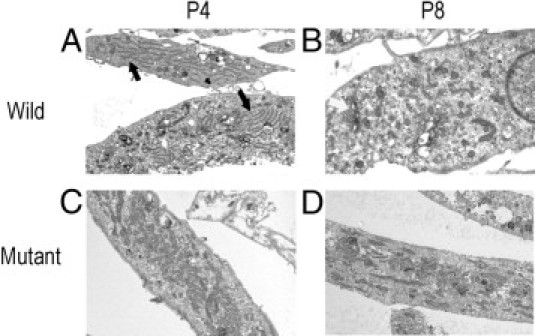
Electron microscopic images of cultured corneal fibroblasts. Wild-type cultured fibroblasts contain small round mitochondria and other cellular components such as endoplasmic reticulum (black arrow) in passage 4 (A) and Golgi complex (white arrow) in passage 8 (B). C: Homozygote cultured fibroblasts demonstrate elongated and swollen mitochondria with increasing numbers throughout the cytoplasm and limited numbers of other intracellular organelles. D: In late-passage homozygous mutant corneal fibroblasts, disorganized and dilated mitochondria with decreased numbers are observed.
Decreased Mitochondrial Activity in Late-Passage Mutant Cells
Compared with wild-type corneal fibroblasts, passage 4 mutant corneal fibroblasts exhibited strong MitoTracker and cytochrome c staining, which detect mitochondrial activity. Wide enlarged cellular contours were also observed in the mutant cells. In passage 8, MitoTracker and cytochrome c staining intensity of mutant corneal fibroblasts decreased, whereas staining in the wild-type corneal fibroblasts was unchanged. The intensity of staining was analyzed, and in both staining procedures, a statistically significant decrease was observed in mutant cells in passage 8 compared with mutant cells in passage 4 (Figure 3).
Figure 3.
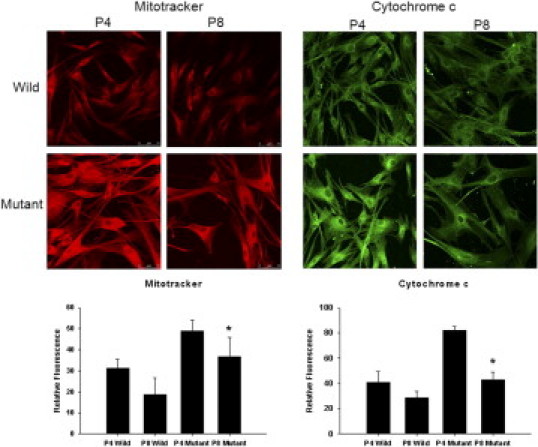
MitoTracker and cytochrome c staining of wild-type and mutant corneal fibroblasts from passages 4 and 8. Wild-type cells demonstrate similar intensity of MitoTracker and cytochrome c staining in passages 4 and 8. Mutant corneal fibroblasts exhibit strong intensity of MitoTracker and cytochrome c staining in passage 4. However, in passage 8, the intensity of both markers is decreased significantly. *P < 0.05; Student's t-test, passage 4 versus passage 8.
Decreased Mitochondrial Depolarization in Late-Passage Mutant Corneal Fibroblasts
When the mitochondrial membrane potential (ΔΨm) is high, JC-1 accumulates in the mitochondrial matrix by forming J-aggregates with red fluorescence. However, when the mitochondrial membrane potential is low, JC-1 is a monomer with green fluorescence. A dot plot of red fluorescence versus green fluorescence differentiates live cells with intact mitochondrial membrane potential from apoptotic and dead cells with lost mitochondrial membrane potential. The ratio of red to green fluorescence did not change in wild-type corneal fibroblasts; however, it was decreased in mutant corneal fibroblasts, in particular in passage 8 (Figure 4).
Figure 4.
