

Abstract
Rotenone exposure has emerged as an environmental risk factor for inflammation-associated neurodegenerative diseases. However, the underlying mechanisms responsible for the harmful effects of rotenone in the brain remain poorly understood. Herein, we report that myeloperoxidase (MPO) may have a potential regulatory role in rotenone-exposed brain-resident immune cells. We show that microglia, unlike neurons, do not undergo death; instead, they exhibit distinctive activated properties under rotenone-exposed conditions. Once activated by rotenone, microglia show increased production of reactive oxygen species, particularly HOCl. Notably, MPO, an HOCl-producing enzyme that is undetectable under normal conditions, is significantly increased after exposure to rotenone. MPO-exposed glial cells also display characteristics of activated cells, producing proinflammatory cytokines and increasing their phagocytic activity. Interestingly, our studies with MPO inhibitors and MPO-knockout mice reveal that MPO deficiency potentiates, rather than inhibits, the rotenone-induced activated state of glia and promotes glial cell death. Furthermore, rotenone-triggered neuronal injury was more apparent in co-cultures with glial cells from Mpo−/− mice than in those from wild-type mice. Collectively, our data provide evidence that MPO has dual functionality under rotenone-exposed conditions, playing a critical regulatory role in modulating pathological and protective events in the brain.
Pesticide exposure has received considerable attention as an environmental risk factor for several diseases, including Parkinson's disease (PD). An increasing number of epidemiological and biochemical studies1,2 have implicated the oxidative properties of pesticides in the pathogenesis of such diseases. Rotenone, one of the most widely used herbicides, is a ketone compound extracted from the roots of Lonchocarpus species.3 It is a highly lipophilic pesticide that readily crosses the blood-brain barrier and accumulates throughout the brain.4,5 Rotenone exposure has disrupted cell membranes and caused damage to proteins, lipids, and DNA, ultimately leading to neuronal cell death. Indeed, there is increasing evidence that long-term exposure to rotenone causes significant degenerative diseases.6–9
Myeloperoxidase (MPO) is a heme-containing protein that catalyzes the formation of the potent oxidant HOCl and other chlorinating species derived from H2O2. MPO and MPO-derived oxidants could mediate inflammatory responses at sites of inflammation, thereby contributing to the defense system against pathogens.10 Reports11,12 indicate that MPO levels are significantly increased in various disease states, such as infection, ischemia, atherosclerosis, and acute myeloid leukemia. Increased MPO levels are widely considered characteristic of systemic inflammatory diseases. Recently, several interesting reports13,14 have revealed that MPO has catalytic activity and exhibits cytokine-like properties, activating and modulating inflammatory signaling cascades. MPO has been closely involved in stimulating mitogen-activated protein kinase activity, cell growth, and protease activity, thereby influencing the immune responses and the progression of several inflammation-associated diseases.10,15–19
Until recently, phagocytic blood cells were thought to be the only cellular sources of MPO. However, recent studies18,20,21 have shown that several cell types, including neuronal cells, express MPO under certain pathological conditions. Also, MPO is not expressed in healthy brain parenchyma but is expressed in several neurodegenerative diseases, such as Alzheimer's disease and PD.20,22,23 However, the precise roles of MPO and the underlying mechanisms responsible for its action have not been determined.
Immune and inflammatory responses in the central nervous system (CNS) are mainly coordinated by the interaction of the brain-resident immune cells, microglia, and astrocytes with neurons. Thus, we questioned how glial cells respond to rotenone exposure and whether glial cells play a role in the pathophysiological consequences of rotenone exposure. In the present study, we investigated the responses of glial cells and their potential roles in combating against rotenone-induced damage in the CNS. Intriguingly, we found that MPO may act as an essential modulator, regulating the activation of glia and affecting neuronal injury under rotenone-exposed conditions. Our data provide new insights into the cellular responses associated with MPO in the rotenone-exposed brain and suggest a potential target for the development of a therapeutic intervention in diseases associated with rotenone exposure.
Materials and Methods
Reagents and Antibodies
Rotenone and human MPO were obtained from Calbiochem (La Jolla, CA); minimal essential medium, Life Technologies, Inc. (Gaithersburg, MD); Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum, Hyclone (Logan, UT); salicyl hydroxamic acid and (D+)-mannose, Sigma-Aldrich (St Louis, MO); and 4-aminobenzoylhydrazide (ABAH), Calbiochem (San Diego, CA). The antibodies used in this study included the following: mouse anti-α-tubulin (Sigma-Aldrich), anti-MPO (Dako, Glostrup, Denmark), anti-glial fibrillary acidic protein (GFAP; Cell Signaling, Beverly, MA), anti-CD11b (Serotec, Oxford, UK), anti-manganese-containing superoxide dismutase (MnSOD) (Upstate, Lake Placid, NY), anti-cyclooxygenase (COX)-2 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-inducible nitric oxide synthase (iNOS) (Upstate), anti-interferon-γ receptor (IFNγR) (Santa Cruz Biotechnology), anti-arginase-1 (BD Biosciences, San Jose, CA), anti-CD16/CD32 (Fc Block; BD Biosciences), and anti-tyrosine hydroxylase (TH; Abcam, Cambridge, MA). Fluorophore-conjugated secondary antibodies (Alexa Fluor 488 and 546) were obtained from Molecular Probes (Eugene, OR), and horseradish peroxidase–conjugated secondary antibodies were obtained from Bio-Rad (Hercules, CA).
Animals
Sprague-Dawley rats were obtained from SamTako Bio Korea (Osan, Korea), and C57BL/6 and B6.129 × 1-Mpotm1Lus/J mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Adult timed-pregnant Sprague-Dawley rats and CrljOri:CD1 mice were obtained from ORIENT BIO (Sungnam, Korea). All animal procedures were performed according to the National Cancer Center guidelines for the care and use of laboratory animals.
Glial Cell Culture
Glial cells isolated from the cerebral cortex of 1- to 3-day-old Sprague-Dawley rats were triturated into single-cell suspensions, plated in 75-cm2 T-flasks (0.5 hemispheres per flask), and cultured in minimal essential medium containing 10% fetal bovine serum for 2 weeks.24 The microglia were detached from the flasks by mild shaking and applied to a nylon mesh to remove astrocytes and cell clumps. Cells were plated in 60-mm2 dishes (8 × 105 cells per dish) or 100-mm2 dishes (2 × 106 cells per dish). One hour later, the cells were washed to remove unattached cells before being used in experiments. After removal of the microglia, primary astrocytes were prepared using trypsinization.25 Murine BV2 microglial cells were maintained in DMEM supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a humidified incubator under 5% CO2.
Neuron-Enriched Mesencephalic Cultures
Ventral mesencephalic tissues were dissected from embryonic day 14 or 15 Sprague-Dawley rats or embryonic day 12 CrljOri:CD1 mice and dissociated enzymatically (0.1% trypsin) and mechanically.26 Cells were seeded onto six-well plates (2 × 106 cells per well) or 24-well plates (5 × 105 cells per well) precoated with poly-d-lysine (5 mg/mL) and laminin (0.2 mg/mL). Rat neurons were maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin-streptomycin (P/S) at 37°C in a humidified 5% CO2 incubator. On the following day, the medium was replaced with a chemically defined serum-free medium containing 50% DMEM; 50% Ham's F12 media; 1% insulin, transferring, selenium; and 1% P/S. Then, it was incubated for 48 hours before treatment. Mouse neurons were resuspended in Neurobasal medium (Invitrogen, Carlsbad, CA) containing 1× B27 supplement (Invitrogen), 0.5 mmol/L glutamine, and 1% P/S at 37°C in a humidified 5% CO2 incubator. During the subsequent 4 to 5 days, cells were refed every 2 days and replaced with Neurobasal medium without B27 for the lactate dehydrogenase (LDH) assay.
Phagocytosis Assay
Microglia were plated in 60-mm2 dishes (2.5 × 104 cells per dish) and then treated or left untreated with rotenone. The phagocytic capacity was measured by incubating cells with fluorescein isothiocyanate (FITC)–conjugated phagocytic beads (8 × 106 beads/mL; FluoSpheres polystyrene microspheres; Molecular Probes) at 37°C for 3 hours. Cells were then washed three times with PBS and then gently removed from the wells using cell scrapers for fluorescence-activated cell sorter (FACS) analysis. To examine Fcγ receptor–mediated phagocytosis, BV2 cells were blocked using 10 μg/mL Fc Block (BD Biosciences) directed against FcγRIII(CD32)/FcγRII(CD16). Fc Block also binds the FcγI receptor (CD64) via its Fc domain.
MPO Activity Assay
MPO activity was measured using an EnzChek Myeloperoxidase Activity Assay Kit, as described by the manufacturer (Molecular Probes or Invitrogen). Briefly, primary microglia and astrocytes were lysed using a standard freeze-thaw method and suspended in 50 μL of PBS. Lysates (50 μL) and supernatant were incubated for 30 minutes at room temperature in the working solution, according to the manufacturer's instructions. Fluorescence was measured at 590 nm after excitation at 530 nm using a Spectra-Max Gemini fluorometer (Molecular Devices, Sunnyvale, CA) at room temperature.
GSH Measurement
Total intracellular γ-glutamyl-cysteinyl-glycine (GSH) content was measured using a kit from Cayman, according to the manufacturer's instructions. In brief, primary microglia were scraped from 60-mm2 dishes (8 × 105 cells per dish), homogenized in 0.1 mL cold buffer, and then centrifuged at 10,000 × g for 15 minutes at 4°C. The supernatant was collected and deproteinized by mixing with metaphosphoric acid before GSH content measurement.
Confocal Microscopy
Cells grown on coverslips were fixed in ice-cold methanol and permeabilized in 0.1% Triton X-100/PBS for 10 minutes. Cells were then blocked with 10% bovine serum albumin/0.1% Triton X-100/PBS for 30 minutes at room temperature, and the coverslips were washed twice with 0.1% Triton X-100/PBS. Fluorescent images were acquired with a confocal laser scanning microscopy system (model LSM510 meta; Carl Zeiss, Jena, Germany) and Axio Observer Z1 (Carl Zeiss) using rhodamine, fluorescein, and DAPI filters. The confocal system software and Axiovision software were used to capture and store the images.
