

Abstract
T helper (Th)17 cells might contribute to immune-mediated renal injury. Thus, we sought to define the time course of IL-17A-induced kidney damage and examined the relation between Th17 and Th1 cells in a model of crescentic anti-glomerular basement membrane glomerulonephritis. Renal injury and immune responses were assessed in wild-type and in IL-17A-deficient mice on days 6, 14, and 21 of disease development. On day 6, when mild glomerulonephritis developed, IL-17A-deficient mice were protected from renal injury. On day 14, when more severe disease developed, protection from renal injury due to IL-17A deficiency was less evident. On day 21, when crescentic glomerulonephritis was fully established, disease was enhanced in IL-17A−/− mice, with increased glomerular T-cell accumulation and fibrin deposition, and augmented Th1 responses. Mice lacking the Th17-promoting cytokine, IL-23 (p19), also developed more severe disease than wild-type animals on day 21. In contrast, mice deficient in the key Th1-promoting cytokine, IL-12 (p35), had decreased Th1 and increased Th17 responses and developed less severe crescentic glomerulonephritis than wild-type animals. These studies show that IL-17A contributes to early glomerular injury, but it attenuates established crescentic glomerulonephritis by suppressing Th1 responses. They provide further evidence that Th1 cells mediate crescentic injury in this model and that Th1 and Th17 cells counterregulate each other during disease development.
Some forms of proliferative glomerulonephritis (GN) are complicated by glomerular crescent formation, indicating severe and rapidly progressive renal injury. One of the best characterized and most widely used models of crescentic GN is the 21-day, autologous phase, anti-glomerular basement membrane (GBM) GN in which heterologous anti-GBM Ig acts as a planted foreign antigen in glomeruli. Crescentic anti-GBM GN is mediated by CD4+ T cells, macrophages, and fibrin.1–4 Observations from patients with crescentic GN suggest that similar events may occur in humans.5 Autologous antibody is not required for the development of crescentic injury in this model,6 highlighting the importance of cellular immunity in inducing glomerular injury.
A large number of studies have clearly shown that crescentic anti-GBM GN is driven by T helper (Th)1 responses, whereas Th2 responses attenuate disease severity. Studies that used interferon (IFN)-γ deficient (−/−) mice or neutralizing IFN-γ antibodies have shown that IFN-γ (the main Th1 cytokine) is critical for the development of crescentic anti-GBM GN.4,7,8 Similarly, mice lacking T-bet, the key transcription factor promoting Th1 differentiation,9 are protected from disease development.10 In contrast, Th2-related cytokines, including IL-4 and IL-10, inhibit Th1 responses and attenuate disease severity.11–13
More recently, a new subset of Th cells has been identified and termed Th17, characterized by their production of IL-17A.14 Th17 cells also secrete a number of other cytokines, including IL-17F, IL-21, IL-22, IL-6, and tumor necrosis factor.14,15 IL-6 and transforming growth factor-β drive early Th17 differentiation in mice, whereas IL-23 plays a key role in the subsequent maintenance and expansion of Th17 cells.14–16 IL-17A plays a critical role in host defense by mobilizing and activating neutrophils14 and has a main role in the development of injury in models of autoimmune diseases such as experimental autoimmune encephalomyelitis, experimental autoimmune uveitis (EAU), and collagen-induced arthritis.17–20
The relation between Th1 and Th17 cells is important and more complex than first thought, as shown by an increasing number of reports. For example, both Th1 and Th17 effector cells are capable of inducing autoimmune-mediated injury in models such as EAU.18 Studies in experimental autoimmune encephalomyelitis and lung inflammation have shown that Th1 and Th17 cells can cooperate to promote disease development.21,22 In contrast, experiments in EAU, colitis, and graft-versus-host disease have shown that these two Th pathways can suppress each other to regulate immune-mediated diseases.18,20,23,24
Recent evidence has implicated the Th17 pathway in the development of GN. IL-23/IL-17A have been shown to promote renal injury in autoimmune models of GN, including Goodpasture's disease and anti-myeloperoxidase GN.25,26 Furthermore, proof-of-concept experiments have shown that, when transferred alone, Th1- or Th17-polarized cells can both induce GN in recombination activating gene-1 (Rag1)−/− recipient mice (which lack adaptive immunity).27 In anti-GBM GN, IL-23/IL-17A contribute to early kidney injury,28 consistent with reports showing that transferred Th17 cells cause early tissue damage in models of GN and EAU, in contrast to Th1 cells which induce later, but ultimately more, severe disease.18,27
Although Th17 cells can contribute to the development of renal injury, many unresolved issues still remain about the role of IL-17A-producing cells in crescentic GN. For example, the time course of IL-17A-mediated kidney damage and the relation (whether synergistic or antagonistic) between Th17 and Th1 subsets, both of which are capable of inducing renal injury, have not been defined. In addition, the recent evidence that Th17 cells contribute to the development of crescentic anti-GBM GN, which has previously been shown to be Th1 dependent, invites a re-examination of the role of the Th1 pathway in crescentic GN. The current studies were undertaken to answer these important questions with the use of the best-characterized model of crescentic GN, anti-GBM GN, and mice genetically deficient in either Th17- [IL-17A and IL-23(p19)] or Th1-specific [IL-12(p35)] cytokines.
Here, we demonstrate that IL-17A contributes to early kidney injury in anti-GBM GN, but it, paradoxically, attenuates the severity of fully established crescentic disease by limiting injurious Th1 responses. We also provide further evidence that Th1 responses mediate severe crescentic injury in this model and that Th1 and Th17 responses inhibit each other during the development of crescentic anti-GBM GN.
Materials and Methods
Animals and Induction of Disease
Eight- to 10-week-old male mice were used for experiments. C57BL/6 wild-type (WT) mice were obtained from Monash University Animal Services (Melbourne, Australia). IL-17A−/− mice (provided by Y. Iwakura, Tokyo, Japan), IL-23p19−/− mice (provided by N. Ghilardi and F. De Sauvage, San Francisco, CA), and IL-12p35−/− mice (The Jackson Laboratory, Bar Harbor, ME) were all on C57BL/6 background. All mice were bred and kept at the Monash Medical Centre Animal Facility (Clayton, Australia). GN was induced by intravenous administration of sheep anti-mouse GBM Ig (15 mg/mouse) on day 0. Disease development and immune responses were assessed on days 6, 14, and 21. All experiments were performed at least twice with a minimum of eight mice per group. Data presented are representative of at least two independent experiments.
Renal Injury, Leukocyte Accumulation, and Fibrin Deposition
Glomerular abnormalities, including crescent formation (defined as two or more cell layers in Bowman's space), hypercellularity, segmental necrosis, hyalinosis, segmental proliferation, and capillary wall thickening, as well as tubulointersitital injury, were assessed on periodic acid Schiff–stained, Bouin's-fixed kidney sections. Tubulointerstitial injury (defined as tubular dilation, tubular atrophy, sloughing of tubular epithelial cells, or leukocyte infiltration in the interstitium) was scored from 0 to 3 as follows: 0, no injury; 1, mild injury; 2, moderate injury; and 3, severe injury. At least 50 glomeruli or 10 randomly selected cortical interstitial high-power fields per animal were counted to assess glomerular abnormalities and interstitial injury, respectively. Proteinuria (in mg/24 hours) was measured by a modified Bradford's method on urine collected during the final 24 hours of experiments. Serum creatinine concentrations (in μmol/L) were measured by an autoanalyser.
Renal macrophages, neutrophils, CD4+ T cells, and CD8+ T cells were shown by immunohistochemistry as described,29 using anti-CD68 (FA/11), anti-Gr-1 (RB6-8C5), anti-CD4 (GK1.5), and anti-CD8 (53–6.7) antibodies, respectively. At least 20 glomeruli or 10 high-power fields per animal were assessed, and results are expressed as cells per glomerular cross section (gcs) or cells per high-power field.
Glomerular fibrin deposition was shown by immunohistochemistry as described30 and was scored from 0 to 3 as follows: 0, background staining; 1, positive staining in <25% of glomerular cross section; 2, positive staining in 25% to 50% of glomerular cross section; and 3, positive staining in >50% of glomerular cross section. At least 20 glomeruli per animal were assessed to determine the average score.