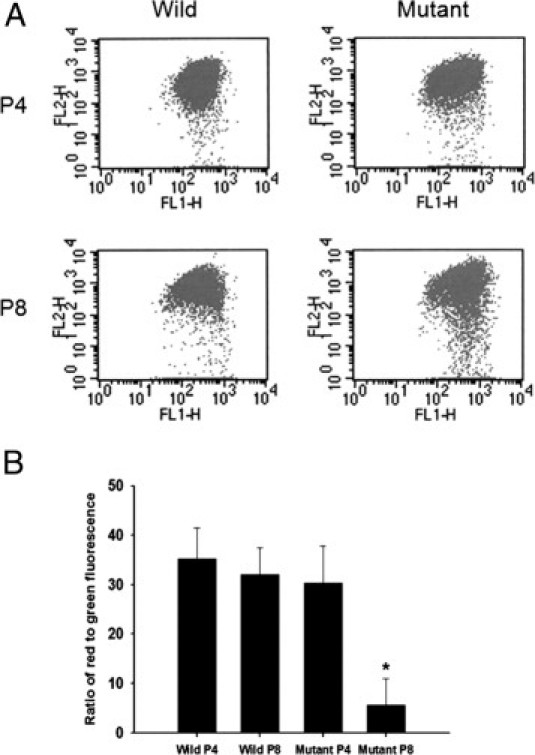
JC-1 assay of wild-type and mutant corneal fibroblasts in passages 4 and 8. A: Dot plot demonstrates increasing numbers of dots in the lower part in passage 8 mutant cells. B: Graph indicates the ratio of red to green fluorescence, which is decreased in mutant corneal fibroblasts, especially in passage 8. *P < 0.05; Student's t-test, passage 4 versus passage 8.
Decreased Viability of Mutant Corneal Fibroblasts
Wild-type corneal fibroblasts in passages 4 and 8 and mutant cells in passage 4 demonstrated time-dependent increases in detection of MTS and CCK-8. However, the rate of increase in MTS and CCK-8 detection was slowed in mutant corneal fibroblasts in passage 8 at 24 and 48 hours (Figure 5A). At annexin V staining, cytoplasmic shrinkage and perimembranous stippling were observed more frequently in the GCD2 homozygote cells in both passages, whereas such staining was less apparent in wild-type cells in both passages (Figure 5B).
Figure 5.
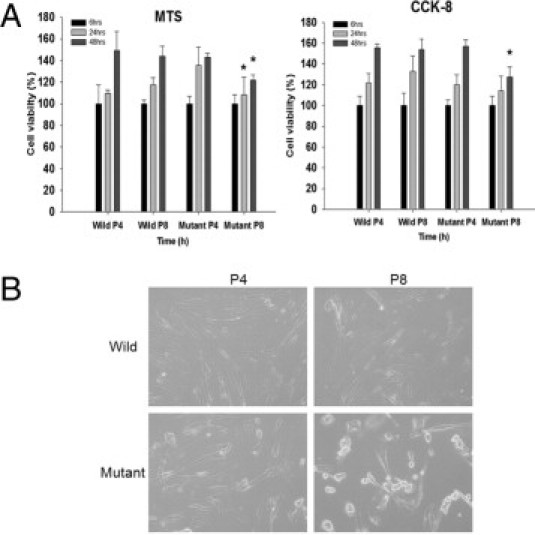
MTS and CCK-8 assays of wild-type and mutant corneal fibroblasts in passages 4 and 8. A: Wild-type corneal fibroblasts in passages 4 and 8 and mutant cells in passage 4 demonstrate time-dependent proliferation in both the MTS and CCK-8 assays. However, the column of mutant corneal fibroblasts in passage 8 after 48 hours is significantly smaller in both assays. *P < 0.05; analysis of variance, passage 4 versus passage 8. B: At annexin V staining, cells undergoing apoptosis experienced cytoplasmic shrinkage and perimembranous stippling. GCD2 homozygote cells demonstrate typical characteristics of apoptosis in both passages, whereas such staining is less apparent in both passages of wild-type cells.
Expression of Mitochondrial Enzymes and Proteins Related to Mitochondrial Function Is Altered in Late-Passage Mutant Corneal Fibroblasts
To determine whether alterations occur in the mitochondrial protein content in the mutant corneal fibroblasts, the levels of randomly selected subunits of complexes I to V of the respiratory chain were measured using Western blot analysis. Statistically significant reductions in complexes I, II, and IV were observed in late-passage mutant corneal fibroblasts; whereas the level of complex III was increased in late-passage wild-type corneal fibroblasts, and the levels of complexes I, II, IV, and V were unchanged (Figure 6).