RT-PCR Analysis
Total RNA was isolated using Easy-Blue (iNtRON, Daejeon, Korea), and cDNA was synthesized using avian myeloblastosis virus reverse transcriptase (TaKaRa, Dalian City, Japan), according to the manufacturer's instructions. PCRs were performed with 35 cycles of sequential reactions. Oligonucleotide primers were obtained from Bioneer (Seoul, Korea). RT-PCR analysis was performed using previously reported primers27,28 and the following primers: rat catalase, 5′-TTATGTTACCTCACAGCCTGGT-3′ (forward) and 5′-GTGTTGTGTGTTCTGTGTGTGTAG-3′ (reverse); rat COX-2, 5′-ACACTCTATCACTGGCATCC-3′ (forward) and 5′-GAAGGGACACCCTTTCACAT-3′ (reverse); rat glutathione peroxidase (GPx)-1, 5′-TGAGAAGTGCGAGGTGAATG-3′ (forward) and 5′-AACACCGTCTGGACCTACCA-3′ (reverse); rat GPx-2, 5′-TGCCCTACCCTTATGACGAC-3′ (forward) and 5′-GGAGATTCCTAGGCTGAGCA-3′ (reverse); rat matrix metalloproteinase (MMP)-3, 5′-CTGGAATGGTCTTGGCTCAT-3′ (forward) and 5′-CTGACTGCATCGAAGGACAA-3′ (reverse); rat MnSOD, 5′-AACGCGCAGATCATGCAGCTGC-3′ (forward) and 5′-ACATTCTCCCAGTTGATTACAT-3′ (reverse); rat MPO, 5′-GTATCGAACCATCACTGGAC-3′ (forward) and 5′-AGCTGGTCTCACAGTTGAGT-3′ (reverse); rat neuronal NOS, 5′-GGCACTGGCATCGCACCCTT-3′ (forward) and 5′-CTTTGGCCTGTCCGGTTCCC-3′ (reverse); and mouse tumor necrosis factor (TNF)-α, 5′-ATGAGCACAGAAAGCATGATC-3′ (forward) and 5′-TACAGGCTTGTCACTCGAATT-3′ (reverse).
ELISA
Enzyme-linked immunosorbent assay (ELISA) kits were used according to the manufacturer's protocols. After treatment with stimuli, 100 μL of conditioned media was collected and assayed using ELISA kits for rat TNF-α (eBioscience, San Diego, CA), IL-4, IL-6, and IL-13 (BioSource International, Comarillo, CA).
Western Blot Analysis
Cells were washed twice with cold PBS and lysed in ice-cold modified radioimmunoprecipitation assay buffer (50 mmol/L Tris-HCl, pH 7.4; 1% Nonidet P-40; 0.25% Na-deoxycholate; 150 mmol/L NaCl; and 10 mmol/L Na2HPO4) containing protease inhibitors (2 mmol/L phenyl-methyl sulfonyl fluoride, 10 μg/mL leupeptin, 10 μg/mL pepstatin, 0.5 mmol/L Na3VO4, 0.5 mol/L NaF, and 2 mmol/L EDTA). The proteins in the medium were further fractionated by using the Rapid-Con Protein concentration kit (Elpis Biotech, Daejeon, Korea), according to the manufacturer's protocol. The lysate was centrifuged for 20 minutes at 13,000 rpm at 4°C, and supernatant proteins were separated by SDS-PAGE on 8% gels and transferred to nitrocellulose membranes. The membranes were incubated with primary antibodies and horseradish peroxidase–conjugated secondary antibodies and then visualized using an enhanced chemiluminescence system.
Flow Cytometric Analysis
Cells cultured according to standard procedures were dissociated from the culture plates by pipetting and washed twice with PBS. Antibody incubations were performed for 30 minutes at 4°C. Cells were washed by sedimenting at 450 × g for 10 minutes at 4°C. Flow cytometric measurements were performed using a Becton Dickinson FACSCalibur system (Becton Dickinson, Mansfield, MA), and data were analyzed using FlowJo software (Treestar, Inc., San Carlos, CA).
Measurement of ROS
Cells were suspended in 5 μmol/L CM-H2DCFDA (DCF; Molecular Probes) or 10 μmol/L aminophenyl fluorescein (APF; Molecular Probes) for 30 minutes at 37°C in the dark. After incubation, the cells were washed twice with PBS and suspended in PBS. The green emission of DCF, APF, and rhodamine-123 was measured using an FACSCalibur flow cytometer (BD Biosciences). DCF detects H2O2, hydroxyl radical, peroxyl radical, and peroxynitrite anion, whereas APF reacts preferentially with HOCl. To evaluate oxidative stress using rhodamine-123 (Molecular Probes), the cells were incubated with 1 μg/mL rhodamine-123 for 10 minutes in culture media at 37°C.
LDH Assay
LDH released into the supernatant by damaged cells was measured by collecting 50 μL of cell-free supernatant into 96-well plates (n = 3) and then adding 125 μL of NADH solution and 25 μL of pyruvate solution. The LDH level was determined immediately by measuring absorbance at a wavelength of 340 nm in kinetics mode for 5 minutes on a microplate reader (Molecular Devices). The percentage cytotoxicity was calculated as follows (using total cellular LDH as a low control): Cytotoxicity (%) = [(Experimental Value-Low Control)/(High Control-Low Control)] × 100%.
Cell Viability Assay
Cell viability was determined using the Live/Dead Viability Cytotoxicity Kit (Molecular Probes) or the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan), according to the manufacturer's instruction. For the Live/Dead Viability Cytotoxicity assay, cells were grown in 24-well plates (primary microglia, 8 × 104 cells per well; primary mesencephalic neurons, 5 × 105 cells per well) and incubated with the indicated stimuli. Viability was assessed by staining cells according to the manufacturer's protocol and analyzing by fluorescence microscopy (Axio Observer Z1). The percentage cell viability was defined in each image as the percentage of live and dead cells versus the total number of cells, counting at least 350 cells per image. For the CCK-8 assay, viable cells were counted by absorbance measurements at 450 nm using a Versamax microplate reader (Molecular Devices) at room temperature.
Measurement of Cell Death by PI and Annexin V Staining
The cytotoxic effects of rotenone were assessed by flow cytometry after staining the cells with propidium iodide (PI). Briefly, 1 × 106 cells per sample were washed twice with cold PBS and fixed in ice-cold 75% ethanol at 4°C. The cells were then washed twice with PBS and incubated with 50 μg/mL RNase A and 40 μg/mL PI for 30 minutes at 4°C. For measurement of cell death using annexin V, cells were harvested and washed in binding buffer, then incubated with annexin V–FITC (BD Pharmingen, San Diego, CA) and 7-amino-actinomycin D for 15 minutes at room temperature in the dark. Cells were then immediately analyzed by flow cytometry using an FACSCalibur flow cytometer (Becton Dickinson). Gating was defined using untreated control samples to exclude aggregates and to determine the appropriate quadrants.
Data Analysis
All data were expressed as the mean ± SEM and analyzed by one-way analysis of variance, followed by post hoc comparisons (Student-Newman-Keuls test) using the Statistical Package for Social Sciences 8.0 (SPSS, Chicago, IL).
Results
Rotenone Exposure Does Not Trigger Microglial Cell Death
Studies29,30 have suggested a potential role for microglia in modulating pathophysiological events in the rotenone-exposed brain. In an effort to precisely understand the distinct responses of glial cells to rotenone exposure, we first examined the cell viability of rotenone-treated primary microglia cultures compared with those of mesencephalic neuron-enriched cultures. Rat primary microglia and neurons were mock treated or treated with rotenone for 3 days, after which the degree of cell death was determined using the LDH assay. Interestingly, rotenone (1 to 100 nmol/L) did not significantly affect the viability of primary microglia but did enhance neuronal cell death in mesencephalic neuron-enriched cultures (Figure 1A; see also Supplemental Figure S1A at http://ajp.amjpathol.org). To further determine the effects of rotenone on microglial cell viability in the brain, we separately cultured rat primary microglia and neurons using transwell chambers. The cells were mock treated or treated with 30 nmol/L rotenone for 3 days, and the viability of each cell type was determined by FACS analysis (Figure 1B). As was the case with the microglia alone culture, microglial cell death was not detected, but neuronal cell death was increased compared with mock-treated controls. Similar results as previously described were obtained by fluorescence microscopy using a Live/Dead Viability Cytotoxicity kit and FACS analysis (Figure 1C; see also Supplemental Figure S1, A and B, at http://ajp.amjpathol.org). These findings indicate that rotenone does not trigger microglial cell death in the absence or presence of neurons.
Figure 1.
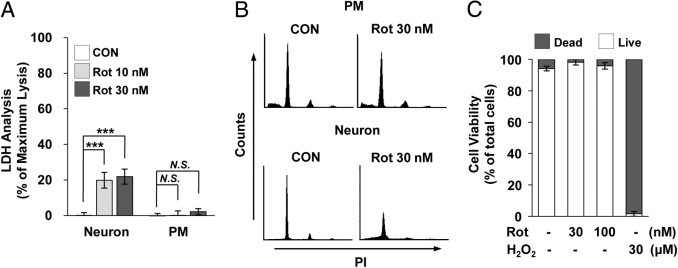
Effects of rotenone (Rot) on the viability of primary microglia. A: Rat primary microglia (PM) and mesencephalic neurons were cultured with the indicated concentrations of Rot for 3 days, and the degree of cell death was assessed by measuring LDH leakage into the culture medium. Data are representative of four independent experiments. ***P < 0.001 compared with mock-treated controls (CONs). N.S. indicates no significant difference. B: PM and mesencephalic neurons were co-cultured using transwell inserts and treated with 30 nmol/L Rot for 3 days. Cells were stained with PI, and the viability of each cell type was measured by FACS analysis. C: Primary microglia were incubated with or without the indicated concentrations of Rot for 3 days. Cell viability was analyzed using the Live/Dead Viability Cytotoxicity kit, as described in Materials and Methods.
Microglia Display Typical Activated Properties under Rotenone-Exposed Conditions
Although rotenone did not trigger microglial cell death, rat primary microglia displayed typical activated phenotypes under rotenone-exposed conditions, indicating that microglia could actively respond to rotenone exposure (see Supplemental Figure S1C at http://ajp.amjpathol.org). Therefore, we carefully examined the characteristics of rotenone-exposed microglia by analyzing the expression profile of genes associated with microglial activation. As shown in Figure 2, A and B, rotenone exposure significantly increased the expression of markers representative of classic (inflammatory M1) activation in rat primary microglia.31,32 Treatment of microglia with rotenone obviously enhanced the production of several proinflammatory mediators, including TNF-α, IL-1β, IL-6, IL-12p40, iNOS, IFN-γ, and IFNγR. To better characterize the properties of activated microglia under conditions of rotenone exposure, we further examined the expression of anti-inflammatory M2 activation markers.31,33 In contrast to previous descriptions, we did not observe any noticeable changes in the level of M2 activation markers, including IL-4, IL-13, IL-10, transforming growth factor-β, and arginase-1, under the same culture condition (Figure 2C). These results indicate that rotenone exposure primes microglia to enter a classically activated state, expressing proinflammatory receptors and mediators.
Figure 2.