Adhesion Molecule and mRNA Expression
Glomerular expression of P-selectin and intercellular adhesion molecule 1 (ICAM-1) was shown by immunofluorescence as described31 and was scored from 0 to 3 as follows: 0, background staining; 1, positive staining in <25% of glomerular cross section; 2, positive staining in 25% to 50% of glomerular cross section; and 3, positive staining in >50% of glomerular cross section. At least 20 glomeruli per animal were assessed to determine the average score. Expression of mRNA was measured by real-time polymerase chain reaction, as described.10 Results are expressed relative to the housekeeping gene 18s as fold change over the mean of the WT with GN group. Primer sequences are available on request.
T-Cell Responses, DC Activation, and Splenocyte Cytokine Production
Splenic CD4+ T-cell apoptosis and proliferation were measured by flow cytometric analysis of Annexin-V staining and intracellular bromodeoxyuridine incorporation, respectively, as described.32 Regulatory T cells were identified by flow cytometry as foxp3+CD25+CD4+ cells, as published.25 Activation of dendritic cells (DCs; identified as CD11chi cells) was measured by flow cytometric analysis of CD86 and expression of major histocompatibility complex class II, as described.33 Concentrations of IFN-γ, IL-4, and IL-17A in supernatant fluids from sheep globulin (10 μg/mL) stimulated splenocytes (4 × 106 cells/mL; 72 hours) were measured by ELISA, as published.34,35
Statistical Analyses
Results are expressed as the mean ± SEM. Unpaired t-test (for parametric data), Mann Whitney test (for non-parametric data), or one-way analysis of variance followed by Dunnett's multiple comparison test was used for statistical analyses (GraphPad Prism; GraphPad Software Inc., San Diego, CA). Differences were considered to be statistically significant if P < 0.05.
Results
To examine how IL-17A affects the development of GN throughout the course of the disease, renal injury and immune responses were assessed in WT and IL-17A−/− mice on days 6, 14, and 21 after anti-GBM globulin injection.
IL-17A Deficiency Decreases Renal Injury on Day 6 of GN
By day 6, WT mice had developed relatively mild GN with glomerular abnormalities that included hypercellularity, segmental necrosis, hyalinosis, segmental proliferation, capillary wall thickening, and only occasional crescent formation (3.5% ± 0.6%). IL-17A−/− mice developed less severe renal injury than WT animals at this early time point with fewer abnormal glomeruli (Figure 1, A, G, and H) and decreased crescent formation (Figure 1B). Attenuated renal injury in IL-17A−/− mice correlated with diminished glomerular accumulation of cellular effectors: CD4+ T cells, macrophages, and neutrophils (Figure 1C).
Figure 1.
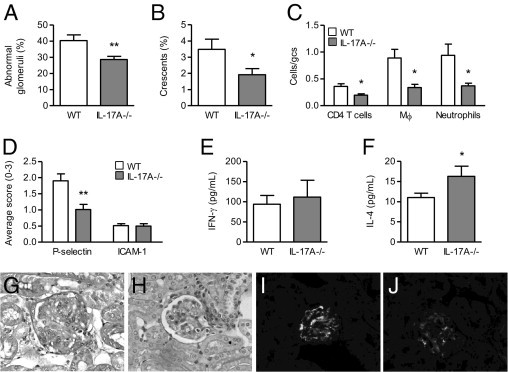
The effect of IL-17A deficiency on kidney injury and Th1/Th2 responses on day 6 of GN. Six days after receiving anti-GBM globulin, IL-17A−/− mice had fewer abnormal glomeruli (A); decreased crescent formation (B); and attenuated glomerular accumulation of CD4+ T cells, macrophages, and neutrophils (C) compared with WT animals. D: Expression of P-selectin was down-regulated in glomeruli of IL-17A−/− compared with WT animals with GN. ICAM-1 was expressed at low levels in glomeruli of normal mice without GN (0.5 ± 0.01 average score) but was not up-regulated in WT or IL-17A−/− animals on day 6 of GN. IL-17A deficiency had no effect on the production of IFN-γ (E) by antigen-stimulated splenocytes, but it did increase the production of IL-4 (F). G and H: Representative photomicrographs of glomerular injury on day 6 of GN, showing hypercellularity, segmental proliferation/necrosis, and capillary wall/basement membrane thickening in WT animals (G) and less severe disease in IL-17A−/− mice (H), as assessed on Bouin's-fixed paraffin-embedded periodic acid Schiff–stained kidney sections. I and J: Representative photomicrographs of glomerular P-selectin expression on day 6 of GN, showing moderate expression in WT mice (I) and decreased expression in IL-17A−/− animals (J), as assessed by immunofluorescence on frozen kidney sections. Original magnification, ×400 (G–J). gcs, glomerular cross section. Mϕ, macrophages. *P < 0.05, **P < 0.01.
Infiltration of leukocytes into the kidney is affected by renal adhesion molecules and chemokines.36–39 We examined whether IL-17A affected the expression of these mediators. P-selectin was moderately expressed in glomeruli of WT mice on day 6, whereas it was absent in glomeruli of normal mice without GN. ICAM-1 was expressed constitutively at low levels in glomeruli and the interstitium of normal mice without GN and was not up-regulated in WT mice on day 6 of disease induction. P-selectin expression was reduced in glomeruli of IL-17A−/− compared with WT animals, whereas glomerular ICAM-1 expression was not affected by the lack of IL-17A at this time point (Figure 1, D, I, and J). Expression of several chemokines [regulated on activation normal T cell expressed and secreted (RANTES), macrophage inflammatory protein 2, IFN-inducible protein 10 (IP-10), T-cell activation-3] was up-regulated in kidneys of WT animals on day 6 compared with normal mice without GN, whereas monocyte chemoattractant protein 1 (MCP-1) was not increased above normal (Table 1). RANTES expression was reduced to normal levels in kidneys of IL-17A−/− compared with WT mice (Table 1). Other chemokines were not altered, but a trend was observed toward decreased expression of IP-10 because of IL-17A deficiency (Table 1).
Table 1.
Renal Chemokine mRNA Expression in WT and IL-17A-Deficient Mice on Day 6 of GN
| WT | IL-17A−/− | Normal (no GN) | |
|---|---|---|---|
| RANTES | 1.02 ± 0.08 | 0.45 ± 0.08⁎ | 0.56 ± 0.11 |
| MCP-1 | 1.01 ± 0.07 | 1.07 ± 0.49 | 0.86 ± 0.30 |
| IP-10 | 1.24 ± 0.28 | 0.69 ± 0.10⁎⁎ | 0.53 ± 0.13 |
| TCA-3 | 1.05 ± 0.14 | 1.30 ± 0.30 | 0.66 ± 0.11 |
| MIP-2 | 1.23 ± 0.28 | 1.13 ± 0.42 | 0.18 ± 0.12 |
Results are expressed relative to 18s as fold change over the mean of the WT group.
MIP-2, macrophage inflammatory protein 2; TCA-3, T-cell activation-3.
P < 0.001,
P = 0.09 versus WT.
To examine how IL-17A deficiency affected Th1/Th2 responses on day 6, we measured splenocyte production of IFN-γ and IL-4. At this early stage of the immune response, production of IFN-γ was not significantly affected by the lack of IL-17A (Figure 1E). However, IL-17A deficiency increased the production of IL-4 (1.5-fold increase; Figure 1F).
Protection from Renal Injury due to IL-17A Deficiency Is Less Evident on Day 14 of GN
Fourteen days after anti-GBM globulin administration, WT mice developed severe GN with an average of 35% ± 4% of glomeruli affected by crescent formation. Protection from renal injury due to the lack of IL-17A was less apparent at this time point than on day 6, with no significant decrease in crescent formation observed in IL-17A−/− animals (Figure 2A; P = 0.1). Accumulation of CD4+ T cells and neutrophils in glomeruli was similar between WT and IL-17A−/− mice on day 14 (Figure 2B), in contrast to day 6 when infiltration of these cells was reduced by the lack of IL-17A. Macrophage accumulation in glomeruli was still reduced in IL-17A−/− mice on day 14 (1.5-fold reduction; Figure 2B) but to a lesser degree than on day 6 (2.6-fold reduction).
Figure 2.
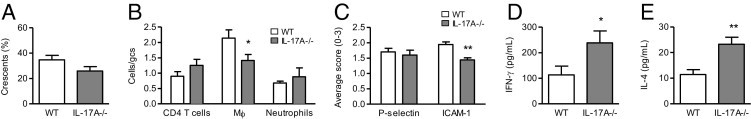
GN development and Th1/Th2 responses in WT and IL-17A−/− mice on day 14. Fourteen days after injection of anti-GBM globulin, IL-17A−/− mice had a trend toward decreased glomerular crescent formation (A) compared with WT animals. B: Glomerular accumulation of CD4+ T cells and neutrophils was similar between WT and IL-17A−/− mice, whereas macrophage infiltration was reduced by IL-17A deficiency. C: P-selectin expression in glomeruli was similar between WT and IL-17A−/− mice. ICAM-1 was up-regulated in glomeruli of WT mice on day 14 of GN compared with normal animals without the disease (0.5 ± 0.01 average score) and was reduced in IL-17A−/− compared with WT mice. Production of IFN-γ (D) and IL-4 (E) by antigen-challenged splenocytes was increased in IL-17A−/− compared with WT mice. gcs, glomerular cross section. Mϕ, macrophages. *P < 0.05, **P < 0.01.
