

Abstract
Senile plaques are a prominent pathological feature of Alzheimer's disease (AD), but little is understood about the association of glial cells with plaques or about the dynamics of glial responses through the disease course. We investigated the progression of reactive glial cells and their relationship with AD pathological hallmarks to test whether glial cells are linked only to amyloid deposits or also to tangle deposition, thus integrating both lesions as a marker of disease severity. We conducted a quantitative stereology-based post-mortem study on the temporal neocortex of 15 control subjects without dementia and 91 patients with AD, including measures of amyloid load, neurofibrillary tangles, reactive astrocytes, and activated microglia. We also addressed the progression of glial responses in the vicinity (≤50 μm) of dense-core plaques and tangles. Although the amyloid load reached a plateau early after symptom onset, astrocytosis and microgliosis increased linearly throughout the disease course. Moreover, glial responses correlated positively with tangle burden, whereas astrocytosis correlated negatively with cortical thickness. However, neither correlated with amyloid load. Glial responses increased linearly around existing plaques and in the vicinity of tangles. These results indicate that the progression of astrocytosis and microgliosis diverges from that of amyloid deposition, arguing against a straightforward relationship between glial cells and plaques. They also suggest that reactive glia might contribute to the ongoing neurodegeneration.
Activated glia is a prominent feature of Alzheimer's disease (AD) neuropathological features, with both reactive astrocytes and activated microglia clustering around and within dense-core amyloid plaques (ie, thioflavin-S–positive plaques).1 A better understanding of how these reactive glial cells accrue during the disease course and how they relate to the classic AD pathological hallmarks [ie, amyloid plaques and neurofibrillary tangles (NFTs)] is crucial for the following reasons: i) a body of preclinical evidence implicates these glial cells in AD pathophysiological features; ii) new positron emission tomographic (PET) radiotracers for amyloid plaques, NFTs, and, particularly, activated glial cells are being developed as diagnostic and progression biomarkers; and iii) clinical trials with anti-inflammatory therapies, ranging from nonsteroidal anti-inflammatory drugs (NSAIDs) to i.v. Ig, are under development.
In a previous quantitative neuropathological study,2 we observed a positive linear correlation between astrocytosis in the temporal neocortex, as measured with a glial fibrillary acidic protein (GFAP) enzyme-linked immunosorbent assay, and the duration of the disease from the onset of cognitive symptoms, despite the plaque burden remaining stable throughout the course of the disease. We hypothesized that a certain threshold of amyloid burden might be needed to trigger glial responses within a particular region of the cortex and that, once triggered, glial responses would reflect a pathogenic cascade increasingly independent of plaques. In the present study, we sought to extend that observation and test the hypothesis that, although initially linked to plaques, glial responses increasingly reflect the widespread ongoing neurodegenerative process. We quantified the number of reactive astrocytes and activated microglial cells in the temporal neocortex of a large cohort of controls without dementia and subjects with AD at different stages of the disease and investigated both their apparent progression throughout the disease course and their relation to the local burden of amyloid plaques and NFTs. Although glial association with amyloid plaques has long been assumed, we found a dissociation between these pathological features, with a linear increase of reactive glia despite a relatively stable plaque burden. The magnitude of these glial changes correlated with the burden of NFTs. A closer analysis in a subset of subjects with AD revealed that reactive glial cells increased both in the proximity of dense-core plaques and near NFTs, thus supporting a previously not described association between glial responses and neurofibrillary degeneration.
Materials and Methods
Brain Specimens and Immunohistochemical Studies
Formalin-fixed, paraffin-embedded tissue specimens from the temporal association isocortex (Brodmann area 38) of 91 patients with AD and 15 controls without dementia were obtained from the Massachusetts Alzheimer Disease Research Center Brain Bank. They were consecutively selected by tissue availability. All of the study subjects or their next of kin gave written informed consent for the brain donation, and the Massachusetts General Hospital Institutional Review Board approved the study protocol. The demographic characteristics of both groups are depicted in Table 1. All of the patients with AD fulfilled the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Associations criteria for probable AD3 and the National Institute on Aging–Reagan criteria for high likelihood of AD.4 Cases with cerebrovascular disease considered severe enough to contribute to the dementia syndrome were excluded because cerebrovascular disease is a major cause of focal gliosis. Cases with Lewy body pathological features were also excluded. Sections (8-μm thick) were deparaffinized for immunohistochemistry by standard methods. Primary and secondary antibodies, pretreatments for antigen retrieval, and visualization strategies are summarized in Table 2. For stereological quantitative studies, immunostained sections were developed with 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA), lightly counterstained with Mayer's hematoxylin, dehydrated with increasing concentrations of ethanol, cleared with xylene, and cover-slipped with Permount mounting media (Fisher Scientific, Fair Lawn, NJ). Nearly adjacent temporal sections from a subset of 40 subjects with AD and six controls without dementia were immunostained using fluorescently labeled secondary antibodies, counterstained with 0.05% thioflavin-S (Sigma, St Louis, MO) in 50% ethanol for 8 minutes, and cover-slipped with Vectashield mounting media with 4',6-diamidino-2-phenylindole (DAPI) (Vector Laboratories).