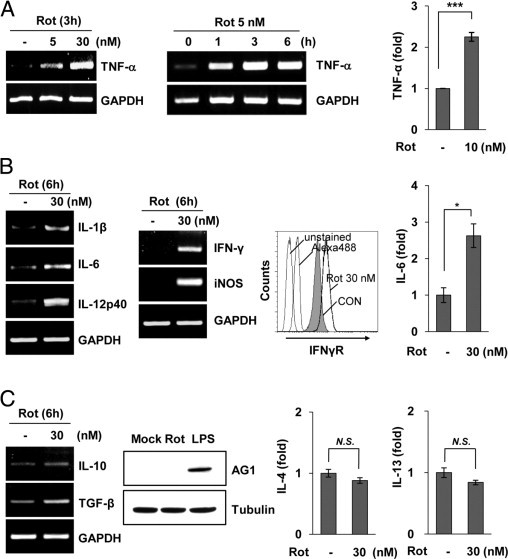
Rotenone (Rot) exposure primes microglia to enter a classically activated state. A: Primary microglia were stimulated with the indicated concentrations of Rot, after which the expression of TNF-α was determined by RT-PCR analysis (left). TNF-α protein was also measured by ELISA in rat primary microglia treated with 10 nmol/L Rot for 24 hours (right). ***P < 0.001 compared with mock-treated controls (CONs). B: The mRNA levels of IL-1β, IL-6, IL-12p40, IFN-γ, and iNOS were determined by RT-PCR (left). The level of IFNγR was observed by flow cytometric analysis using an anti-IFNγR antibody (middle). The level of IL-6 secreted into the media was measured by ELISA in primary microglia treated with 30 nmol/L Rot for 24 hours (right). *P < 0.05 compared with mock-treated CONs. C: The mRNA levels of IL-10 and transforming growth factor-β were determined by RT-PCR (left). Primary microglia were cultured with 30 nmol/L Rot or 10 ng/mL lipopolysaccharide for 24 hours, and the levels of arginase (AG)-1 were measured using Western blot analysis (middle). The levels of IL-4 or IL-13 were assayed by ELISA (right). GAPDH indicates glyceraldehyde-3-phosphate dehydrogenase; N.S., no significant difference.
To better define the state of rotenone-exposed microglia, we examined the phagocytic activity of microglia, one of the representative characteristic properties of activated microglia. FACS analysis showed that treatment of BV2 microglia with rotenone considerably augmented the cellular uptake of fluorescent beads (Figure 3A). Next, we investigated whether rotenone-induced phagocytic activity was dependent of Fcγ receptors (FcγRs) because FcγRs on microglia contribute to diverse cellular functions and FcγR-mediated phagocytosis is associated with the inflammatory activity of microglia.34,35 FACS analysis using a rat anti-mouse CD16/CD32 antibody indicated that FcγRs on BV2 microglial cells were up-regulated after exposure to rotenone (Figure 3B and data not shown). Moreover, rotenone-triggered phagocytic activity was markedly reduced by blocking FcγRs using Fc Block directed against such receptors (Figure 3C). These results suggest that rotenone is capable of promoting FcγR-mediated phagocytic activity, resulting in inflammatory conditions in the brain. Taken together, our results indicate that microglia are not killed by rotenone; rather, they act as effector cells in the brain.
Figure 3.
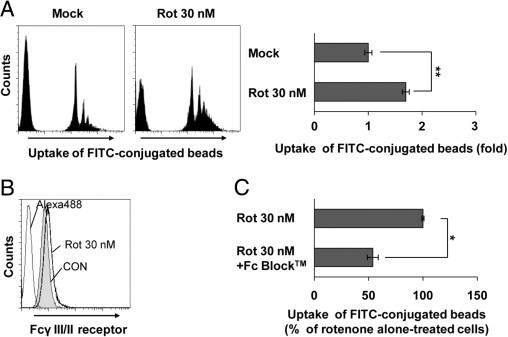
Rotenone (Rot) exposure triggers phagocytic activity in microglia. A: BV2 microglia were cultured in the presence or absence of 30 nmol/L Rot for 24 hours, and phagocytosis of FITC-conjugated fluorescent beads was determined by flow cytometric analysis, as described in Materials and Methods. **P < 0.005 when compared with mock-treated cells. B: BV2 microglia were treated with 30 nmol/L Rot for 24 hours, after which expression of Fcγ receptors was determined by FACS analysis using an anti-CD16/CD32 antibody directed against FcγIII/II receptors. C: BV2 microglia were incubated with 10 μg/mL Fc Block (BD Biosciences) for 1 hour at 37°C and followed by exposure to 30 nmol/L Rot for 18 hours. The phagocytic activity was analyzed by FACS using FITC-conjugated fluorescent beads. *P < 0.05 when compared with Rot alone–treated samples. CON indicates control.
Rotenone Stimulates MPO Generation in the Microglia
Oxidative stresses caused by reactive oxygen species (ROS) have been intimately implicated as critical mediators of many cellular processes and can lead to diverse physiological and pathological outcomes. To further investigate the events that occur in microglia after exposure to rotenone, we monitored the generation of intracellular ROS. The levels of DCF fluorescence were much greater in rotenone-exposed rat primary microglia than in mock-treated cells (Figure 4A), indicating an increase in intracellular ROS. Notably, rotenone exposure significantly increased the fluorescence of APF, a highly sensitive probe that selectively detects hydroxyl radicals and hypochloride anions (Figure 4A).36
Figure 4.
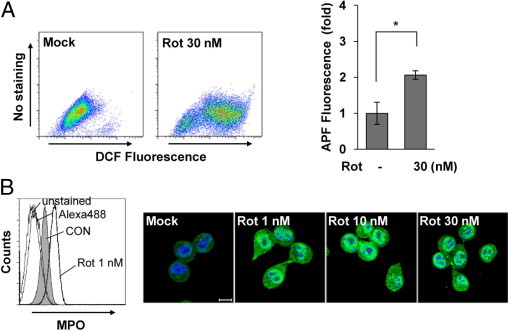
Rotenone (Rot) exposure significantly enhances ROS generation and MPO expression in microglia. A: Primary microglia were cultured with or without 30 nmol/L Rot for 3 hours and then incubated with 5 μmol/L DCF for 30 minutes at 37°C (left). DCF fluorescence was measured by FACS analysis. The data are representative of three independent experiments with similar results. Primary microglia were treated with or without Rot for 3 hours and then stained with 10 μmol/L APF for 30 minutes at 37°C and analyzed by FACS (right). The graph represents the fold changes in mean ± SEM fluorescence intensity from three independent experiments. *P < 0.05 when compared with mock-treated controls (CONs). B: Primary microglia were mock treated or treated with 1 nmol/L Rot for 18 hours, after which the intracellular levels of MPO were analyzed by FACS using an anti-MPO antibody (left). After exposure of BV2 cells to Rot for 12 hours, MPO expression was observed by confocal microscopy (right). MPO, green; DAPI, blue. Scale bar = 10 μm. The data shown are representative of at least three independent experiments.
The formation of hypochloride is catalyzed by MPO from chloride ions and H2O2. Therefore, we considered the possibility that MPO could be involved in rotenone-induced activation of microglia and subsequent pathophysiological events. FACS analyses showed that treatment of rat primary microglia with rotenone resulted in a significant induction of MPO expression. In particular, this increase in MPO expression was detected in cells treated with a concentration of rotenone as low as 1 nmol/L (Figure 4B). By using confocal microscopy, we confirmed the rotenone-stimulated increase of MPO expression in BV2 microglial cells. MPO expression was low in mock-treated BV2 microglial cells and was significantly increased by rotenone at all concentrations tested (Figure 4B). Interestingly, an increase of MPO levels by rotenone is much more prominent compared with those by other microglial activators, such as lipopolysaccharide, IFN-γ, Gmix, and sulfatide (data not shown). These findings suggest the interesting possibility that rotenone exposure characteristically increases MPO expression in microglia.
Microglia Actively Respond to Rotenone-Triggered Oxidative Stress by Producing Pro-Oxidative and Antioxidative Enzymes
To further address the effects of rotenone on oxidative events in microglia, we examined the expression levels of representative pro-oxidative and antioxidative enzymes under rotenone-exposed conditions. As shown in Figure 5A, rotenone exposure significantly induced the expression of iNOS, COX-2, and MPO in rat primary microglia. Interestingly, under the same experimental conditions, we also observed that the levels of antioxidative enzymes, such as MnSOD, catalase, GPx-1, and GPx-2, became elevated in rat primary microglia within 3 hours after treatment with rotenone (Figure 5B), indicating that rotenone exposure may trigger the antioxidant defense system of microglia.37 These results suggest that rotenone exposure enhanced the levels of pro-oxidative and antioxidative enzymes in glial cells. In addition, 24 hours after exposure to rotenone, the levels of glutathione (ie, GSH), a prominent antioxidant, were higher in rotenone-treated microglia than in mock-treated cells (Figure 5C). Overall, our results indicate that microglia actively respond to rotenone-triggered oxidative stress by simultaneously modulating pro-oxidative and antioxidative enzymes, especially MPO, thereby affecting inflammatory and defense systems in the brain.
Figure 5.
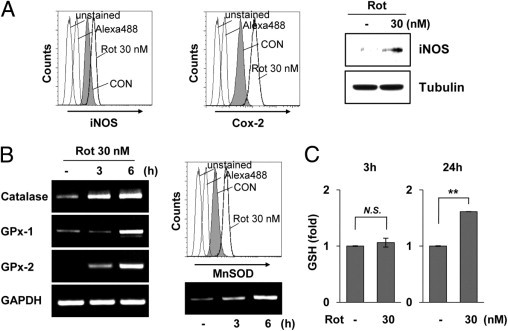
Rotenone (Rot) exposure affects the levels of pro-oxidative and antioxidative enzymes. A: BV2 microglia were mock treated or treated with 30 nmol/L Rot for 24 hours, and intracellular expression levels of iNOS and COX-2 were determined by FACS (left). Rat primary microglia were mock treated or treated with 30 nmol/L Rot for 24 hours, after which iNOS levels were examined using Western blot analysis (right). B: The transcript levels of catalase, GPx-1, and GPx-2 (left) and MnSOD (bottom right) were determined by RT-PCR in primary microglia. The intracellular levels of MnSOD were determined by FACS analysis (top right). C: Primary microglial cells were mock treated or treated with 30 nmol/L Rot for the indicated times, and the total intracellular GSH content was measured using a kit from Cayman, according to the manufacturer's protocol. **P < 0.005 when compared with mock-treated control (CON) cells. GAPDH indicates glyceraldehyde-3-phosphate dehydrogenase; N.S., no significant difference.