As on day 6, P-selectin was up-regulated in glomeruli of WT mice with GN on day 14 compared with normal mice without disease. However, although reduced on day 6, glomerular expression of P-selectin was not different between WT and IL-17A−/− mice on day 14 (Figure 2C). ICAM-1, although not up-regulated on day 6, was increased in kidneys of WT mice on day 14 compared with normal mice without GN. The absence of IL-17A reduced glomerular expression of ICAM-1 on day 14 (Figure 2C). All of the chemokines measured, including MCP-1 (which was not increased on day 6) were up-regulated in kidneys of WT mice on day 14 compared with normal animals without GN (Table 2). At this time point, renal expression of MCP-1 was reduced by the absence of IL-17A (Table 2). However, RANTES, although reduced on day 6, showed a tendency toward increased expression in IL-17A−/− compared with WT mice on day 14 (Table 2). A trend was also observed toward elevated expression of IP-10 due to the absence of IL-17A on day 14 (Table 2), in contrast to day 6 when IP-10 tended to be reduced. Other chemokines (macrophage inflammatory protein 2, T-cell activation-3) were not altered by the lack of IL-17A (Table 2).
Table 2.
Renal Chemokine mRNA Expression in WT and IL-17A-Deficient Mice on Day 14 of GN
| WT | IL-17A−/− | Normal (no GN) | |
|---|---|---|---|
| RANTES | 1.11 ± 0.20 | 1.58 ± 0.23⁎ | 0.68 ± 0.13 |
| MCP-1 | 1.15 ± 0.21 | 0.57 ± 0.07⁎⁎ | 0.07 ± 0.03 |
| IP-10 | 1.12 ± 0.21 | 1.75 ± 0.26⁎⁎⁎ | 0.73 ± 0.03 |
| TCA-3 | 1.59 ± 0.44 | 1.75 ± 0.26 | 0.73 ± 0.03 |
| MIP-2 | 1.16 ± 0.27 | 0.96 ± 0.18 | 0.04 ± 0.02 |
Results are expressed relative to 18s as fold change over the mean of the WT group.
MIP-2, macrophage inflammatory protein 2; TCA-3, T-cell activation-3.
P = 0.1,
P = 0.02, and
P = 0.08 versus WT.
Although unaltered on day 6, IFN-γ production was significantly increased in IL-17A−/− compared with WT mice on day 14 (Figure 2D). IL-4 was also increased by IL-17A deficiency (Figure 2E). At this time point in the immune response, IFN-γ and IL-4 were up-regulated in IL-17A−/− mice to a similar extent (IFN-γ, 2.1-fold increase; IL-4, 2.0-fold increase).
IL-17A Deficiency Causes More Severe Cresentic GN on Day 21
On day 21, WT mice had fully established, severe GN with extensive tubulointerstitial injury and an average of 43% ± 3.1% of glomeruli affected by crescent formation, as well as elevated proteinuria and serum creatinine levels. At this time point in disease development, renal injury was more severe in IL-17A−/− compared with WT mice, as shown by enhanced glomerular crescent formation (Figure 3, A and D–G) and tubulointerstitial injury (Figure 3C), as well as a trend toward increased proteinuria (Figure 3B; P = 0.1). Serum creatinine levels were not different between WT (25 ± 2 μmol/L) and IL-17A−/− (26 ± 2 μmol/L) mice.
Figure 3.

Renal injury in WT and IL-17A−/− mice on day 21 of anti-GBM GN. Twenty-one days after anti-GBM globulin administration, IL-17A−/− mice developed more severe crescentic GN than WT animals, as shown by significantly enhanced glomerular crescent formation (A) and interstitial injury (C), as well as a trend toward increased proteinuria (B; urine collected during the final 24 hours). D–G: Representative photomicrographs of glomerular injury (assessed on Bouin's-fixed paraffin-embedded periodic acid Schiff–stained kidney sections) on day 21, showing severe crescent formation in WT mice (D and F) and enhanced disease in IL-17A−/− animals (E and G). Original magnification: ×200 (D and E); ×400 (F and G). *P < 0.01, **P < 0.001.
In contrast to earlier time points, IL-17A−/− mice had increased glomerular and interstitial accumulations of CD4+ T cells compared with WT animals on day 21 (Figure 4, A, B, D, and E), correlating with enhanced disease. Although reduced on day 6 and to a lesser extent on day 14, infiltration of macrophages in glomeruli and the interstitium was not significantly different between WT and IL-17A−/− mice on day 21 (Figure 4, A and B). Glomerular accumulation of neutrophils (Figure 4A) or CD8+ T cells (WT, 0.16 ± 0.02 cells/gcs; IL-17A−/− mice, 0.15 ± 0.02 cells/gcs) was not altered by IL-17A deficiency.
Figure 4.
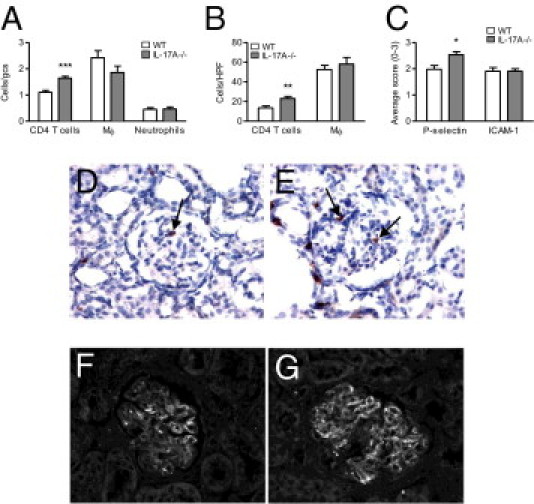
The effect of IL-17A deficiency on accumulation of cells and expression of adhesion molecules on day 21 of GN. A: Compared with WT animals, IL-17A−/− mice had similar numbers of macrophages and neutrophils but increased numbers of CD4+ T cells in glomeruli. B: In the interstitium, accumulation of CD4+ T cells was increased, whereas macrophage infiltration was unaffected by the lack of IL-17A. C: Glomerular P-selectin expression was up-regulated, whereas ICAM-1 was not affected in IL-17A−/− compared with WT mice. D and E: Representative photomicrographs of glomerular CD4+ T cells (brown, indicated by arrows), shown by immunoperoxidase staining of periodate lysine paraformaldehyde-fixed frozen kidney sections with the use of diaminobenzidine to detect positive cells (hematoxylin counterstain, blue), showing enhanced accumulation of CD4+ T cells in IL-17A−/− (E) compared with WT mice (D). F and G: Representative photomicrographs of glomerular P-selectin expression on day 21 of GN, showing moderate expression in WT mice (F) and increased expression in IL-17A−/− animals (G), as assessed by immunofluorescence on frozen kidney sections. Original magnification: ×400 (D–G). gcs, glomerular cross section; HPF, high-power field; Mϕ, macrophages. *P < 0.05, **P < 0.01, and ***P < 0.001.
On day 21, P-selectin expression was significantly enhanced in glomeruli of IL-17A−/− compared with WT mice (Figure 4, C, F, and G), in contrast to the earlier time points. ICAM-1, although reduced on day 14, was not altered by IL-17A deficiency on day 21 (Figure 4C). None of the chemokines measured were significantly different between WT and IL-17A−/− mice on day 21 (Table 3). RANTES and MCP-1, which were significantly reduced on days 6 and 14, respectively, were either not different (RANTES) or showed a trend toward increased expression (MCP-1) on day 21 due to the absence of IL-17A (Table 3).
Table 3.