Figure 6.
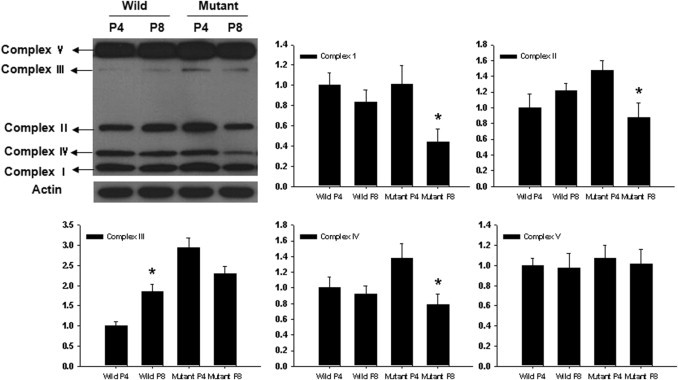
Western blot analysis of complexes I to V of wild-type and mutant corneal fibroblasts in passages 4 and 8. Expression of complexes I, II, and IV was statistically significantly reduced in late passages of mutant corneal fibroblasts, whereas that of complex III was increased in the late passage of wild-type. Levels of complexes I, II, IV, and V were unchanged in wild-type. The y axis of each graph indicates the relative ratio considering the expression level of wild-type fibroblasts in passage 4 as 1.0.
Expression of ANT-1 demonstrated a statistically significant decrease in late-passages mutant cells, and PGC-1α, p-Akt (Ser 473), p-Akt (Thr 308), and p-mTOR (Ser 2481) exhibited a statistically significant decrease in late passages of both wild-type and mutant cells (Figure 7A). To test the effect of PGC-1α expression on NF-κB in nulcei, the expression of both was analyzed using Western blot analysis in nuclear extract. The expression pattern of PGC-1α in cellular extract did not demonstrate the direct correlation with NF-κB expression in nuclei; however, expression patterns of PGC-1α and NF-κB in nuclear extract were similar (Figure 7B).
Figure 7.
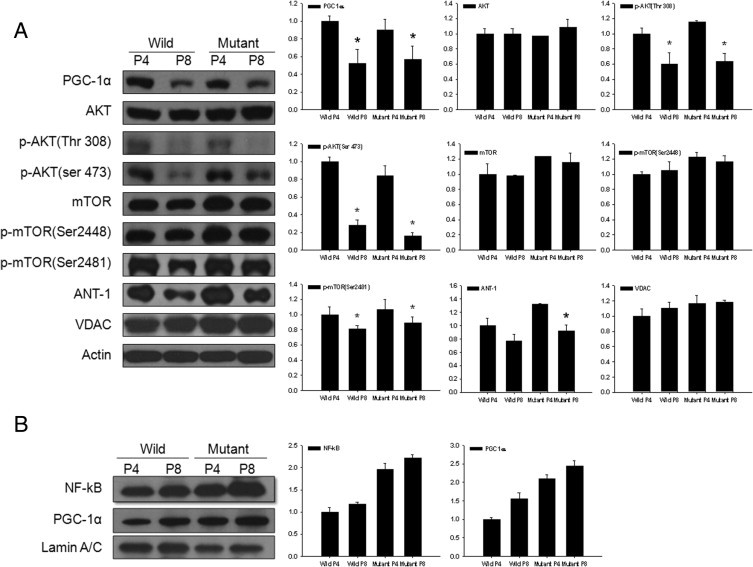
Western blot analysis of proteins involved in mitochondrial function in wild-type and mutant corneal fibroblasts in passages 4 and 8. A: Expression of ANT-1 demonstrates a statistically significant decrease in late passages of mutant cells, and PGC-1α, p-Akt (Ser 473), p-Akt (Thr 308), and p-mTOR (Ser 2481) demonstrated a statistically significant decrease in late passages of both wild-type and mutant cells. B: Expression of PGC-1α and NF-κB in nuclear extract increased in late-passage or mutant fibroblasts in similar pattern. The y axis of each graph indicates the relative ratio considering the expression level of wild-type fibroblasts in passage 4 as 1.0.