MPO Enhances Production of ROS and Inflammation-Associated Molecules
MPO is an oxidative free radical–producing enzyme that regulates oxidative stress at sites of inflammation.18,38,39 Because rotenone exposure caused a notable increase in MPO expression, we sought to determine whether MPO could influence the ability of microglia to mediate cellular events in the rotenone-exposed brain. First, we investigated the effects of MPO on the viability of microglia. At all concentrations tested (ranging from 1 ng/mL to 1 μg/mL), MPO had no effect on BV2 microglial viability, indicating that MPO, like rotenone, does not directly induce microglial cell death (Figure 6A and data not shown). Second, we examined the impact of MPO on the activation state of microglia. Primary microglia were treated with vehicle or 100 ng/mL MPO for 3 hours, after which DCF fluorescence was measured by FACS analysis. As expected, treatment with exogenous MPO resulted in a marked increase in ROS generation, indicating that MPO is able to activate microglia (Figure 6B). We also measured the phagocytic activity of MPO-exposed BV2 microglia and examined the expression levels of representative microglial activation-associated molecules that have previously been associated with inflammation and neurodegeneration (Figure 6C).18,40 MPO exposure considerably augmented phagocytosis and significantly increased the production of several inflammatory mediators, including IL-6, IL-12p40, TNF-α, and matrix metalloproteinase-3 (Figure 6, D–G). Moreover, the MPO transcript level was also significantly increased by MPO, indicating that MPO may regulate its own transcription in microglia (Figure 6E). However, we did not observe an MPO-dependent transcriptional change of representative anti-inflammatory M2 markers that we tested (Figure 6G and data not shown). Collectively, these findings demonstrate that MPO activates microglia, thereby further enhancing production of inflammation-associated molecules and affecting the pathological environment in the brain.
Figure 6.
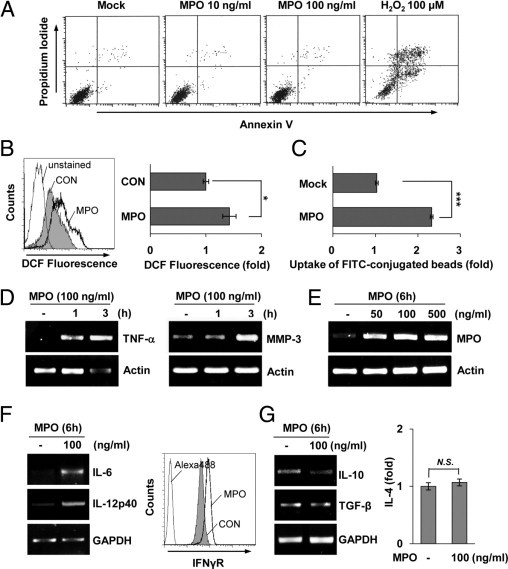
MPO promotes the induction of several inflammation-associated molecules and ROS generation in microglia. A: BV2 microglia were cultured in the presence or absence of MPO for 24 hours, after which the cells were stained with PI and annexin V. Cell viability was measured by FACS analysis. B: Primary microglia were mock treated or treated with 100 ng/mL of MPO for 3 hours, and DCF fluorescence was measured by FACS (left). The graph represents the fold changes in mean ± SEM fluorescence intensity from three independent experiments (right). *P < 0.05 when compared with mock-treated control (CON) cells. C: BV2 microglia were cultured in the presence or absence of 100 ng/mL MPO for 24 hours, and phagocytosis of FITC-conjugated fluorescent beads was determined by flow cytometric analysis. The graph shows the mean fold change in the percentage of phagocytic cells. ***P < 0.001 when compared with mock-treated cells. D and E: Primary microglia were treated with indicated concentrations of MPO for the indicated times, and mRNA levels of TNF-α, matrix metalloproteinase-3, and MPO were determined using an RT-PCR–based assay. F and G: The mRNA levels of IL-6, IL-12p40, IL-10, and transforming growth factor (TGF)-β were determined by RT-PCR analysis (left). At 24 hours after primary microglia were treated with 100 ng/mL MPO, the expression level of IFNγR was observed by flow cytometric analysis (F, right) and the level of IL-4 secreted into the media was measured by ELISA (G, right). GAPDH indicates glyceraldehyde-3-phosphate dehydrogenase; MMP, matrix metalloproteinase; N.S., no significant difference.
MPO Augments Its Own Expression and Activity in Microglia and Astrocytes
After showing that MPO increased the level of its own transcription in microglia, we investigated whether MPO could influence its expression at the protein level in rat primary microglia. As shown in Figure 7A, MPO protein was not detectable in mock-treated primary microglia. However, MPO protein levels were rapidly increased in a dose- and time-dependent manner on exposure to MPO (Figure 7A and data not shown). In addition, concomitant treatment with MPO and cycloheximide, an inhibitor of protein synthesis, considerably reduced the MPO-stimulated expression of MPO protein, supporting that MPO augments its own expression (Figure 7A). Confocal analyses also clearly demonstrated that the MPO level was markedly increased by MPO in rat primary microglia (data not shown). Taken together, these results suggest that MPO is capable of regulating its own expression at both the mRNA and protein levels in microglia.
Figure 7.
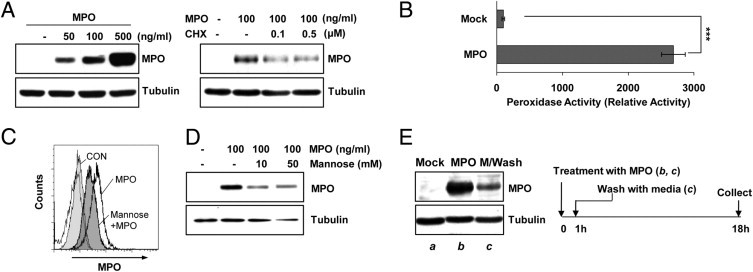
MPO-exposed glial cells enhance their MPO levels by both uptaking extracellular MPO and augmenting MPO expression. A: Primary microglia were treated with the indicated concentrations of MPO for 18 hours, followed by Western blot analysis using MPO and α-tubulin antibodies (left). Primary astrocytes were pretreated with cycloheximide (CHX) for 1 hour, followed by incubation with 100 ng/mL MPO for 18 hours; then, MPO levels were examined by using Western blot analysis (right). B: Primary microglia were treated with or without 100 ng/mL MPO for 24 hours, and cell lysates were collected. MPO activity was measured fluorometrically (SpectraMax Gemini EM spectrofluorometer; Molecular Devices) using an EnzChek Myeloperoxidase activity assay kit. Data are representative of four independent experiments. ***P < 0.001 compared with mock-treated cells. C: Primary microglia were preincubated with 50 mmol/L (D+)-mannose for 1 hour, followed by MPO exposure for 24 hours. The cells were indirectly immunostained using anti-MPO antibody for FACS analysis. D: Primary astrocytes were pretreated with (D+)-mannose (10 to 50 mmol/L) for 1 hour, followed by 100 ng/mL MPO for 24 hours. Cell lysates were prepared, and MPO levels were examined by using Western blot analysis. E: Primary astrocytes were mock treated (a) or treated with 100 ng/mL MPO for 1 hour (b and c), after which MPO-treated cells were washed three times with media (b) or unwashed (c). The cells were further incubated for 17 hours; then, MPO levels were examined by using Western blot analysis. The data shown are representative of at least three independent experiments.
Next, we used a peroxidase activity-based colorimetric MPO activity assay to examine the effect of MPO exposure on MPO activity in glial cells. Rat primary microglial cells were mock treated or treated with 100 ng/mL MPO, and the cell extracts were analyzed for MPO enzymatic activity. As shown in Figure 7B, the peroxidase activity of MPO was markedly elevated in microglial cell extracts after treatment with MPO. These results indicate that the increase in MPO levels induced by MPO was associated with marked augmentation of MPO enzymatic activity, which could potentially influence cellular targets in the brain.
MPO expression was previously demonstrated in astrocytes from patients with PD and in a mouse model of PD.18 Thus, we examined whether MPO could have a role in rotenone- or MPO-exposed astrocytes, another glial cell type vital to brain immune responses. Consistent with the results in microglia, rotenone also clearly enhanced ROS generation in rat primary astrocytes (see Supplemental Figure S2A at http://ajp.amjpathol.org). In addition, treatment of primary astrocytes with MPO markedly increased intracellular MPO levels. As shown in the merged image from confocal microscopy results in Supplemental Figure S2B (available at http://ajp.amjpathol.org), MPO protein was not detected in GFAP-positive astrocytes in cultures treated with vehicle but was abundantly expressed in all GFAP-positive cells in the MPO-treated group. We also observed an increase of intracellular MPO using FACS analysis (see Supplemental Figure S2B at http://ajp.amjpathol.org). These results indicate that MPO might augment its own expression and activity in both microglia and astrocytes.
Glial Cells Amplify Levels of MPO through Uptake of Extracellular MPO and Augmentation of Their MPO Expression
MPO has been absorbed by macrophages through the macrophage mannose receptor.41 Thus, we examined the possibility that uptake of MPO by glial cells might partly contribute to enhance intracellular MPO levels under MPO-exposed conditions. By using FACS and Western blot analyses, we observed that an MPO-induced increase of the intracellular MPO level was meaningfully reduced in the presence of (D+)-mannose, a macrophage mannose receptor ligand. These results suggest that primary glia are able to uptake MPO through a macrophage mannose receptor and that the internalized MPO might be a part of the enhanced intracellular MPO in glial cells (Figure 7, C and D).
To further validate the previous results, we examined the intracellular levels of MPO after washing exogenous MPO with serum-free medium in glia. Primary astrocytes were incubated with 100 ng/mL MPO for 1 hour, washed three times with serum-free media, and then further incubated using the indicated times. MPO-dependent enhancement of MPO levels was still observed even after washing three times with serum-free media (Figure 7E), demonstrating that MPO promotes enhancement of its own expression. However, MPO levels were significantly reduced compared with those in unwashed cells (Figure 7E), demonstrating that glial cells are able to uptake extracellular MPO. Similar results were observed in primary microglia (data not shown). Collectively, these findings indicate that MPO-exposed glial cells augment their expression of MPO and enhance the uptake of extracellular MPO, thus amplifying levels of MPO in the brain.
Blockade of MPO Activity Augments, Rather than Inhibits, Rotenone-Induced ROS Generation and Causes Glial Cell Death
The previous data indicate that MPO might contribute to the activation of glia and subsequent events in the rotenone-exposed brain. Therefore, we examined whether inhibition of MPO activity could affect the response of glial cells to rotenone exposure. BV2 microglial cells were pretreated with 10 μmol/L ABAH, a representative MPO inhibitor, for 30 minutes and then incubated with or without 5 nmol/L rotenone for 1 hour. Changes in ROS levels were measured by monitoring DCF fluorescence using FACS. Unexpectedly, inhibition of microglial MPO by treatment with ABAH noticeably increased rotenone-induced DCF fluorescence compared with that seen in rotenone alone–treated controls (Figure 8A). A similar pattern of increase in ROS levels was detected in rotenone-exposed primary microglial cells in the presence of ABAH (Figure 8A). In addition, both NO release and COX-2 expression were considerably enhanced when primary microglia were treated with rotenone and 10 μmol/L ABAH compared with cells treated with rotenone alone (Figure 8A).
Figure 8.