Renal Chemokine mRNA Expression in WT and IL-17A-Deficient Mice on Day 21 of GN
| WT | IL-17A−/− | Normal (no GN) | |
|---|---|---|---|
| RANTES | 0.61 ± 0.19 | 0.72 ± 0.10 | 0.45 ± 0.35 |
| MCP-1 | 0.80 ± 0.16 | 1.29 ± 0.23⁎ | 0.28 ± 0.22 |
| IP-10 | 1.02 ± 0.15 | 1.17 ± 0.04 | 0.38 ± 0.25 |
| TCA-3 | 0.68 ± 0.11 | 0.86 ± 0.10 | 0.19 ± 0.08 |
| MIP-2 | 1.40 ± 0.26 | 1.23 ± 0.17 | 0.11 ± 0.01 |
Results are expressed relative to 18s as fold change over the mean of the WT group.
MIP-2, macrophage inflammatory protein 2; TCA-3, T-cell activation-3.
P = 0.1 versus WT.
In addition to cellular effectors, fibrin is an important mediator of severe crescent formation in this model.2 Compared with WT mice, deposition of fibrin was significantly increased in glomeruli of IL-17A−/− animals on day 21 (Figure 5, A, and C–F). MMP-9, the production of which is stimulated by IL-17A,40 plays a protective role in this model by degrading fibrin.41 Correlating with increased fibrin deposition, IL-17A−/− mice had decreased MMP-9 expression in the kidney (Figure 5B).
Figure 5.
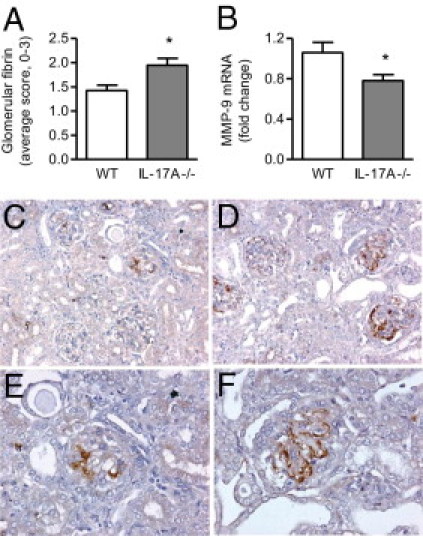
Deposition of fibrin and expression of matrix metalloproteinase (MMP)-9 in kidneys of WT and IL-17A−/− mice on day 21 of GN. IL-17A deficiency enhanced fibrin deposition (A) in glomeruli and decreased renal expression of MMP-9 mRNA (B). C–F: Representative photomicrographs of glomerular fibrin deposition in WT (C and E) and IL-17A−/− mice (D and F), as assessed by immunoperoxidase staining of Bouin's-fixed paraffin-embedded kidney sections with the use of diaminobenzidine to show positive staining (brown). Original magnification: ×200 (C and D); ×400 (E and F). *P < 0.05.
On day 21, IL-17A deficiency caused a marked up-regulation in IFN-γ production (12.1-fold increase; Figure 6A) and a smaller increase in IL-4 secretion (3.0-fold increase; Figure 6B) by antigen-stimulated splenocytes, thus shifting the Th1/Th2 (IFN-γ/IL-4) balance toward Th1 in IL-17A−/− mice (Figure 6C). To examine potential mechanisms by which IL-17A affected the development of Th1 and Th2 responses in secondary lymphoid tissue, we measured splenic expression of the Th1- and Th2-promoting transcription factors, T-bet9 and GATA-3,9 respectively. Consistent with IFN-γ and IL-4 data, deletion of IL-17A increased the expression of T-bet and, to a lesser extent, GATA-3 (Figure 6D).
Figure 6.
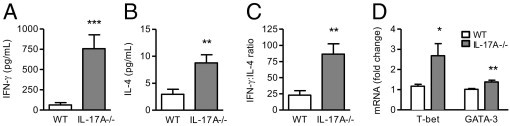
The effect of IL-17A deficiency on day 21 Th1/Th2 responses and transcription factors. Compared with WT animals, IL-17A−/− mice had enhanced production of IFN-γ (A) and, to a lesser extent, IL-4 (B) by antigen-stimulated splenocytes, resulting in an increased IFN-γ/IL-4 (Th1/Th2) ratio (C). D: IL-17A−/− mice had augmented splenic mRNA expression of T-bet and, to a smaller degree, GATA-3 on day 6. *P < 0.05, **P < 0.01, and ***P < 0.001.
IL-23(p19) Deficiency Exacerbates Crescentic GN on Day 21
To confirm that reduced Th17 (IL-17A) responses lead to augmentation of disease on day 21, we assessed GN outcome in mice lacking IL-23(p19), the cytokine that plays a critical role in the maintenance and expansion of Th17 cells.15,16 IL-23p19−/− mice developed more severe crescentic GN than WT mice on day 21 with enhanced crescent formation (Figure 7, A and D–G) and a trend toward increased proteinuria (Figure 7B; P = 0.06). Serum creatinine concentration was not different between WT (23 ± 1 μmol/L) and IL-23p19−/− (26 ± 2 μmol/L) mice. IL-23p19−/− mice had enhanced numbers of macrophages present in glomeruli compared with WT mice (Figure 7C). Glomerular accumulation of CD4+ T cells and neutrophils (Figure 7C), as well as CD8+ T cells (not shown), was not affected by the lack of IL-23(p19).
Figure 7.
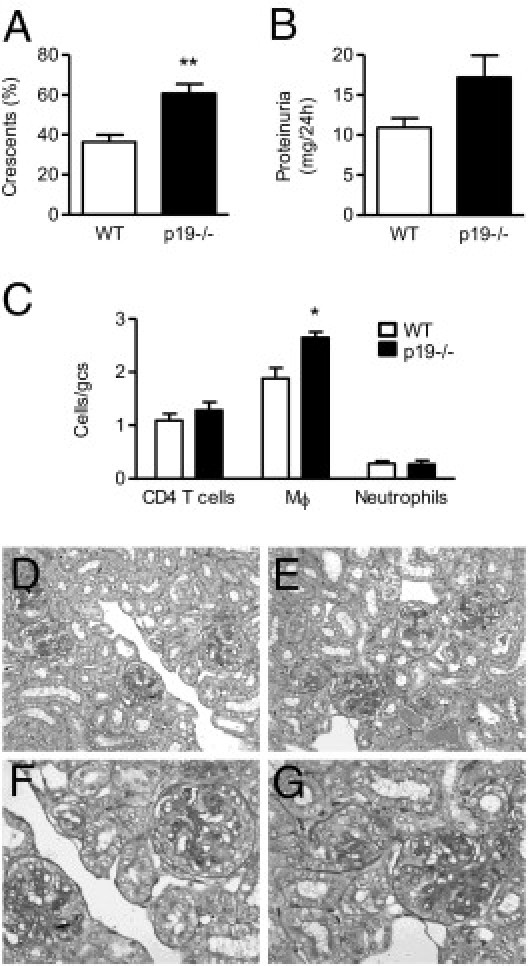
The development of GN in WT and IL-23p19−/− mice on day 21. Renal injury was enhanced by IL-23(p19) deficiency, as shown by significantly increased glomerular crescent formation (A) and a trend toward augmented proteinuria (B). C: Glomerular accumulation of CD4+ T cells and neutrophils was not affected, whereas macrophage infiltration was enhanced by the lack of IL-23(p19). D–G Representative photomicrographs of glomerular injury (assessed on Bouin's-fixed paraffin-embedded periodic acid Schiff–stained kidney sections) on day 21, showing severe crescent formation in WT mice (D and F) and enhanced disease in IL-23(p19)−/− animals (E and G). Original magnification: ×200 (D and E); ×400 (F and G). gcs, glomerular cross section; Mϕ, macrophages. *P < 0.01, **P < 0.001.
To show that IL-23 promotes Th17 responses in this model, we measured IL-17A expression in WT and IL-23p19−/− animals on day 21. IL-23(p19) deficiency decreased the production of IL-17A by antigen-stimulated splenocytes (Figure 8A). In the kidney, IL-17A mRNA was not detected in normal animals without GN but was up-regulated in WT mice on day 21 of disease development. Mice lacking IL-23(p19) had significantly decreased renal IL-17A mRNA expression compared with WT animals on day 21 (Figure 8B).
Figure 8.
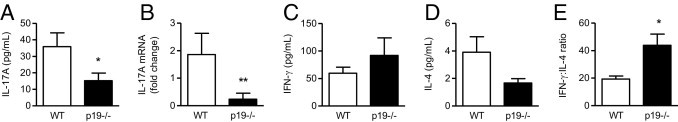
Th1, Th2, and Th17 responses in WT and IL-23p19−/− mice on day 21. IL-23(p19)−/− mice had decreased production of IL-17A by antigen-challenged splenocytes (A) and reduced expression of IL-17A mRNA in the kidney (B) compared with WT animals with GN. IL-17A mRNA was not detected in kidneys of normal animals without the disease. The lack of IL-23(p19) caused a trend toward increased IFN-γ (C) and decreased IL-4 (D) production by antigen-stimulated splenocytes, resulting in an increased IFN-γ/IL-4 ratio (E). *P < 0.05, **P < 0.01.