Change in Intracellular ROS and Proliferation before and after Application of BHA
The intracellular ROS level increased in late- passage wild-type cells and decreased in late-passage mutant cells. However, after treatment with 100 μmol/L BHA, the intracellular ROS level showed similar levels in all types of cells (Figure 8A). In addition, at 24 hours after application of 10 or 100 μmol/L BHA, cellular proliferation of passage 8 mutant cells measured with MTS increased significantly (Figure 8B).
Figure 8.
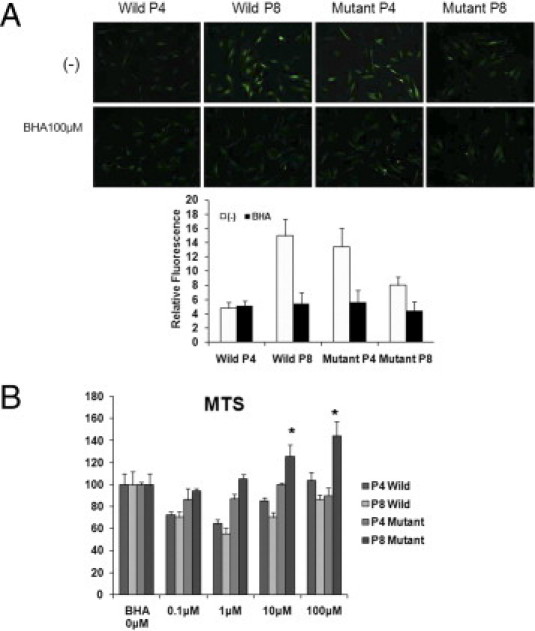
Intracellular ROS staining and MTS assay after application of BHA. Intensity of intracellular ROS staining increased in late passage of wild-type cells, and decreased in late passage of mutant cells. A: After treatment with 100 μmol/L BHA, the intensity of intracellular ROS decreased in all types of cells. B: At 24 hours after application of 10 or 100 μmol/L BHA, cellular proliferation of passage 8 mutant cells increased statistically significantly in MTS assay.
Discussion
GCD2 is an abnormal condition of granular and lattice-type corneal deposits that interfere with vision. The disease can be diagnosed at the molecular level by the presence of specific mutations.13–15 Patients heterozygous for GCD2 mutations exhibit generally mild corneal opacities that slowly progress until they are in their thirties, and initial clinical impairment in these patients usually is not remarkable. Although individual cases vary, severity of the corneal deposits increases with age. Recently, a refractive procedure that induces rapidly progressive severe corneal opacities that cannot be controlled without amputation of the flap or graft replacement has been reported for treatment of GCD2.16,17 Because of the unexpected progression of corneal opacity and the relatively high incidence in Asian countries,18 where a large number of refractive procedures are performed, GCD2 is gaining attention in ophthalmology. Clinicians now attempt to prevent serious deterioration by using meticulous preoperative corneal evaluation for diagnosis of GCD2 and are making an effort to understand the major factors that exacerbate the natural course of corneal dystrophy.19
Whereas heterozygous cornea demonstrate a naturally slow progression of abnormalities, homozygous cornea are characterized by significant visual impairment caused by white dense deposits from early childhood.20,21 Thus, the homozygous R124H point mutation of the gene encoding the TGFBI protein may increase formation of deposits or have higher toxicity than does the heterozygous mutation of the inducing protein.
Previously, the pathogenesis of GCD2 was thought to be caused by numerous electron-dense fusiform deposits identified as hyaline or amyloid at histochemistry and electron microscopy.22 However, after refractive procedures, even the mutated state did not change the component of the mutant protein, and other factors such as surgical trauma or cutting the edge of the collagen structure abruptly resulted in deteriorated corneal deposits. Our group recently reported that increased intracellular ROS and oxidative damage induced by decreased catalase is involved in GCD2 pathogenesis.23 In addition, increased oxidative stress led to a more sensitive response in homozygous corneal fibroblasts. In the present study, keratocytes located near the stromal deposit demonstrated degenerative change and damaged intracellular structure on electron microscopic images. In seems that in addition to the deposits themselves, the increased cellular stress caused by mutated protein production, secretion, and absorption and the altered metabolism induced by increased oxidative stress may be involved in progression of the disease.