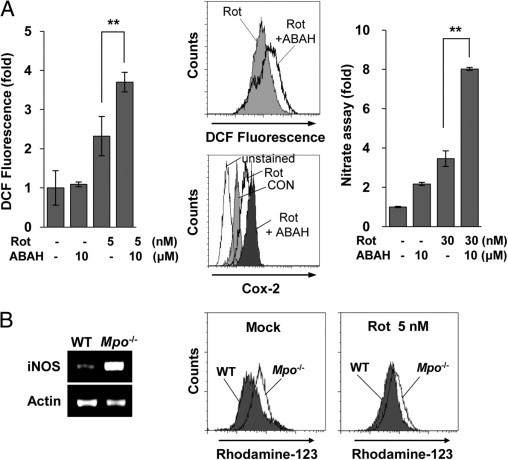
Blockade of MPO activity unexpectedly enhances rotenone (Rot)-induced ROS generation and cell death. A: BV2 microglia were pretreated with ABAH (10 μmol/L) for 30 minutes and then mock treated or treated with 5 nmol/L Rot for 1 hour. Left: The cells were harvested and stained with 5 μmol/L DCF for 30 minutes at 37°C to measure ROS generation. The graph represents the fold changes in mean ± SEM fluorescence intensity from three independent experiments. **P < 0.005 compared with mock-treated cells. Middle: Primary microglia were pretreated with ABAH (10 μmol/L) for 30 minutes and then treated with 30 nmol/L Rot for 3 or 24 hours, after which ROS levels (top) and intracellular COX-2 levels (bottom) were measured by FACS analysis using DCF and COX-2 antibody, respectively. The data shown are representative of at least three independent experiments. Right: Primary microglia were mock treated or treated with 30 nmol/L Rot in the presence or absence of 10 μmol/L ABAH for 2 days, and supernatants were assayed for nitrate concentration, as described in Materials and Methods. **P < 0.005 compared with Rot-treated cells. B: Primary glial cells were cultured from Mpo−/− mice (Mpo−/−) or B6 WT mice (WT), and the cells were incubated with or without 5 nmol/L Rot for 6 hours. The transcript levels of iNOS were determined by RT-PCR (left), and ROS generation was measured by FACS using rhodamine-123 (right).
To confirm these results, we assessed the effects of rotenone on primary glial cells from Mpo−/− mice. ROS levels were examined in primary microglia from Mpo−/− mice and wild-type (WT) controls cultured with or without rotenone for 1 hour. Consistent with the results obtained using MPO inhibitors, the basal levels of ROS and iNOS were considerably higher in Mpo−/− mice than in WT mice (Figure 8B). In addition, rotenone-induced ROS generation was markedly increased in Mpo−/− mice compared with that in WT controls (Figure 8B). These results show that MPO deficiency leads to an augmentation in rotenone-induced ROS generation in microglia, suggesting that MPO activates microglia, promotes ROS generation, and may have a defensive function against rotenone by regulating ROS generation in the brain.
Primary Glial Cells from MPO-Deficient Mice Exhibit Impaired Responses to Rotenone Exposure
To further investigate the response of glial cells from Mpo−/− mice under rotenone-exposed conditions, we carefully examined the viability of primary glia after exposure to rotenone. Primary cultures of microglial cells from Mpo−/− and WT mice were incubated in the presence or absence of 30 nmol/L rotenone for 3 days. Intriguingly, the viability of microglia from MPO-deficient mice was significantly reduced after exposure to rotenone, whereas the microglia from WT mice were not affected (Figure 9A). Similar results were observed in primary astrocytes from Mpo−/− mice (Figure 9B). These results led us to examine the expression of representative inflammation-associated molecules in microglia from Mpo−/− and WT mice. RT-PCR analysis showed that the rotenone-induced increase in IL-1β expression was greater in Mpo−/− mice than in WT mice (Figure 9C). In addition, the basal levels of COX-2 and TNF-α mRNA were high in microglia from Mpo−/− mice and undetectable in those from WT mice (Figure 9C). NO release was also significantly increased in glial cells from Mpo−/− mice compared with that in WT mice (Figure 9D). However, we did not detect any difference of anti-inflammatory cytokine message levels between Mpo−/− and WT mice (data not shown). Collectively, our findings show that MPO-deficient mice produce increased levels of several inflammatory mediators and exhibit excessive glial cell death, suggesting that MPO deficiency impairs the regulatory defense system of glial cells against rotenone exposure in the brain.
Figure 9.
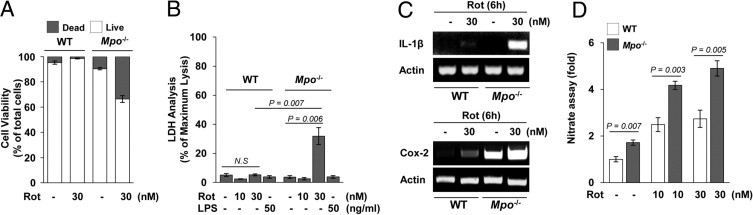
Glial cells from Mpo−/− mice exhibit responses to rotenone (Rot) that are distinct from those of WT mice. A: Primary microglia from Mpo−/− mice (Mpo−/−) or normal WT B6 mice (WT) were mock treated or treated with 30 nmol/L Rot for 3 days. Cell viability was measured by fluorescence microscopy, using the Live/Dead Viability Cytotoxicity kit. B: Primary astrocytes from Mpo−/− or WT mice were mock treated or treated with the indicated concentrations of Rot or 50 ng/mL lipopolysaccharide (LPS) as a representative inflammatory stimulus, for 3 days and then subjected to LDH assay. C: The levels of IL-1β and COX-2 mRNA in primary microglia from Mpo−/− or WT mice were determined by RT-PCR–based analysis. D: Primary glial cells from Mpo−/− or WT control mice were mock treated or treated with the indicated concentrations of Rot for 1 day, and supernatants were assayed for nitrate concentration, as described in Materials and Methods. N.S. indicates no significant difference.
MPO Influences the Neuronal State but Does Not Directly Cause Cell Death or Phenotypic Change
Next, we questioned whether MPO could influence the neuronal phenotype and properties, as it does the microglial and astrocytic properties. We first examined the viability of rat primary mesencephalic neurons after exposure to MPO. Rat primary neurons were mock treated or treated with MPO, 10 to 500 ng/mL, for 5 days, after which neuronal cell viability was observed using a Live/Dead Viability Cytotoxicity assay. Consistent with the results obtained from microglia, MPO did not significantly affect the viability of primary mesencephalic neurons (Figure 10A). Thus, we explored whether MPO could influence the morphological features of neurons by phase-contrast microscopy. As shown in Figure 10A, treatment of mesencephalic neurons with MPO did not cause any significant change in neuronal phenotype. To further assess the effects of MPO on neurons, we carefully examined the phenotype of dopaminergic neurons by immunostaining with anti-TH in the presence or absence of MPO. Rotenone exposure resulted in a decrease in dendrite length and in the number of TH-stained neurons; however, MPO did not cause any noticeable changes in either parameter (Figure 10A). Similar results were obtained in primary mesencephalic neurons from WT mice (data not shown). In addition, we examined whether MPO could influence the response of neurons to rotenone. However, we found no synergistic neurotoxicity of rotenone and MPO in neuronal cell viability using a CCK-8 (Figure 10B) and a Live/Dead Viability Cytotoxicity kit at all concentrations and times tested (Figure 10B). Moreover, we did not observe any significant differences in phenotype and number of TH-stained neurons between cells with rotenone alone and cells with rotenone plus MPO (Figure 10C).
Figure 10.
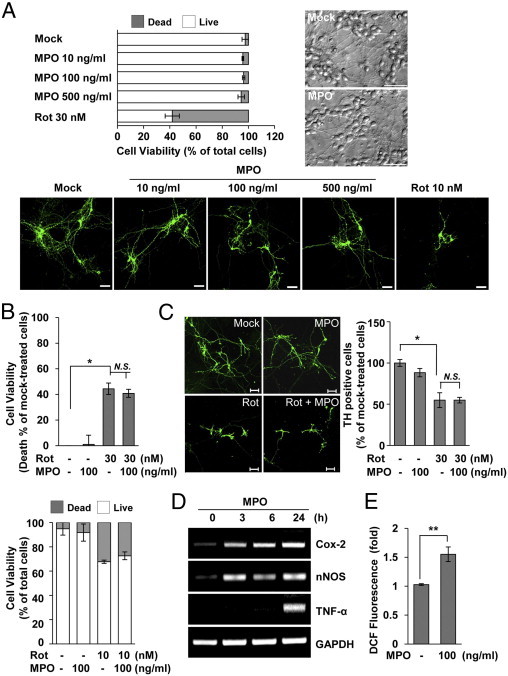
MPO influences the neuronal state but does not directly cause cell death or phenotypic change. A: Rat primary mesencephalic neurons were mock treated or treated with the indicated doses of MPO or rotenone (Rot) for 5 days. Cell viability was measured by a Live/Dead Viability Cytotoxicity kit (top left), and morphological features were observed by phase-contrast microscopy (top right). Bottom: Rat primary mesencephalic neurons were incubated with the indicated concentrations of Rot and/or MPO for 5 days, and dopaminergic neuronal cells were evaluated immunocytochemically using antibodies specific for TH (green). Scale bar = 50 μm. B: Rat primary mesencephalic neurons were incubated with the indicated concentrations of Rot and/or MPO for 3 days. Cell viability was determined using the CCK-8 assay (top) and a Live/Dead Viability Cytotoxicity kit (bottom), as described in Materials and Methods. Data were expressed as the percentage of cell death relative to mock-treated cells. *P < 0.05 compared with mock-treated cells. C: TH-stained neurons were examined in rat primary mesencephalic neurons treated with Rot (30 nmol/L) and/or MPO (100 ng/mL) for 3 days (left). Scale bar = 20 μm. The results for TH-positive cells are expressed as a percentage of the mock-treated control cultures (right). *P < 0.05 compared with mock-treated cells. D: Rat primary mesencephalic neurons were incubated with 100 ng/mL MPO for the indicated times, and the message levels of COX-2, neuronal NOS (nNOS), and TNF-α were determined by RT-PCR analysis. E: Rat primary mesencephalic neurons were treated with 100 ng/mL MPO for 24 hours, and the cells were stained with 5 μmol/L DCF to measure ROS generation before FACS analysis. **P < 0.005 compared with mock-treated cells. GAPDH indicates glyceraldehyde-3-phosphate dehydrogenase; N.S., no significant difference.
To better define the effects of MPO on neurons, we explored whether MPO could influence the expression of genes associated with inflammation and neurodegeneration. Interestingly, treatment of mesencephalic neurons with MPO significantly increased the expression levels of several genes, including COX-2, neuronal NOS, and TNF-α (Figure 10D). In addition, MPO exposure considerably enhanced the generation of ROS by neurons (Figure 10E). Overall, these results suggest that MPO may influence the neuronal state but does not directly cause cell death or phenotypic change.