IL-23p19 deficiency caused a trend toward increased IFN-γ (Figure 8C) and decreased IL-4 production by antigen-challenged splenocytes on day 21 (Figure 8D), shifting the IFN-γ/IL-4 (Th1/Th2) ratio in IL-23p19−/− mice toward Th1(Figure 8E).
Lack of IL-23 or IL-17A Enhances DC Activation and T-Cell Survival
DCs play a key role in the induction of T-cell immunity, and IL-23 and IL-17A can affect DC function.42,43 Therefore, we examined how these cytokines affected DC activation and T-cell responses in the spleen. DC activation was significantly increased by the absence of IL-23(p19) and, to a greater extent, IL-17A, as indicated by the expression of major histocompatibility complex class II (Figure 9A) and CD86 (Figure 9B). The proportion of DCs in the spleen was not altered by the absence of IL-23 or IL-17A (WT, 1.5% ± 0.8% DCs; IL-23p19−/− mice, 1.4% ± 1.0% DCs; IL-17A−/− mice, 1.4% ± 0.1% DCs).
Figure 9.
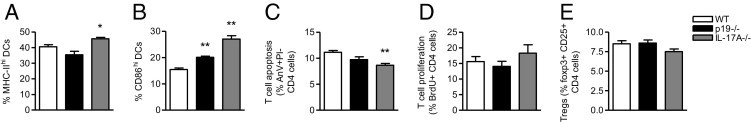
The effect of IL-17A or IL-23p19 deficiency on DCs and CD4+ T-cell responses. DC activation was increased by the lack of IL-23p19 and, to a greater extent, IL-17A, as shown by the proportion of DCs in the spleen expressing major histocompatibility complex class II (A) and CD86 (B) 6 days after anti-GBM globulin injection. C: Compared with WT animals, IL-17A−/− mice had decreased CD4+ T-cell apoptosis, whereas a trend toward reduced apoptosis was observed in IL-23p19−/− mice. CD4+ T-cell proliferation (D) and the proportion of Tregs (E) were not significantly affected by IL-23p19 or IL-17A deficiency. AnV, Annexin V; BrdU, bromodeoxyuridine; MHC-II, major histocompatibility complex class II; PI, propidium iodide; Tregs, regulatory T cells. *P < 0.05 versus WT; **P < 0.01 versus WT.
In correlation with DC expression of CD86, IL-17A−/− mice had decreased CD4+ T-cell apoptosis, whereas a trend toward reduced T-cell apoptosis was observed in IL-23p19−/− animals (Figure 9C). T-cell proliferation (Figure 9D) and proportion of regulatory T cells (Figure 9E) were not significantly affected by IL-23 or IL-17A deficiency.
Absence of IL-12(p35) Attenuates Crescentic GN
Our results suggest that IL-17A deficiency exacerbates established crescentic GN by enhancing Th1 responses. As further evidence that Th1 responses mediate crescentic injury in this model, we examined how disease was affected in mice lacking IL-12(p35), the key Th1-promoting cytokine.44 IL-12p35−/− mice developed less severe crescentic GN than WT animals on day 21 with reduced glomerular crescent formation (Figure 10, A and D–G) and serum creatinine levels (Figure 10C), as well as a trend toward decreased proteinuria (Figure 10B; P = 0.06). IL-12p35−/− mice also had significantly reduced renal injury on day 14 as assessed by proteinuria (WT mice, 14.8 ± 1.5 mg/24 hours; IL-12p35−/− mice, 8.5 ± 1.1 mg/24 hours; P < 0.01).
Figure 10.
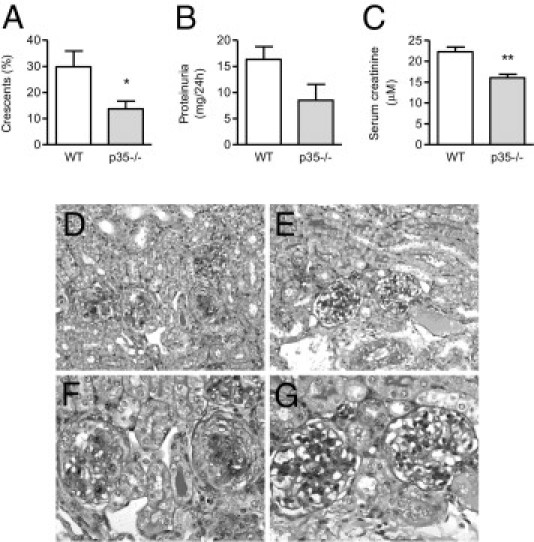
Renal injury in WT and IL-12p35−/− mice on day 21 of GN. Renal injury was diminished by the lack of IL-12(p35), as shown by significantly decreased glomerular crescent formation (A) and serum creatinine levels (C), as well as a trend toward reduced proteinuria (B). D–G: Representative photomicrographs of glomerular injury (assessed on Bouin's-fixed paraffin-embedded periodic acid Schiff–stained kidney sections) on day 21, showing severe crescent formation in WT mice (D and F) and reduced disease in IL-12(p35)−/− animals (E and G). Original magnification: ×200 (D and E); ×400 (F and G). *P < 0.05, **P < 0.01.
IFN-γ production by antigen-stimulated splenocytes was significantly decreased in IL-12p35−/− compared with WT mice (Figure 11A). In contrast, IL-4 production was up-regulated in the absence of IL-12p35 (Figure 11B). This caused a marked reduction in the IFN-γ/IL-4 (Th1/Th2) ratio in IL-12p35−/− compared with WT mice (Figure 11C).
Figure 11.
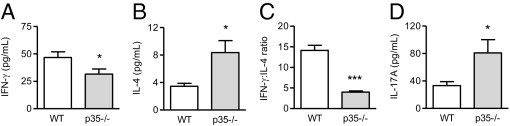
The effect of IL-12(p35) deficiency on Th1, Th2, and Th17 responses. Compared with WT animals, IL-12p35−/− mice had decreased IFN-γ (A) but increased IL-4 (B) production by antigen-challenged splenocytes, resulting in a diminished IFN-γ/IL-4 ratio (C). IL-17A (D) production was up-regulated by the lack of IL-12(p35).
To assess how IL-12(p35) affected Th17 responses, we measured IL-17A secretion by antigen-challenged splenocytes on day 21. As shown in Figure 11D, IL-17A production was significantly increased by IL-12(p35) deficiency.
Discussion
Crescentic anti-GBM GN has been previously shown to be mediated by Th1 cells.4,7,8,10,27 More recently, the Th17 (IL-17A-producing) pathway has also been implicated in immune-mediated kidney injury.26–28,45 The current studies provide novel insights into the role of IL-17A during the development of crescentic GN. They define the time course of IL-17A-mediated kidney damage and delineate the relationship between Th1 and Th17 cells in crescentic GN.
Here, we showed that during the early stage of anti-GBM GN when the disease is still relatively mild (day 6), IL-17A−/− mice developed less severe renal injury than WT animals. This correlated with decreased renal expression of adhesion molecules (P-selectin) and chemokines (RANTES, IP-10) and, subsequently, reduced glomerular infiltration of CD4+ T cells, macrophages, and neutrophils. These results, showing that IL-17A promotes early kidney injury in anti-GBM GN by up-regulating renal adhesion molecules/chemokines and, consequently, accumulation of cellular effectors, are consistent with our recently published studies that used IL-17A−/− mice or adoptively transferred Th17 cells which showed that the Th17 pathway promotes early glomerular injury in autoimmune and planted antigen models of GN.26,27 They are also supported by a previous report showing that IL-17A−/− mice are protected from early (day 10) renal injury in a similar model of anti-GBM GN.28
However, the present studies show that, as the disease evolved, protection from kidney injury was less evident (on day 14) and eventually (on day 21) disease became more severe in IL-17A−/− mice. This correlated with a progressive increase in glomerular accumulation of cellular effectors (CD4+ T cells and macrophages) and renal expression of adhesion molecules/chemokines as time went on. On day 14, when more severe (crescentic) disease developed and renal injury only tended to be reduced in IL-17A−/− mice, glomerular infiltration of CD4+ T cells and neutrophils (which had been reduced on day 6) was now similar between WT and IL-17A−/− mice, correlating with renal expression of adhesion molecules (P-selectin) and chemokines (RANTES, IP-10) which attract those cells into the kidney.37–39 Macrophage accumulation in glomeruli was still reduced by the lack of IL-17A on day 14, consistent with reduced renal expression of macrophage-attracting adhesion molecules (ICAM-1) and chemokines (MCP-1).36,37 However, in association with decreased protection from renal injury, macrophage infiltration was reduced in IL-17A−/− mice on day 14 to a much lesser extent than on day 6. Further in disease development, when crescentic GN was fully established (day 21), injury not only had completely caught up but also was more severe in IL-17A−/− mice. This was associated with increased glomerular accumulation of effector CD4+ T cells (correlating with enhanced glomerular expression of P-selectin) and now similar numbers of glomerular macrophages (correlating with renal expression of ICAM-1 and MCP-1 which were now similar or tended to be up-regulated, respectively, in IL-17A−/− animals). Collectively, these results show that IL-17A promotes early renal injury but that its contribution to kidney damage becomes less prominent as crescentic GN develops and, eventually, it attenuates established disease.