In the present study, we focused on and evaluated the state of mitochondria in GCD2 mutant corneal fibroblasts, in tissue, and under cultured conditions. The keratocytes surrounding the corneal deposits of GCD2 heterozygous cornea demonstrated degenerative changes including swollen and damaged mitochondria. To simulate the aging condition in our in vitro culture system, we compared corneal fibroblasts from an early passage4 and a late passage.8 To avert the age-related changes in mitochondrial function in the primary culture cells, we used healthy corneas from 20-year-old individuals and mutant cells from 8-year-old patients. Passage 8 was selected as a late passage because apparent retardation of cellular growth was observed in this passage in several preliminary trials (data not shown), and the halfway point (passage 4) was chosen as the early passage. In contrast to the small round mitochondria in wild-type cells in the early and late passages, a large number of elongated mitochondria were observed in early-passage mutant cells, whereas in late-passage mutant cells, the number of mitochondria was decreased.
In experiments performed to observe mitochondrial activity using MitoTraker and cytochrome c staining, mutant cells demonstrated increased detection of both markers of mitochondrial function, specifically in the early passage. However, the signal was abruptly decreased in the late passage, whereas wild-type cells maintained the same staining intensity.
The loss of mitochondrial membrane potential is a hallmark of apoptosis. It is an early event that precedes phosphatidylserine externalization and coincides with caspase activation.24,25 In nonapoptotic cells, JC-1 exists as a monomer in the cytosol (green) and accumulates as aggregates in the mitochondria, which stain red. In apoptotic and necrotic cells, JC-1 exists in monomeric form and stains the cytosol green. Compared with other cells, late-passage mutant cells demonstrated a statistically significant reduction in the percentage of red fluorescence. Delayed proliferation and cellular viability was observed in mutant cells in MTS and CCK-8 assays. Cell viability was maintained for 6 hours under culture conditions, but from 24 hours significantly decreased levels of each marker were observed. Annexin V stain demonstrated typical apoptotic feature in mutant cells, in particular in the late passage. All of these findings suggest that the mitochondria of corneal fibroblasts may be impaired by mutation of GCD2, in particular after multiple passages.
The membrane-bound mitochondrial complexes I to V pathway establishes a proton gradient across the mitochondrial membrane, producing a thermodynamic state with potential to do work. A small percentage of electrons prematurely leak oxygen, resulting in formation of the toxic free-radical superoxide, a molecule thought to contribute to a number of diseases and aging.26,27 Wild-type corneal fibroblasts demonstrated unchanged or increased activity of complexes I to V. In contrast, mutant cells exhibited decreased expression of complexes I to V in the late passages. This indicates that mutant cells lose their mitochondrial function to generate energy and redox reactions in later passages.
Mammalian target of rapamycin (mTOR) is a large serine/threonine protein kinase of approximately 280 kDa that forms multiple subunit complexes with numerous protein partners. In mammalian cells, two distinct complexes have been identified: mTORC1, in which mTOR is bound to the protein partner raptor, and mTORC2, in which mTOR is bound to another protein partner, rector.28,29 These protein complexes have distinct biological functions such as a role in metabolism during aging and a role in the Akt/mTOR pathway during the life span, determined by regulating mitochondrial oxygen consumption and oxidative capacity.30 Transcriptional complexes that contain PGC−1α control mitochondrial oxidative function to maintain energy homeostasis in response to nutrient and hormone signals. Expression of PGC-1α relates to the NF-κB pathway.31 To evaluate the correlation of each factor, expression of NF-κB and PGC-1α in nuclei was analyzed. Expression of PGC-1α in the cellular component decreased in late passages of both cell types. However, in the nuclear extraction component, expression of NF-κB and PGC-1α increased in mutant cells. Therefore, the correlation between NF-κB and PGC-1α expression was observed in the nuclear component.