Rotenone-Triggered Neuronal Injury Is More Apparent in Co-Cultures with Glial Cells from Mpo−/− Mice than in Those from WT Mice
Recently, much attention has been focused on the reciprocal interactions between glia and neurons during the pathogenesis of inflammatory and neurodegenerative disease.42,43 Thus, we determined the neuronal response to rotenone exposure in the presence of primary glia with or without MPO. Primary mesencephalic neuron-enriched cultures from WT mice were incubated with glial cells from Mpo−/− or WT mice using transwell chambers, after which the extent of neuronal cell death was determined using LDH analysis and CCK-8. Rotenone-triggered neuronal cell death in co-cultures with glia from Mpo−/− mice was significantly increased compared with that seen in co-cultures with glia from WT mice (Figure 11A). Similar results were obtained by inhibition of MPO activity using the MPO inhibitor, ABAH, in both rat and mouse primary cells (Figure 11B). These results suggest the possibility that MPO deficiency contributes to the response of glial cells to rotenone, thus affecting neuronal injury. Taken together, our results indicate that microglia influence the neuronal response to rotenone via MPO activity, thus modulating pathophysiological events in the brain.
Figure 11.
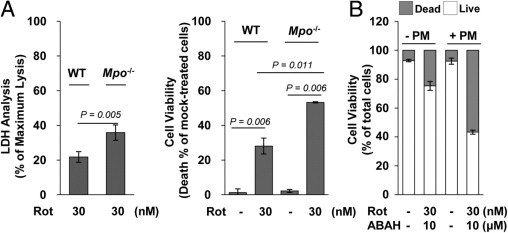
Rotenone (Rot)-triggered neuronal injury in co-cultures with glial cells from Mpo−/− or WT mice. A: Mouse primary mesencephalic neurons were incubated with primary glial cells from Mpo−/− mice (Mpo−/−) or WT control mice using transwell chambers, after which cells were mock treated or treated with 30 nmol/L Rot for 3 days. The extent of neuronal cell death was determined by LDH analysis (left) and CCK-8 assay (right). All data are presented as the mean ± SEM of at least three independent experiments. B: Rat primary mesencephalic (PM) neurons were incubated with rat primary glial cells using transwell chambers and mock treated or treated with 30 nmol/L Rot in the presence or absence of ABAH for 3 days. Neuronal viability was determined using a Live/Dead Viability Cytotoxicity kit.
Discussion
Microglia and astrocytes are major immune cells that serve as the first line of defense against tissue injury or pathogens in the CNS. They are sensitive to even subtle alterations in the CNS and rapidly undergo a variety of changes in immunological function that prevent neuronal damage from pathological stimuli.25,27,44–46 Accumulating evidence has revealed that glial cells perform distinct responses in different pathological states, thereby setting in motion an appropriate defense system. However, inappropriate or excessive responses of glia can cause additional damage that contributes to diverse disease in the CNS.
Environmental toxins, such as pesticides, have emerged as an important underlying risk factor for the development of various diseases. Rotenone, a common herbicide, specifically may provoke critical neuronal damage that ultimately leads to neurodegenerative diseases.6–8 In particular, rotenone exposure has been implicated as a potential risk factor for PD. Studies have shown that mice and rats given various concentrations of rotenone, from 2 to 30 mg/kg, exhibited clinical and pathological features of PD, such as loss of TH-positive dopaminergic neurons and/or motor dysfunction. In addition, several reports have described the toxic effects of rotenone on neurons. Notably, Gao et al29 reported that rotenone-induced neurotoxicity is greater in the presence of microglia than in neurons cultured alone, highlighting the potential capacity of microglia to have a role in the pathophysiological consequences of rotenone exposure. These reports raise the question of what events might be mediated by microglia under rotenone-exposed conditions. In the present study, we investigated the response of glial cells and their potential roles in combating rotenone-induced damage in the CNS.
Once rotenone enters the body, it easily crosses the blood-brain barrier and is distributed throughout the brain.1 After systemic administration of 2 to 3 mg/kg per day, the concentration of free rotenone in the brain is approximately 20 to 30 nmol/L.4,5 In addition, Talpade et al4 suggested that 1 to 30 nmol/L of rotenone may be a reasonable concentration in the living rat brain under rotenone-triggered pathological conditions. Based on these reports, we used 1 to 30 nmol/L of rotenone in our present study. Neurons have been shown to undergo death after rotenone exposure; therefore, we first examined the effects of rotenone on the viability of microglia as part of our efforts to assess the glial responses to rotenone exposure in the brain. In our experiments, neuronal death was detected in mesencephalic neurons cultured with physiologically relevant concentrations of rotenone, consistent with previous reports (Figure 1). Unlike neurons, exposure to rotenone was not toxic to microglia, in either the presence or the absence of neurons; however, rotenone-treated microglia adopted a distinct activated form, indicating that microglia could actively respond to rotenone exposure. Studies37,47 have shown that activated microglia release numerous inflammatory mediators, such as NO and superoxide, and produce antioxidants to successfully defend the CNS against threats to the brain. Microglia are equipped with efficient antioxidative defense mechanisms to prevent oxidative damage that would compromise their function.37 In this experiment, we found that rotenone exposure enhanced the levels of pro-oxidative and antioxidative enzymes, including SOD, catalase, and GPxs, showing that the response to rotenone-triggered oxidative stress is active (Figure 5). In addition, GSH levels were higher in rotenone-exposed microglia than in mock-treated cells. Based on previous reports and on our findings, it is likely that rotenone exposure microglial antioxidant systems protect the cells from self-damage potentially caused by rotenone and microglia-released oxidants, contributing to the efficacy of microglia as effector cells under rotenone-exposed conditions.
MPO is an enzyme that functions as a key molecular component of the host defense reaction against inflammatory stimulators.38,48 Inflammatory sites, as the result of microorganism infections or injured tissues, are characterized by increased levels of MPO. Indeed, studies have increasingly suggested potential links between MPO and the development of diverse diseases. Interestingly, we found that a distinguishing feature of rotenone-exposed microglia is an increase in the expression level of MPO (Figure 4). Therefore, we hypothesized that MPO might be associated with pathophysiological events in the brain under rotenone-exposed conditions. MPO is mainly present in neutrophils, monocytes, and macrophages; thus, it has been used as a marker for infiltrated neutrophils at sites of injury.49,50 Recently, MPO was reported to be present in brain-resident cells under pathological conditions.18,23 The results of the present study using primary glial cell cultures strongly support the hypothesis that glial cells are among the MPO-expressing cells of the brain. The distinct increase in MPO levels in microglia by rotenone exposure led us to focus our research on the characteristics of MPO in rotenone-exposed glia. Therefore, we further examined the effects of MPO on glial cells using purified MPO. Several reports15 have shown that purified MPO has its activity when added to cultured cells or animals. Although relatively little is known of the amounts of MPO in the CNS, the plasma levels of MPO in patients with vascular diseases have been described.51,52 In addition, we detected approximately 25 ng/mL MPO in rotenone-exposed primary microglia (data not shown). On the basis of these reports and our own results, we chose to use 1 to 1000 ng/mL MPO in the present study. In microglia and astrocytes, MPO did not cause cell death at any concentration tested; instead, it exhibited cytokinelike properties. MPO significantly increased the expression of several inflammation-associated molecules, including TNF-α, and promoted the production of ROS. Notably, MPO also augmented its own expression at the mRNA and protein levels, resulting in increased MPO secretion into the extracellular space (Figures 6 and 7). These observations indicate that MPO may regulate its own expression and activity in an autocrine and paracrine manner in the brain.
Our previous findings lend credence to the interesting notion that MPO could take an active part in cellular events that respond against rotenone in the CNS. To evaluate the influence of MPO on the rotenone-exposed brain, we used MPO-knockout mice and pharmacological inhibitors of MPO. Intriguingly, treatment of glia with MPO inhibitors further enhanced, rather than reduced, the rotenone-stimulated production of ROS. Similarly, rotenone-induced generation of ROS was greater in microglia from Mpo−/− mice than in those from WT mice (Figure 8). These unexpected results indicate that MPO deficiency could impair modulation of rotenone-stimulated ROS generation. Therefore, we further investigated the characteristics of glial cells from Mpo−/− mice after exposure to rotenone. In primary astrocytes and microglia from Mpo−/− mice, rotenone induced increased levels of several inflammatory mediators compared with normal WT control. In addition, cell viability was significantly reduced in glia from Mpo−/− mice but not in those from healthy WT control mice. These observations show that MPO deficiency exacerbates rotenone-induced inflammatory responses and causes dysregulation of ROS generation, thus leading to abnormal outcomes in glial cells.
To better assess the action of MPO in the rotenone-exposed CNS, we examined the neuronal response to rotenone in the presence of glial cells with or without MPO. More interestingly, rotenone-induced neuronal cell death in co-cultures with glia from Mpo−/− mice was significantly enhanced compared with that seen in cultures with glia from healthy mice (Figure 11). We obtained similar results using MPO inhibitors. These findings indicate that MPO is required for appropriate responses of the glial defense system against rotenone exposure in the brain. There are several reports that MPO deficiency can contribute to pathological conditions in animal models and patients. Mice lacking MPO have a significantly increased incidence of experimental autoimmune encephalomyelitis53 and show increased atherosclerosis and enhanced lung inflammation.54,55 Similarly, there is a greater occurrence of severe infections and chronic inflammatory processes among MPO-deficient patients.56 In contrast, blockade of MPO activity in mouse models and in humans can reduce the pathological response in diseases such as PD and in pathological conditions such as renal ischemia.11,18 Considering our results in the context of these conflicting reports, it seems that MPO may act with dual functionality, having both pathological and protective functions under abnormal conditions.
MPO has been shown to act as a direct and significant mediator of decreased NO bioavailability. MPO can oxidize NO, thereby inhibiting NO-dependent signaling and modulating reduction-oxidation–sensitive signaling cascades during inflammation.14,57 One possible explanation for our findings is that MPO-deficient glial cells inappropriately regulate ROS and reactive nitrogen species under rotenone-exposed conditions, perhaps because of increased NO bioavailability (Figures 8 and 9). Several lines of evidences indicate that MPO may be a metabolic “sink” for several types of ROS, including superoxide and H2O2, and may, therefore, compromise NO bioavailability.13,58 Previous reports58,59 have associated MPO deficiency with increased levels of pulmonary iNOS expression and NO production. Also, iNOS expression is considerably greater in Mpo−/− mice than in healthy mice (Figure 8). Based on these previous reports and our findings, it is likely that MPO may have a regulatory influence over the activation state of immune and inflammatory cells by affecting the production of ROS and the expression of inflammatory mediators. MPO has been shown to act as a physiologically relevant regulator of inflammatory response by oxidatively limiting tissue-degrading protease activity.16,60 These reports raise the possibility that MPO might contribute to protection of the host from pathogens via diverse mechanisms associated with the activity of inflammatory mediators. Such a regulatory effect of MPO on inflammatory mediators might also help to explain our observations in Mpo−/− mice. To definitely address these possibilities, we are undertaking a thorough characterization of MPO-deficient mice and investigating the underlying basis of MPO actions under rotenone-exposed conditions.