To explore mechanisms that may explain these effects of IL-17A on the development of crescentic GN, we assessed Th1 (and Th2) responses in WT and IL-17A−/− mice throughout the course of the disease. Th1 and Th2 responses, which are well known to drive and attenuate crescentic anti-GBM GN, respectively,7,12 can be affected by Th17 (IL-17A-producing) cells.23,24 In the present study, the absence of IL-17A did not affect Th1 responses, but it did increase protective Th2 responses early on (day 6), which may partly contribute to disease-attenuating effects of IL-17A deficiency during the early phase of anti-GBM GN. However, as time went on, the lack of IL-17A enhanced Th1 (and to a lesser extent Th2) responses at an increasing rate, correlating with effects on disease outcome. The Th1/Th2 balance was eventually shifted toward the injurious Th1 on day 21, resulting in exacerbated disease. These data suggest that IL-17A promotes kidney injury without affecting Th1 immunity during the early phase of anti-GBM GN. However, IL-17A suppresses injurious Th1 responses later on in the development of nephritogenic immunity, resulting in attenuation of established crescentic disease. These results were supported by data from animals deficient in IL-23(p19), a cytokine that promotes Th17 maintenance,15,16 which showed that decreased levels of IL-17A (both systemically in the spleen and locally in the kidney) in IL-23p19−/− mice were associated with a shift toward Th1 responses and more severe crescentic GN on day 21. Mechanistically, we showed that IL-17A limits nephritogenic Th1 and, to a lesser degree, Th2 responses by down-regulating the expression of the transcription factors, T-bet and GATA-3, respectively, that promote the development of those Th cells. These findings, indicating that IL-17A contributes to early, but attenuates late GN by down-regulating Th1 responses, are reminiscent of our recently published studies showing that another cytokine, IL-27, has a similar (biphasic) role to IL-17A in anti-GBM GN, promoting earlier, but suppressing later GN through inhibition of Th1 responses.46
IL-17A is also likely to have opposing roles in early and established GN through its effects on neutrophils. It is known that IL-17A stimulates initial neutrophil mobilization and recruitment to inflamed tissues.47 In this model, neutrophils are likely to promote renal injury more during the early than the later phases of the disease because they were more abundant in glomeruli on day 6 than on days 14 and 21. On day 6, but not on day 14 or 21, IL-17A−/− mice had less neutrophils in glomeruli than WT animals, suggesting that IL-17A promotes initial kidney injury, in part, by enhancing early neutrophil recruitment to the kidney. These observations are consistent with published reports showing that transferred Th17-polarized cells cause early tissue inflammation associated with a predominant neutrophilic infiltrate in models of GN, EAU, and experimental autoimmune encephalomyelitis.18,27,48 Similarly, we have shown, in a model of anti-myeloperoxidase GN, that IL-17A stimulates initial anti-GBM Ig-triggered neutrophil recruitment to glomeruli and causes injury.26
In contrast to the current experiments, previous studies that have shown a pathogenic role for the Th17 pathway in experimental GN failed to identify a protective effect of Th17 cells in kidney damage. In the two previous studies in anti-GBM GN with the use of IL-17A−/−28 or retinoic acid-related orphan receptor γt–deficient mice,45 IL-17A/Th17 cells were found to promote renal injury on day 10 or days 11 to 15, which is consistent with the findings of our study. However, neither of those two previous studies assessed immune responses and disease development later on in disease development (day 21), the time at which the protective effects of Th17 cells were identified by the current investigations. Similarly, in a model of anti-myeloperoxidase GN,26 the protective role of IL-17A-producing cells was not shown, most probably, because disease severity was assessed only 4 days after anti-GBM Ig injection. In addition, in the experiments that examined the role of retinoic acid-related orphan receptor γt (Th17-defining transcription factor) in anti-GBM GN,45 mice were pre-immunized with heterologous sheep globulin before administration of sheep anti-mouse GBM Ig. In contrast to the current studies, this “telescoped” model brings the heterologous (neutrophil-mediated) and autologous (T-cell/macrophage-mediated) phases of anti-GBM GN into overlap at the start of disease induction. Because IL-17A contributes to renal injury by playing a main role in promoting initial anti-GBM Ig-triggered neutrophil recruitment to glomeruli,26 it may be harder to separate the innate and adaptive effects of IL-17A on disease development in an accelerated (pre-immunized) model of anti-GBM GN. Furthermore, in another planted antigen model of GN,27 in vitro-differentiated/pre-activated Th17 or Th1 cells were shown to induce glomerular injury when transferred to Rag1−/− mice (which lack T and B cells). The protective role of IL-17A-producing T cells was not identified in those studies because the Th17 cells were transferred alone and, therefore, could not exert their regulatory effects on Th1 cells. Those adoptive transfer studies,27 however, do provide supportive evidence for our findings by showing that Th17 cells cause renal injury earlier than Th1 cells that develop and act later in disease development.
Our results suggest that IL-17A inhibits established crescentic anti-GBM GN by attenuating Th1 responses. Numerous studies have shown that crescentic injury in this model is driven by Th1 (and attenuated by Th2) responses. Deficiency or blockade of IFN-γ, as well as the lack of T-bet, resulted in suppression of disease.4,7,8,10 In contrast, IL-4 and IL-10 attenuated Th1 responses and crescentic GN.11–13 In another planted antigen model of GN, we have shown that adoptively transferred antigen-specific Th1 cells into Rag1−/− mice cause crescentic injury.27 The present studies provide further evidence that Th1 responses mediate crescentic GN in this model by showing that mice deficient in IL-12(p35), the key Th1-promoting cytokine,44 have reduced Th1 responses and are protected from disease development. In addition, increased production of IL-17A in IL-12p35−/− mice shows that Th1 responses counterregulate Th17 responses during the development of anti-GBM GN.
Our findings showing that IL-17A suppresses nephritogenic Th1 responses are consistent with several studies in other organ systems. IL-17A−/− mice developing experimental autoimmune encephalomyelitis had increased numbers of Th1 cells.20 Th1 responses and the severity of colitis and graft-versus-host disease were also increased in recipients of IL-17A-deficient T cells.23,24 Moreover, increased Th1 responses in recipients of IL-17A−/− T cells were observed at a later, but not early, time point,23 supporting our results that show that IL-17A inhibits Th1 responses later in the generation of nephritogenic immunity. IL-23 has also been reported to inhibit Th1 responses and to attenuate colitis.42 In addition, studies that used mice lacking IL-23 or IL-17A showed that these cytokines acted as negative regulators of Th1 responses in immunity against fungi.49 Th1 responses were also augmented when IL-23 or IL-17A were blocked with antibodies,49 showing that increased Th1 immunity in IL-23p19−/− and IL-17A−/− mice is not observed only in genetically deficient animals. Because IL-17A also inhibits Th1 differentiation in humans,50 our data suggest that neutralization of IL-17A in patients might augment the development of Th1-mediated diseases, including Th1-driven forms of GN.