In a previous study, mTOR regulated mitochondrial gene expression and oxygen consumption by controlling the gene expression of the mitochondrial transcriptional regulator PGC-1α and other nuclear respiratory factors.32 ANT was observed to be the only protein in the mitochondrial membrane that exhibits a detectable age-associated increase in carbonyls. The age-related increase in molecular oxidative damage to mitochondrial membrane proteins is primarily due to the severe loss of ANT activity.33 VDAC has a central role in mitochondria-mediated apoptosis. Cytochrome c and other small molecules pass through the mitochondrial permeability transition pore, which comprises VDAC in the outer membrane, ANT in the inner membrane, and several auxiliary proteins that include the Bcl-2 family involved in apoptosis.34,35 With the exception of VDAC, expression of these proteins demonstrated a passage-dependent decrease in both wild-type and mutant cells.
Intracellular ROS was measured directly using CM-H2DCFDA by obtaining DCF fluorescence images. Intracellular ROS increased in late passage of wild-type cells and early passage of mutant cells. The different levels of intracellular ROS were equalized by application of the known antioxidant BHA. Not only the level of intracellular ROS but cellular proliferation measured using MTS changed significantly in late passage of mutant cells after application of BHA. Thus, increased mitochondrial stress and altered cellular viability of mutant cells were modulated by application of the proper antioxidant.
Our results suggest that mutation of GCD2 may affect mitochondrial activity via abnormal protein production or increased oxidative stress23 rather than a direct change in functional mitochondrial proteins. Thus, antioxidants may be an applicable modulator for rescue of GCD2-induced cellular toxicity. Any effort to reduce mitochondrial stress in the cornea, for example, averting mechanical trauma or UV light, would be helpful.
In conclusion, the GCD2 mutation is associated with altered mitochondrial structure and activity and delayed cellular proliferation in aged cells. Though it cannot be concluded that the mitochondrial changes are the cause of the disease or a byproduct of the mutated GCD2 protein, it seems that the decreased mitochondrial activity is related more to the mutation than to aging. Therefore, alteration of mitochondrial function is critical for understanding the pathogenesis and potential treatment of GCD2.
Footnotes
Supported in part by grant MEST No. 2009-0075333 from the National Research Foundation of Korea and by a grant from KRIBB Research Initiative Program.
References
- 1.El-Ashry M.F., Abd El-Aziz M.M., Larkin D.F., Clarke B., Cree I.A., Hardcastle A.J., Bhattacharya S.S., Ebenezer N.D. A clinical, histopathological, and genetic study of Avellino corneal dystrophy in British families. Br J Ophthalmol. 2003;87:839–842. doi: 10.1136/bjo.87.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellies P., Renard G., Valleix S., Boelle P.Y., Dighiero P. Clinical outcome of eight BIGH3-linked corneal dystrophies. Ophthalmology. 2002;109:793–797. doi: 10.1016/s0161-6420(01)01025-9. [DOI] [PubMed] [Google Scholar]
- 3.Munier F.L., Korvatska E., Djemaï A., Le Paslier D., Zografos L., Pescia G., Schorderet D.F. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet. 1997;15:247–251. doi: 10.1038/ng0397-247. [DOI] [PubMed] [Google Scholar]
- 4.Klintworth G.K. Advances in the molecular genetics of corneal dystrophies. Am J Ophthalmol. 1999;128:747–754. doi: 10.1016/s0002-9394(99)00358-x. [DOI] [PubMed] [Google Scholar]
- 5.Klintworth G.K., Valnickova Z., Enghild J.J. Accumulation of βig-h3 gene product in corneas with granular dystrophy. Am J Pathol. 1998;152:743–748. [PMC free article] [PubMed] [Google Scholar]