Appropriate inflammatory responses and efficient resolution are essential attributes of an effective defense system against pathogens and environmental stimuli. MPO is a characteristic of inflammation, and its aberrant expression is presumed to be a detrimental factor in disease. Our present findings suggest that MPO may be associated with pathological outcomes and with protective events in the brain-resident immune cells under rotenone-exposed conditions. We have shown that MPO deficiency exacerbates rotenone-induced generation of ROS in glia and have demonstrated that MPO-null mice display inflammatory responses that are distinct from those of WT mice. Although the origin of adult microglia and microglial progenitors still remains controversial, microglia are widely considered as myeloid lineage–derived cells.61–63 Recently, we observed that rotenone exposure led to increased MPO levels in peripheral blood mononuclear cells and peritoneal macrophages, as well as in microglia (unpublished data). These results further support the notion that rotenone exposure affects levels of MPO. The results of this study further expand our current understanding of the characteristics and roles of MPO under inflammatory conditions and indicate that the balance of MPO activity could be decisive for efficient resolution of the rotenone-induced pathological state in the brain. MPO has emerged as a regulatory target for the treatment of diverse diseases. Our results provide new insights into the responses associated with MPO and rotenone in the CNS and should inform the development of novel targeted therapies for reducing inflammation.
Acknowledgments
We thank Mi Ae Kim of the Microscopy Core and Tae Sik Kim of the Flow Cytometry Core (National Cancer Center) for their expert assistance and helpful suggestions.
Footnotes
Supported by the Korea Health Industry Development Institute (grant A090570-1002-0000100).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.04.033.
Supplementary data
Rotenone (Rot) does not influence the viability but not morphology of microglia. A: Primary microglia were mock-treated or treated with the indicated concentrations of Rot for 3 days. Cell viability was analyzed using the Live/Dead Viability Cytotoxicity kit, as described in Materials and Methods. B: BV2 cells were cultured in the presence or absence of Rot for 1 day, after which the cells were stained with PI and annexin V. Cell viability was measured by FACS analysis. The data shown are representative of three independent experiments. C: Primary microglial cells were mock treated or treated with 1 nmol/L Rot for 1 day, after which the cells were stained for CD11b (green). The images were captured with confocal laser scanning. Scale bar = 20 μm. CON indicates control.
Primary astrocytes show similar responses to rotenone and MPO exposure. A: Primary astrocytes were treated with or without 5 nmol/L Rot for 3 days, and the amount of ROS generation was determined by FACS using rhodamine-123. B: Primary astrocytes (top) and microglia (bottom) were incubated with 100 ng/mL MPO. For confocal microscopy, the astrocytes were stained with anti-GFAP and anti-MPO antibodies and with DAPI; and microglia were stained with anti-MPO antibody and with DAPI (left). Scale bar = 20 μm. Primary astrocytes were treated with 100 ng/mL MPO for 18 hours, and intracellular MPO levels were examined by FACS analysis (right). The data shown are representative of at least three independent experiments. CON indicates control.
References
- 1.Miller R.L., James-Kracke M., Sun G.Y., Sun A.Y. Oxidative and inflammatory pathways in Parkinson's disease. Neurochem Res. 2009;34:55–65. doi: 10.1007/s11064-008-9656-2. [DOI] [PubMed] [Google Scholar]
- 2.Drechsel D.A., Patel M. Role of reactive oxygen species in the neurotoxicity of environmental agents implicated in Parkinson's disease. Free Radic Biol Med. 2008;44:1873–1886. doi: 10.1016/j.freeradbiomed.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betarbet R., Sherer T.B., MacKenzie G., Garcia-Osuna M., Panov A.V., Greenamyre J.T. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 4.Talpade D.J., Greene J.G., Higgins D.S., Jr, Greenamyre J.T. In vivo labeling of mitochondrial complex I (NADH: ubiquinone oxidoreductase) in rat brain using [(3)H]dihydrorotenone. J Neurochem. 2000;75:2611–2621. doi: 10.1046/j.1471-4159.2000.0752611.x. [DOI] [PubMed] [Google Scholar]
- 5.Fleming S.M., Zhu C., Fernagut P.O., Mehta A., DiCarlo C.D., Seaman R.L., Chesselet M.F. Behavioral and immunohistochemical effects of chronic intravenous and subcutaneous infusions of varying doses of rotenone. Exp Neurol. 2004;187:418–429. doi: 10.1016/j.expneurol.2004.01.023. [DOI] [PubMed] [Google Scholar]
- 6.Betarbet R., Porter R.H., Greenamyre J.T. GluR1 glutamate receptor subunit is regulated differentially in the primate basal ganglia following nigrostriatal dopamine denervation. J Neurochem. 2000;74:1166–1174. doi: 10.1046/j.1471-4159.2000.741166.x. [DOI] [PubMed] [Google Scholar]
- 7.Gao H.M., Liu B., Hong J.S. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2003;23:6181–6187. doi: 10.1523/JNEUROSCI.23-15-06181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alam M., Schmidt W.J. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav Brain Res. 2002;136:317–324. doi: 10.1016/s0166-4328(02)00180-8. [DOI] [PubMed] [Google Scholar]
- 9.Moore D.J., West A.B., Dawson V.L., Dawson T.M. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 10.Zhang R., Brennan M.L., Shen Z., MacPherson J.C., Schmitt D., Molenda C.E., Hazen S.L. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J Biol Chem. 2002;277:46116–46122. doi: 10.1074/jbc.M209124200. [DOI] [PubMed] [Google Scholar]
- 11.Matthijsen R.A., Huugen D., Hoebers N.T., de Vries B., Peutz-Kootstra C.J., Aratani Y., Daha M.R., Tervaert J.W., Buurman W.A., Heeringa P. Myeloperoxidase is critically involved in the induction of organ damage after renal ischemia reperfusion. Am J Pathol. 2007;171:1743–1752. doi: 10.2353/ajpath.2007.070184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawayama Y., Miyazaki Y., Ando K., Horio K., Tsutsumi C., Imanishi D., Tsushima H., Imaizumi Y., Hata T., Fukushima T., Yoshida S., Onimaru Y., Iwanaga M., Taguchi J., Kuriyama K., Tomonaga M. Expression of myeloperoxidase enhances the chemosensitivity of leukemia cells through the generation of reactive oxygen species and the nitration of protein. Leukemia. 2008;22:956–964. doi: 10.1038/leu.2008.8. [DOI] [PubMed] [Google Scholar]
- 13.Lau D., Mollnau H., Eiserich J.P., Freeman B.A., Daiber A., Gehling U.M., Brummer J., Rudolph V., Munzel T., Heitzer T., Meinertz T., Baldus S. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci U S A. 2005;102:431–436. doi: 10.1073/pnas.0405193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eiserich J.P., Baldus S., Brennan M.L., Ma W., Zhang C., Tousson A., Castro L., Lusis A.J., Nauseef W.M., White C.R., Freeman B.A. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science. 2002;296:2391–2394. doi: 10.1126/science.1106830. [DOI] [PubMed] [Google Scholar]
- 15.El Kebir D., Jozsef L., Pan W., Filep J.G. Myeloperoxidase delays neutrophil apoptosis through CD11b/CD18 integrins and prolongs inflammation. Circ Res. 2008;103:352–359. doi: 10.1161/01.RES.0000326772.76822.7a. [DOI] [PubMed] [Google Scholar]
- 16.Hirche T.O., Gaut J.P., Heinecke J.W., Belaaouaj A. Myeloperoxidase plays critical roles in killing Klebsiella pneumoniae and inactivating neutrophil elastase: effects on host defense. J Immunol. 2005;174:1557–1565. doi: 10.4049/jimmunol.174.3.1557. [DOI] [PubMed] [Google Scholar]
- 17.Daugherty A., Dunn J.L., Rateri D.L., Heinecke J.W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest. 1994;94:437–444. doi: 10.1172/JCI117342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi D.K., Pennathur S., Perier C., Tieu K., Teismann P., Wu D.C., Jackson-Lewis V., Vila M., Vonsattel J.P., Heinecke J.W., Przedborski S. Ablation of the inflammatory enzyme myeloperoxidase mitigates features of Parkinson's disease in mice. J Neurosci. 2005;25:6594–6600. doi: 10.1523/JNEUROSCI.0970-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagra R.M., Becher B., Tourtellotte W.W., Antel J.P., Gold D., Paladino T., Smith R.A., Nelson J.R., Reynolds W.F. Immunohistochemical and genetic evidence of myeloperoxidase involvement in multiple sclerosis. J Neuroimmunol. 1997;78:97–107. doi: 10.1016/s0165-5728(97)00089-1. [DOI] [PubMed] [Google Scholar]
- 20.Green P.S., Mendez A.J., Jacob J.S., Crowley J.R., Growdon W., Hyman B.T., Heinecke J.W. Neuronal expression of myeloperoxidase is increased in Alzheimer's disease. J Neurochem. 2004;90:724–733. doi: 10.1111/j.1471-4159.2004.02527.x. [DOI] [PubMed] [Google Scholar]
- 21.Gray E., Thomas T.L., Betmouni S., Scolding N., Love S. Elevated activity and microglial expression of myeloperoxidase in demyelinated cerebral cortex in multiple sclerosis. Brain Pathol. 2008;18:86–95. doi: 10.1111/j.1750-3639.2007.00110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reynolds W.F., Rhees J., Maciejewski D., Paladino T., Sieburg H., Maki R.A., Masliah E. Myeloperoxidase polymorphism is associated with gender specific risk for Alzheimer's disease. Exp Neurol. 1999;155:31–41. doi: 10.1006/exnr.1998.6977. [DOI] [PubMed] [Google Scholar]
- 23.Maki R.A., Tyurin V.A., Lyon R.C., Hamilton R.L., DeKosky S.T., Kagan V.E., Reynolds W.F. Aberrant expression of myeloperoxidase in astrocytes promotes phospholipid oxidation and memory deficits in a mouse model of Alzheimer disease. J Biol Chem. 2009;284:3158–3169. doi: 10.1074/jbc.M807731200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du Y., Chen C.P., Tseng C.Y., Eisenberg Y., Firestein B.L. Astroglia-mediated effects of uric acid to protect spinal cord neurons from glutamate toxicity. Glia. 2007;55:463–472. doi: 10.1002/glia.20472. [DOI] [PubMed] [Google Scholar]