DCs play a key role in the generation of T-cell responses and can be affected by IL-23 and IL-17A.42,43 Here, we showed that DC expression of CD86 was up-regulated by the absence of IL-23(p19) and, to an even greater extent, IL-17A. These results are supported by previous studies showing that IL-17A and IL-23 can inhibit DC function in models of asthma and colitis.42,43 In association with DC expression of CD86, a costimulatory molecule known to promote T-cell survival,51 IL-17A−/− mice had decreased CD4+ T-cell apoptosis that, together with increased adhesion molecule (P-selectin) expression, correlated with increased accumulation of effector T cells in glomeruli on day 21. These results suggest that another mechanism by which IL-17A attenuates crescentic GN is by inhibiting DC activation which leads to decreased survival and accumulation of CD4+ T cells in the target organ. Although most evidence suggests that IL-17A suppresses T-cell responses by antigen-presenting cells,24,52 IL-17A could have also acted directly on T cells, as has been reported in a model of colitis.23
IL-17A stimulates the production of MMPs, including MMP-9, by various cells.49,53 MMP-9 plays a protective role in anti-GBM GN in mice by degrading fibrin, an important mediator of crescentic injury in this model.2,41 Because intrarenal IL-17A expression is up-regulated on day 21, IL-17A may affect MMP-9 expression in the kidney. Indeed, the lack of IL-17A decreased renal MMP-9 expression which correlated with enhanced glomerular deposition of fibrin. These results suggest that an additional mechanism by which IL-17A attenuates established crescentic GN is by stimulating MMP-9 expression and, therefore, limiting glomerular fibrin deposition.
In summary, the present studies have defined the time course of IL-17A-mediated kidney damage and delineated the relation between Th1 and Th17 cells in anti-GBM GN. We have demonstrated that IL-17A promotes early glomerular injury, but that its contribution to GN development becomes less prominent as time goes on because of its suppressive effects on Th1 responses which eventually result in exacerbation of disease. These studies have also provided further evidence that Th1 cells mediate crescentic injury in this model and that Th1 and Th17 pathways antagonize each other during the development of anti-GBM GN.
Acknowledgments
We thank Alice Wright, Cecilia Lo, Lynelle Jones, and Leon Moussa for technical assistance and James Ngui for help with flow cytometry.
Footnotes
Supported by grants from the National Health and Medical Research Council of Australia.
D.O. and P.-Y.G. contributed equally to this work.
References
- 1.Kitching A.R., Holdsworth S.R., Ploplis V.A., Plow E.F., Collen D., Carmeliet P., Tipping P.G. Plasminogen and plasminogen activators protect against renal injury in crescentic glomerulonephritis. J Exp Med. 1997;185:963–968. doi: 10.1084/jem.185.5.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drew A.F., Tucker H.L., Liu H., Witte D.P., Degen J.L., Tipping P.G. Crescentic glomerulonephritis is diminished in fibrinogen-deficient mice. Am J Physiol Renal Physiol. 2001;281:F1157–F1163. doi: 10.1152/ajprenal.2001.281.6.F1157. [DOI] [PubMed] [Google Scholar]
- 3.Duffield J.S., Tipping P.G., Kipari T., Cailhier J.F., Clay S., Lang R., Bonventre J.V., Hughes J. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol. 2005;167:1207–1219. doi: 10.1016/S0002-9440(10)61209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang X.R., Tipping P.G., Shuo L., Holdsworth S.R. Th1 responsiveness to nephritogenic antigens determines susceptibility to crescentic glomerulonephritis in mice. Kidney Int. 1997;51:94–103. doi: 10.1038/ki.1997.12. [DOI] [PubMed] [Google Scholar]
- 5.Holdsworth S.R., Kitching A.R., Tipping P.G. Th1 and Th2 T helper cell subsets affect patterns of injury and outcomes in glomerulonephritis. Kidney Int. 1999;55:1198–1216. doi: 10.1046/j.1523-1755.1999.00369.x. [DOI] [PubMed] [Google Scholar]
- 6.Li S., Holdsworth S.R., Tipping P.G. Antibody independent crescentic glomerulonephritis in μ-chain deficient mice. Kidney Int. 1997;51:672–678. doi: 10.1038/ki.1997.97. [DOI] [PubMed] [Google Scholar]
- 7.Kitching A.R., Holdsworth S.R., Tipping P.G. IFN-gamma mediates crescent formation and cell-mediated immune injury in murine glomerulonephritis. J Am Soc Nephrol. 1999;10:752–759. doi: 10.1681/ASN.V104752. [DOI] [PubMed] [Google Scholar]
- 8.Timoshanko J., Holdsworth S., Kitching A., Tipping P. IFN-gamma production by intrinsic renal cells and bone-marrow derived cells is required for full expression of crescentic glomerulonephritis in mice. J Immunol. 2002;168:4135–4141. doi: 10.4049/jimmunol.168.8.4135. [DOI] [PubMed] [Google Scholar]
- 9.O'Garra A., Arai N. The molecular basis of T helper 1 and T helper 2 cell differentiation. Trends Cell Biol. 2000;10:542–550. doi: 10.1016/s0962-8924(00)01856-0. [DOI] [PubMed] [Google Scholar]
- 10.Phoon R.K., Kitching A.R., Odobasic D., Jones L.K., Semple T.J., Holdsworth S.R. T-bet deficiency attenuates renal injury in experimental crescentic glomerulonephritis. J Am Soc Nephrol. 2008;19:477–485. doi: 10.1681/ASN.2007030392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitching A.R., Tipping P.G., Huang X.R., Mutch D., Holdsworth S.R. Interluekin-4 and interleukin-10 attenuate established crescentic glomerulonephritis in mice. Kidney Int. 1997;52:52–59. doi: 10.1038/ki.1997.303. [DOI] [PubMed] [Google Scholar]
- 12.Kitching A.R., Tipping P.G., Mutch D., Huang X.R., Holdsworth S.R. Interleukin-4 deficiency enhances Th1 responses and crescentic glomerulonephritis in mice. Kidney Int. 1998;53:112–118. doi: 10.1046/j.1523-1755.1998.00733.x. [DOI] [PubMed] [Google Scholar]
- 13.Kitching A.R., Tipping P.G., Timoshanko J., Holdsworth S.R. Endogenous interleukin-10 regulates Th1 responses that induce crescentic glomerulonephritis. Kidney Int. 2000;57:518–525. doi: 10.1046/j.1523-1755.2000.00872.x. [DOI] [PubMed] [Google Scholar]
- 14.Korn T., Bettelli E., Oukka M., Kuchroo V.K. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 15.Damsker J.M., Hansen A.M., Caspi R.R. Th1 and Th17 cells: adversaries and collaborators. Ann N Y Acad Sci. 2010;1183:211–221. doi: 10.1111/j.1749-6632.2009.05133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stritesky G.L., Yeh N., Kaplan M.H. IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol. 2008;181:5948–5955. doi: 10.4049/jimmunol.181.9.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hofstetter H.H., Ibrahim S.M., Koczan D., Kruse N., Weishaupt A., Toyka K.V., Gold R. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol. 2005;237:123–130. doi: 10.1016/j.cellimm.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Luger D., Silver P.B., Tang J., Cua D., Chen Z., Iwakura Y., Bowman E.P., Sgambellone N.M., Chan C.C., Caspi R.R. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. 2008;205:799–810. doi: 10.1084/jem.20071258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakae S., Nambu A., Sudo K., Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 20.Komiyama Y., Nakae S., Matsuki T., Nambu A., Ishigame H., Kakuta S., Sudo K., Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 21.Khader S.A., Bell G.K., Pearl J.E., Fountain J.J., Rangel-Moreno J., Cilley G.E., Shen F., Eaton S.M., Gaffen S.L., Swain S.L., Locksley R.M., Haynes L., Randall T.D., Cooper A.M. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 22.O'Connor R.A., Prendergast C.T., Sabatos C.A., Lau C.W., Leech M.D., Wraith D.C., Anderton S.M. Cutting edge: Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis. J Immunol. 2008;181:3750–3754. doi: 10.4049/jimmunol.181.6.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Connor W., Jr, Kamanaka M., Booth C.J., Town T., Nakae S., Iwakura Y., Kolls J.K., Flavell R.A. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. 2009;10:603–609. doi: 10.1038/ni.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yi T., Zhao D., Lin C.L., Zhang C., Chen Y., Todorov I., LeBon T., Kandeel F., Forman S., Zeng D. Absence of donor Th17 leads to augmented Th1 differentiation and exacerbated acute graft-versus-host disease. Blood. 