- 6.Streeten B.W., Qi Y., Klintworth G.K., Eagle R.C., Jr, Strauss J.A., Bennett K. Immunolocalization of βigh-3 protein in the 5q31-linked corneal dystrophies and normal corneas. Arch Ophthalmol. 1999;117:67–75. doi: 10.1001/archopht.117.1.67. [DOI] [PubMed] [Google Scholar]
- 7.Kim B.Y., Olzmann J.A., Choi S.I., Ahn S.Y., Kim T.I., Cho H.S., Suh H., Kim E.K. Corneal dystrophy-associated R124H mutation disrupts TGFBI interaction with Periostin and causes mislocalization to the lysosome. J Biol Chem. 2009;284:19580–19591. doi: 10.1074/jbc.M109.013607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skonier J., Neubauer M., Madisen L., Bennett K., Plowman G.D., Purchio A.F. cDNA cloning and sequence analysis of beta ig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-beta. DNA Cell Biol. 1992;11:511–522. doi: 10.1089/dna.1992.11.511. [DOI] [PubMed] [Google Scholar]
- 9.Thapa N., Lee B.H., Kim I.S. TGFBIp/betaig-h3 protein: a versatile matrix molecule induced by TGF-beta. Int J Biochem Cell Biol. 2007;39:2183–2194. doi: 10.1016/j.biocel.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 10.Hipkiss A.R. Mitochondrial dysfunction, proteotoxicity, and aging: causes or effects, and the possible impact of NAD+-controlled protein glycation. Adv Clin Chem. 2010;50:123–150. [PubMed] [Google Scholar]
- 11.Bonda D.J., Wang X., Perry G., Smith M.A., Zhu X. Mitochondrial dynamics in Alzheimer's disease: opportunities for future treatment strategies. Drugs Aging. 2010;27:181–192. doi: 10.2165/11532140-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okada M., Yamamoto S., Inoue Y., Watanabe H., Maeda N., Shimomura Y., Ishii Y., Tano Y. Severe corneal dystrophy phenotype caused by homozygous R124H keratoepithelin mutations. Invest Ophthalmol Vis Sci. 1998;39:1947–1953. [PubMed] [Google Scholar]
- 13.Holland E.J., Daya S.M., Stone E.M., Folberg R., Dobler A.A., Cameron J.D., Doughman D.J. Avellino corneal dystrophy: clinical manifestations and natural history. Ophthalmology. 1992;99:1564–1568. doi: 10.1016/s0161-6420(92)31766-x. [DOI] [PubMed] [Google Scholar]
- 14.Ferry A.P., Benson W.H., Weinberg R.S. Combined granular-lattice (“Avellino”) corneal dystrophy. Trans Am Ophthalmol Soc. 1997;95:61–77. [PMC free article] [PubMed] [Google Scholar]
- 15.Folberg R., Alfonso E., Croxatto J.O., Driezen N.G., Panjwani N., Laibson P.R., Boruchoff S.A., Baum J., Malbran E.S., Fernandez-Meijide R. Clinically atypical granular corneal dystrophy with pathologic features of lattice-like amyloid deposits: a study of these families. Ophthalmology. 1988;95:46–51. doi: 10.1016/s0161-6420(88)33226-4. [DOI] [PubMed] [Google Scholar]
- 16.Kim T.I., Kim T., Kim S.W., Kim E.K. Comparison of corneal deposits after LASIK and PRK in eyes with granular corneal dystrophy type II. J Refract Surg. 2008;24:392–395. doi: 10.3928/1081597X-20080401-13. [DOI] [PubMed] [Google Scholar]
- 17.Jun R.M., Tchah H., Kim T.I., Stulting R.D., Jung S.E., Seo K.Y., Lee D.H., Kim E.K. Avellino corneal dystrophy after LASIK. Ophthalmology. 2004;111:463–468. doi: 10.1016/j.ophtha.2003.06.026. [DOI] [PubMed] [Google Scholar]
- 18.Lee J.H., Cristol S.M., Kim W.C., Chung E.S., Tchah H., Kim M.S., Nam C.M., Cho H.S., Kim E.K. Prevalence of granular corneal dystrophy type 2 (Avellino corneal dystrophy) in the Korean population. Ophthalmic Epidemiol. 2010;17:160–165. doi: 10.3109/09286581003624939. [DOI] [PubMed] [Google Scholar]
- 19.Kim T.I., Roh M.I., Grossniklaus H.E., Kang S.J., Hamilton S.M., Schorderet D.F., Lee W.B., Kim E.K. Deposits of transforming growth factor-beta–induced protein in granular corneal dystrophy type II after LASIK. Cornea. 2008;27:28–32. doi: 10.1097/ICO.0b013e318156d36d. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe H., Hashida Y., Tsujikawa K., Tsujikawa M., Maeda N., Inoue Y., Yamamoto S., Tano Y. Two patterns of opacity in corneal dystrophy caused by the homozygous BIG-H3 R124H mutation. Am J Ophthalmol. 2001;132:211–216. doi: 10.1016/s0002-9394(01)00962-x. [DOI] [PubMed] [Google Scholar]