- 25.Jou I., Lee J.H., Park S.Y., Yoon H.J., Joe E.H., Park E.J. Gangliosides trigger inflammatory responses via TLR4 in brain glia. Am J Pathol. 2006;168:1619–1630. doi: 10.2353/ajpath.2006.050924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han B.S., Hong H.S., Choi W.S., Markelonis G.J., Oh T.H., Oh Y.J. Caspase-dependent and -independent cell death pathways in primary cultures of mesencephalic dopaminergic neurons after neurotoxin treatment. J Neurosci. 2003;23:5069–5078. doi: 10.1523/JNEUROSCI.23-12-05069.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeon S.B., Yoon H.J., Park S.H., Kim I.H., Park E.J. Sulfatide, a major lipid component of myelin sheath, activates inflammatory responses as an endogenous stimulator in brain-resident immune cells. J Immunol. 2008;181:8077–8087. doi: 10.4049/jimmunol.181.11.8077. [DOI] [PubMed] [Google Scholar]
- 28.Jeon S.B., Yoon H.J., Chang C.Y., Koh H.S., Jeon S.H., Park E.J. Galectin-3 exerts cytokine-like regulatory actions through the JAK-STAT pathway. J Immunol. 2010;185:7037–7046. doi: 10.4049/jimmunol.1000154. [DOI] [PubMed] [Google Scholar]
- 29.Gao H.M., Hong J.S., Zhang W., Liu B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2002;22:782–790. doi: 10.1523/JNEUROSCI.22-03-00782.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao H.M., Hong J.S., Zhang W., Liu B. Synergistic dopaminergic neurotoxicity of the pesticide rotenone and inflammogen lipopolysaccharide: relevance to the etiology of Parkinson's disease. J Neurosci. 2003;23:1228–1236. doi: 10.1523/JNEUROSCI.23-04-01228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colton C.A. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kigerl K.A., Gensel J.C., Ankeny D.P., Alexander J.K., Donnelly D.J., Popovich P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biswas S.K., Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 34.Caron E., Hall A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 1998;282:1717–1721. doi: 10.1126/science.282.5394.1717. [DOI] [PubMed] [Google Scholar]
- 35.Neumann H., Kotter M.R., Franklin R.J. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Setsukinai K., Urano Y., Kakinuma K., Majima H.J., Nagano T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J Biol Chem. 2003;278:3170–3175. doi: 10.1074/jbc.M209264200. [DOI] [PubMed] [Google Scholar]
- 37.Dringen R., Pawlowski P.G., Hirrlinger J. Peroxide detoxification by brain cells. J Neurosci Res. 2005;79:157–165. doi: 10.1002/jnr.20280. [DOI] [PubMed] [Google Scholar]
- 38.Klebanoff S.J. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 39.Zhang R., Brennan M.L., Fu X., Aviles R.J., Pearce G.L., Penn M.S., Topol E.J., Sprecher D.L., Hazen S.L. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286:2136–2142. doi: 10.1001/jama.286.17.2136. [DOI] [PubMed] [Google Scholar]
- 40.Fu X., Kao J.L., Bergt C., Kassim S.Y., Huq N.P., d'Avignon A., Parks W.C., Mecham R.P., Heinecke J.W. Oxidative cross-linking of tryptophan to glycine restrains matrix metalloproteinase activity: specific structural motifs control protein oxidation. J Biol Chem. 2004;279:6209–6212. doi: 10.1074/jbc.C300506200. [DOI] [PubMed] [Google Scholar]
- 41.Zabucchi G., Menegazzi R., Soranzo M.R., Patriarca P. Uptake of human eosinophil peroxidase by human neutrophils. Am J Pathol. 1986;124:510–518. [PMC free article] [PubMed] [Google Scholar]
- 42.Tian L., Rauvala H., Gahmberg C.G. Neuronal regulation of immune responses in the central nervous system. Trends Immunol. 2009;30:91–99. doi: 10.1016/j.it.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 43.Biber K., Neumann H., Inoue K., Boddeke H.W. Neuronal “On” and “Off” signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 44.Park E.J., Ji K.A., Jeon S.B., Choi W.H., Han I.O., You H.J., Kim J.H., Jou I., Joe E.H. Rac1 contributes to maximal activation of STAT1 and STAT3 in IFN-gamma-stimulated rat astrocytes. J Immunol. 2004;173:5697–5703. doi: 10.4049/jimmunol.173.9.5697. [DOI] [PubMed] [Google Scholar]
- 45.Nimmerjahn A., Kirchhoff F., Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 46.Perry V.H., Nicoll J.A., Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 47.Innamorato N.G., Lastres-Becker I., Cuadrado A. Role of microglial redox balance in modulation of neuroinflammation. Curr Opin Neurol. 2009;22:308–314. doi: 10.1097/WCO.0b013e32832a3225. [DOI] [PubMed] [Google Scholar]
- 48.Arnhold J., Flemmig J. Human myeloperoxidase in innate and acquired immunity. Arch Biochem Biophys. 2010;500:92–106. doi: 10.1016/j.abb.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 49.Barone F.C., Hillegass L.M., Price W.J., White R.F., Lee E.V., Feuerstein G.Z., Sarau H.M., Clark R.K., Griswold D.E. Polymorphonuclear leukocyte infiltration into cerebral focal ischemic tissue: myeloperoxidase activity assay and histologic verification. J Neurosci Res. 1991;29:336–345. doi: 10.1002/jnr.490290309. [DOI] [PubMed] [Google Scholar]
- 50.Matsuo Y., Onodera H., Shiga Y., Nakamura M., Ninomiya M., Kihara T., Kogure K. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat: effects of neutrophil depletion. Stroke. 1994;25:1469–1475. doi: 10.1161/01.str.25.7.1469. [DOI] [PubMed] [Google Scholar]
- 51.Baldus S., Heeschen C., Meinertz T., Zeiher A.M., Eiserich J.P., Munzel T., Simoons M.L., Hamm C.W. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108:1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- 52.Vita J.A., Brennan M.L., Gokce N., Mann S.A., Goormastic M., Shishehbor M.H., Penn M.S., Keaney J.F., Jr, Hazen S.L. Serum myeloperoxidase levels independently predict endothelial dysfunction in humans. Circulation. 2004;110:1134–1139. doi: 10.1161/01.CIR.0000140262.20831.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brennan M., Gaur A., Pahuja A., Lusis A.J., Reynolds W.F. Mice lacking myeloperoxidase are more susceptible to experimental autoimmune encephalomyelitis. J Neuroimmunol. 2001;112:97–105. doi: 10.1016/s0165-5728(00)00392-1. [DOI] [PubMed] [Google Scholar]
- 54.Brennan M.L., Anderson M.M., Shih D.M., Qu X.D., Wang X., Mehta A.C., Lim L.L., Shi W., Hazen S.L., Jacob J.S., Crowley J.R., Heinecke J.W., Lusis A.J. Increased atherosclerosis in myeloperoxidase-deficient mice. J Clin Invest. 2001;107:419–430. doi: 10.1172/JCI8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Milla C., Yang S., Cornfield D.N., Brennan M.L., Hazen S.L., Panoskaltsis-Mortari A., Blazar B.R., Haddad I.Y. Myeloperoxidase deficiency enhances inflammation after allogeneic marrow transplantation. Am J Physiol Lung Cell Mol Physiol. 2004;287:L706–L714. doi: 10.1152/ajplung.00015.2004. [DOI] [PubMed] [Google Scholar]
- 56.Kutter D., Devaquet P., Vanderstocken G., Paulus J.M., Marchal V., Gothot A. Consequences of total and subtotal myeloperoxidase deficiency: risk or benefit. Acta Haematol. 2000;104:10–15. doi: 10.1159/000041062. [DOI] [PubMed] [Google Scholar]
- 57.Eiserich J.P., Hristova M., Cross C.E., Jones A.D., Freeman B.A., Halliwell B., van der Vliet A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 58.Baldus S., Heitzer T., Eiserich J.P., Lau D., Mollnau H., Ortak M., Petri S., Goldmann B., Duchstein H.J., Berger J., Helmchen U., Freeman B.A., Meinertz T., Munzel T. Myeloperoxidase enhances nitric oxide catabolism during myocardial ischemia and reperfusion. Free Radic Biol Med. 2004;37:902–911. doi: 10.1016/j.freeradbiomed.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 59.Brovkovych V., Gao X.P., Ong E., Brovkovych S., Brennan M.L., Su X., Hazen S.L., Malik A.B., Skidgel R.A. Augmented inducible nitric oxide synthase expression and increased NO production reduce sepsis-induced lung injury and mortality in myeloperoxidase-null mice. Am J Physiol Lung Cell Mol Physiol. 2008;295:L96–L103. doi: 10.1152/ajplung.00450.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clark R.A., Stone P.J., El Hag A., Calore J.D., Franzblau C. Myeloperoxidase-catalyzed inactivation of alpha 1-protease inhibitor by human neutrophils. J Biol Chem. 1981;256:3348–3353. [PubMed] [Google Scholar]
- 61.Ginhoux F., Greter M., Leboeuf M., Nandi S., See P., Gokhan S., Mehler M.F., Conway S.J., Ng L.G., Stanley E.R., Samokhvalov I.M., Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hailer N.P., Heppner F.L., Haas D., Nitsch R. Fluorescent dye prelabelled microglial cells migrate into organotypic hippocampal slice cultures and ramify. Eur J Neurosci. 1997;9:863–866. doi: 10.1111/j.1460-9568.1997.tb01436.x. [DOI] [PubMed] [Google Scholar]
- 63.Schmitz G., Leuthauser-Jaschinski K., Orso E. Are circulating monocytes as microglia orthologues appropriate biomarker targets for neuronal diseases. Cent Nerv Syst Agents Med Chem. 2009;9:307–330. doi: 10.2174/187152409789630424. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Rotenone (Rot) does not influence the viability but not morphology of microglia. A: Primary microglia were mock-treated or treated with the indicated concentrations of Rot for 3 days. Cell viability was analyzed using the Live/Dead Viability Cytotoxicity kit, as described in Materials and Methods. B: BV2 cells were cultured in the presence or absence of Rot for 1 day, after which the cells were stained with PI and annexin V. Cell viability was measured by FACS analysis. The data shown are representative of three independent experiments. C: Primary microglial cells were mock treated or treated with 1 nmol/L Rot for 1 day, after which the cells were stained for CD11b (green). The images were captured with confocal laser scanning. Scale bar = 20 μm. CON indicates control.
Primary astrocytes show similar responses to rotenone and MPO exposure. A: Primary astrocytes were treated with or without 5 nmol/L Rot for 3 days, and the amount of ROS generation was determined by FACS using rhodamine-123. B: Primary astrocytes (top) and microglia (bottom) were incubated with 100 ng/mL MPO. For confocal microscopy, the astrocytes were stained with anti-GFAP and anti-MPO antibodies and with DAPI; and microglia were stained with anti-MPO antibody and with DAPI (left). Scale bar = 20 μm. Primary astrocytes were treated with 100 ng/mL MPO for 18 hours, and intracellular MPO levels were examined by FACS analysis (right). The data shown are representative of at least three independent experiments. CON indicates control.