2008;112:2101–2110. doi: 10.1182/blood-2007-12-126987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ooi J.D., Phoon R.K., Holdsworth S.R., Kitching A.R. IL-23, not IL-12, directs autoimmunity to the Goodpasture antigen. J Am Soc Nephrol. 2009;20:980–989. doi: 10.1681/ASN.2008080891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gan P.Y., Steinmetz O.M., Tan D.S., O'Sullivan K.M., Ooi J.D., Iwakura Y., Kitching A.R., Holdsworth S.R. Th17 cells promote autoimmune anti-myeloperoxidase glomerulonephritis. J Am Soc Nephrol. 2010;21:925–931. doi: 10.1681/ASN.2009070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Summers S.A., Steinmetz O.M., Li M., Kausman J.Y., Semple T., Edgtton K.L., Borza D.B., Braley H., Holdsworth S.R., Kitching A.R. Th1 and Th17 cells induce proliferative glomerulonephritis. J Am Soc Nephrol. 2009;20:2518–2524. doi: 10.1681/ASN.2009030337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paust H.J., Turner J.E., Steinmetz O.M., Peters A., Heymann F., Holscher C., Wolf G., Kurts C., Mittrucker H.W., Stahl R.A., Panzer U. The IL-23/Th17 axis contributes to renal injury in experimental glomerulonephritis. J Am Soc Nephrol. 2009;20:969–979. doi: 10.1681/ASN.2008050556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Odobasic D., Kitching A.R., Semple T.J., Holdsworth S.R. Inducible co-stimulatory molecule ligand is protective during the induction and effector phases of crescentic glomerulonephritis. J Am Soc Nephrol. 2006;17:1044–1053. doi: 10.1681/ASN.2005101022. [DOI] [PubMed] [Google Scholar]
- 30.Moussa L., Apostolopoulos J., Davenport P., Tchongue J., Tipping P.G. Protease-activated receptor-2 augments experimental crescentic glomerulonephritis. Am J Pathol. 2007;171:800–808. doi: 10.2353/ajpath.2007.061155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Odobasic D., Kitching A.R., Semple T.J., Timoshanko J.R., Tipping P.G., Holdsworth S.R. Glomerular expression of CD80 and CD86 is required for leukocyte accumulation and injury in crescentic glomerulonephritis. J Am Soc Nephrol. 2005;16:2012–2022. doi: 10.1681/ASN.2004060437. [DOI] [PubMed] [Google Scholar]
- 32.Odobasic D., Kitching A.R., Tipping P.G., Holdsworth S.R. CD80 and CD86 costimulatory molecules regulate crescentic glomerulonephritis by different mechanisms. Kidney Int. 2005;68:584–594. doi: 10.1111/j.1523-1755.2005.00436.x. [DOI] [PubMed] [Google Scholar]
- 33.Li M., O'Sullivan K.M., Jones L.K., Semple T., Kumanogoh A., Kikutani H., Holdsworth S.R., Kitching A.R. CD100 enhances dendritic cell and CD4+ cell activation leading to pathogenetic humoral responses and immune complex glomerulonephritis. J Immunol. 2006;177:3406–3412. doi: 10.4049/jimmunol.177.5.3406. [DOI] [PubMed] [Google Scholar]
- 34.Odobasic D., Kitching A.R., Semple T.J., Holdsworth S.R. Endogenous myeloperoxidase promotes neutrophil-mediated renal injury, but attenuates T cell immunity inducing crescentic glomerulonephritis. J Am Soc Nephrol. 2007;18:760–770. doi: 10.1681/ASN.2006040375. [DOI] [PubMed] [Google Scholar]
- 35.Odobasic D., Leech M.T., Xue J.R., Holdsworth S.R. Distinct in vivo roles of CD80 and CD86 in the effector T-cell responses inducing antigen-induced arthritis. Immunology. 2008;124:503–513. doi: 10.1111/j.1365-2567.2007.02802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawasaki K., Yaoita E., Yamamoto T., Tamatani T., Miyasaka M., Kihara I. Antibodies against intracellular adhesion molecule-1 and lymphocyte function-associated antigen-1 prevent glomerular injury in rat experimental crescentic glomerulonephritis. J Immunol. 1993;150:1074–1083. [PubMed] [Google Scholar]
- 37.Lloyd C.M., Minto A.W., Dorf M.E., Proudfoot A., Wells T.N.C., Salant D.J., Gutierrez-Ramos J.-C. RANTES and monocyte chemoattractant protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J Exp Med. 1997;185:1371–1380. doi: 10.1084/jem.185.7.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tipping P., Huang X., Berndt M., Holdsworth S. A role for P-selectin in complement-independent neutrophil-mediated glomerular injury. Kidney Int. 1994;46:79–88. doi: 10.1038/ki.1994.246. [DOI] [PubMed] [Google Scholar]
- 39.Tipping P.G., Huang X.R., Berndt M., Holdsworth S.R. P-selectin directs T lymphocyte-mediated injury in delayed-type hypersensitivity responses: studies in glomerulonephritis and cutaneous delayed-type hypersensitivity. Eur J Immunol. 1996;26:454–460. doi: 10.1002/eji.1830260228. [DOI] [PubMed] [Google Scholar]
- 40.Park H., Li Z., Yang X., Chang S., Nurieva R., Wang Y.-H., Wang Y., Hood L., Zhu Z., Tian Q., Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lelongt B., Bengatta S., Delauche M., Lund L.R., Werb Z., Ronco P.M. Matrix metalloproteinase 9 protects mice from anti-glomerular basement membrane nephritis through its fibrinolytic activity. J Exp Med. 2001;193:793–802. doi: 10.1084/jem.193.7.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Becker C., Dornhoff H., Neufert C., Fantini M.C., Wirtz S., Huebner S., Nikolaev A., Lehr H.A., Murphy A.J., Valenzuela D.M., Yancopoulos G.D., Galle P.R., Karow M., Neurath M.F. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J Immunol. 2006;177:2760–2764. doi: 10.4049/jimmunol.177.5.2760. [DOI] [PubMed] [Google Scholar]
- 43.Schnyder-Candrian S., Togbe D., Couillin I., Mercier I., Brombacher F., Quesniaux V., Fossiez F., Ryffel B., Schnyder B. Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med. 2006;203:2715–2725. doi: 10.1084/jem.20061401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Calvani N., Satoh M., Croker B.P., Reeves W.H., Richards H.B. Nephritogenic autoantibodies but absence of nephritis in Il-12p35-deficient mice with pristane-induced lupus. Kidney Int. 2003;64:897–905. doi: 10.1046/j.1523-1755.2003.00178.x. [DOI] [PubMed] [Google Scholar]
- 45.Steinmetz O.M., Summers S.A., Gan P.Y., Semple T., Holdsworth S.R., Kitching A.R. The Th17-defining transcription factor RORgammat promotes glomerulonephritis. J Am Soc Nephrol. 2011;22:472–483. doi: 10.1681/ASN.2010040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Summers S.A., Phoon R.K., Ooi J.D., Holdsworth S.R., Kitching A.R. The IL-27 receptor has biphasic effects in crescentic glomerulonephritis mediated through Th1 responses. Am J Pathol. 2011;178:580–590. doi: 10.1016/j.ajpath.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iwakura Y., Nakae S., Saijo S., Ishigame H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev. 2008;226:57–79. doi: 10.1111/j.1600-065X.2008.00699.x. [DOI] [PubMed] [Google Scholar]
- 48.Kroenke M.A., Carlson T.J., Andjelkovic A.V., Segal B.M. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology: CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zelante T., De Luca A., Bonifazi P., Montagnoli C., Bozza S., Moretti S., Belladonna M.L., Vacca C., Conte C., Mosci P., Bistoni F., Puccetti P., Kastelein R.A., Kopf M., Romani L. IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunol. 2007;37:2695–2706. doi: 10.1002/eji.200737409. [DOI] [PubMed] [Google Scholar]
- 50.Toh M.L., Kawashima M., Zrioual S., Hot A., Miossec P. IL-17 inhibits human Th1 differentiation through IL-12R beta 2 downregulation. Cytokine. 2009;48:226–230. doi: 10.1016/j.cyto.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 51.Collette Y., Benziane A., Razanajaona D., Olive D. Distinct regulation of T-cell death by CD28 depending on both its aggregation and T-cell receptor triggering: a role for Fas-FasL. Blood. 1998;92:1350–1363. [PubMed] [Google Scholar]
- 52.Nakae S., Iwakura Y., Suto H., Galli S.J. Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J Leukoc Biol. 2007;81:1258–1268. doi: 10.1189/jlb.1006610. [DOI] [PubMed] [Google Scholar]
- 53.Sergejeva S., Ivanov S., Lotvall J., Linden A. Interleukin-17 as a recruitment and survival factor for airway macrophages in allergic airway inflammation. Am J Respir Cell Mol Biol. 2005;33:248–253. doi: 10.1165/rcmb.2004-0213OC. [DOI] [PubMed] [Google Scholar]