- 21.Moon J.W., Kim S.W., Kim T.I., Cristol S.M., Chung E.S., Kim E.K. Homozygous granular corneal dystrophy type II (Avellino corneal dystrophy): natural history and progression after treatment. Cornea. 2007;26:1095–1100. doi: 10.1097/ICO.0b013e3181484013. [DOI] [PubMed] [Google Scholar]
- 22.Teng C.C. Granular dystrophy of the cornea: a histochemical and electron microscopic study. Am J Ophthalmol. 1967;63:772–791. doi: 10.1016/0002-9394(67)91305-0. [DOI] [PubMed] [Google Scholar]
- 23.Choi S.I., Kim T.I., Kim K.S., Kim B.Y., Ahn S.Y., Cho H.J., Lee H.K., Cho H.S., Kim E.K. Decreased catalase expression and increased susceptibility to oxidative stress in primary cultured corneal fibroblasts from patients with granular corneal dystrophy type II. Am J Pathol. 2009;175:248–261. doi: 10.2353/ajpath.2009.081001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mantymaa P., Siitonen T., Guttorm T., Saily M., Kinnula V., Savolainen E.R., Koistinen P. Induction of mitochondrial manganese superoxide dismutase confers resistance to apoptosis in acute myeloblastic leukaemia cells exposed to etoposide. Br J Haematol. 2000;108:574–581. doi: 10.1046/j.1365-2141.2000.01852.x. [DOI] [PubMed] [Google Scholar]
- 25.Korper S., Nolte F., Rojewski M.T., Thiel E., Schrezenmeier H. The K+ channel openers diazoxide and NS1619 induce depolarization of mitochondria and have differential effects on cell Ca2+ in CD34+ cell line KG-1a. Exp Hematol. 2003;31:815–823. doi: 10.1016/s0301-472x(03)00199-1. [DOI] [PubMed] [Google Scholar]
- 26.Afanas'ev I.B. Free radical mechanisms of aging processes under physiological conditions. Biogerontology. 2005;6:283–290. doi: 10.1007/s10522-005-2626-z. [DOI] [PubMed] [Google Scholar]
- 27.Matés J.M., Pérez-Gómez C., Núñez de Castro I. Antioxidant enzymes and human diseases. Clin Biochem. 1999;32:595–603. doi: 10.1016/s0009-9120(99)00075-2. [DOI] [PubMed] [Google Scholar]
- 28.Sarbassov D.D., Ali S.M., Sabatini D.M. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 29.Wullschleger S., Loewith R., Hall M.N. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 30.Schieke S.M., Phillips D., McCoy J.P., Jr, Aponte A.M., Shen R.F., Balaban R.S., Finkel T. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem. 2006;281:27643–27652. doi: 10.1074/jbc.M603536200. [DOI] [PubMed] [Google Scholar]
- 31.Palomer X., Alvarez-Guardia D., Rodríguez-Calvo R., Coll T., Laguna J.C., Davidson M.M., Chan T.O., Feldman A.M., Vázquez-Carrera M. TNF-alpha reduces PGC-1alpha expression through NF-kappaB and p38 MAPK leading to increased glucose oxidation in a human cardiac cell model. Cardiovasc Res. 2009;81:703–712. doi: 10.1093/cvr/cvn327. [DOI] [PubMed] [Google Scholar]
- 32.Cunningham J.T., Rodgers J.T., Arlow D.H., Vazquez F., Mootha V.K., Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 33.Yan L.J., Sohal R.S. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc Natl Acad Sci USA. 1998;95:12896–12901. doi: 10.1073/pnas.95.22.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baines C.P., Kaiser R.A., Sheiko T., Craigen W.J., Molkentin J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vyssokikh M., Zorova L., Zorov D., Heimlich G., Jürgensmeier J., Schreiner D., Brdiczka D. The intra-mitochondrial cytochrome c distribution varies correlated to the formation of a complex between VDAC and the adenine nucleotide translocase: this affects Bax-dependent cytochrome c release. Biochim Biophys Acta. 2004;1644:27–36. doi: 10.1016/j.bbamcr.2003.10.007. [DOI] [PubMed] [Google Scholar]