Table 1.
Demographic Characteristics of the Cohorts without Dementia and with AD and Their Corresponding Subsets Included in the Quantitative Neuropathological Study
| Characteristics | Control cohort (n = 15) | AD cohort (n = 91) | P value | Control subset (n = 6) | AD subset (n = 40) | P value |
|---|---|---|---|---|---|---|
| Age at death (years)⁎ | 79.9 ± 13.3 | 79.0 ± 7.8 | NS | 83.7 ± 14.0 | 77.6 ± 8.6 | 0.0429 |
| Female sex† | 10 (66.7) | 58 (63.7) | NS | 4 (66.7) | 26 (65.0) | NS |
| Disease duration (years)‡ | NA | 9.8 (6.8–13.7) | NA | NA | 9.9 (5.4–15.5) | NA |
| APOE genotype† | ||||||
| APOEε4 carriers | 4 (26.7) | 59 (64.8) | 0.0090 | 2 (33.3) | 21 (52.5) | NS |
| APOEε4 alleles§ | 4 (13.3) | 70 (38.5) | 0.0070 | 2 (16.7) | 25 (31.2) | NS |
| Post-mortem interval (hours)⁎ | 22.3 ± 12.8 | 13.9 ± 9.1 | 0.0085 | 21.0 ± 11.1 | 14.1 ± 6.2 | NS |
Information about cause of death was available in only 44 of the 91 subjects with AD because nursing homes are the main source of our brain donation program. Patients with protracted death (mostly aspirative pneumonia and cancer, n = 31) did not differ from patients with sudden death (mostly pulmonary emboli and myocardial infarction, n = 13) regarding their age at death (P = 0.2261), disease duration (P = 0.9898), or amount of astrocytosis (P = 0.1870) and microgliosis (P = 0.9180). Statistically significant P values are boldfaced.
NA, not applicable; NS, not significant.
Data are given as mean ± SD. P values were obtained using the two-tailed Mann-Whitney U-test.
Data are given as number (percentage) of each group. P values were obtained using the two-tailed χ2 test with Fisher's exact test.
Data are given as median (interquartile range).
To obtain percentages, the denominators for this row were doubled.
Table 2.
Antibodies, Antigen Retrieval Protocols, and Visualization Strategies Used in the IHC Studies
| Primary antibody | Host | Dilution | Antigen retrieval⁎ | Secondary antibody† | Visualization strategy |
|---|---|---|---|---|---|
| 10D5 (Elan Pharmaceuticals, Inc.) | Ms | 1:50 | Citrate buffer + MW and 90% formic acid for 5 minutes | HRP anti-Ms (1:200) | DAB (Vector Laboratories) |
| PHF1 (gift from Dr. Peter Davies) | Ms | 1:200 | Citrate buffer + MW | Biotin anti-Ms (1:200) | ABC kit + DAB (Vector Laboratories for both) |
| GFAP (catalogue no. G9269; Sigma) | Rb | 1:1000 | Citrate buffer + MW | i) Biotin anti-Rb (1:200) and ii) Cy3 anti-Rb (1:200) | i) ABC kit + DAB (Vector Laboratories for both) and ii) none |
| CD68 (catalogue no. M0814; Dako, Glostrup, Denmark) | Ms | 1:100 | Citrate buffer + MW | Biotin anti-Ms (1:200) | ABC kit + DAB (Vector Laboratories for both) |
| Iba1 (catalogue no. 019-19741; Wako) | Rb | 1:250 | Citrate buffer + MW | Cy3 anti-Rb (1:200) | None |
| NAB61 (gift from Dr. Virginia Lee) | Ms | 1:500 | None | Biotin anti-Ms (1:200) | ABC kit (Vector Laboratories) + streptavidin-Cy3 (1:200) (Invitrogen) |
ABC, avidin-biotin complex; DAB, 3,3′-diaminobenzidine; HRP, horseradish peroxidase; Ms, mouse; Rb, rabbit.
Citrate buffer + MW indicates 0.01 mol/L citrate buffer (pH 6.0) with 0.05% Tween 20 in a microwave oven at 95°C for 20 minutes.
All secondary antibodies were obtained from Jackson ImmunoResearch Labs (West Grove, PA).
Quantitative Neuropathological Analyses
We took advantage of stereology tools to perform unbiased quantitative neuropathological studies in these brain specimens. All analyses were conducted blinded to disease status.
Cortical Thickness
Cortical thickness was measured in sections stained with Luxol fast blue H&E, as previously described.5 Briefly, the image analysis software CAST (Olympus, Copenhagen, Denmark), mounted on an upright BX51 Olympus microscope (Olympus) and coupled with a motorized stage and a charge-coupled device camera, was used to randomly sample the cortex of the entire section and measure the thickness of the full cortex. The measurements of full cortical thickness in 20 random sites were averaged.
Amyloid Load and Number of Total Amyloid Plaques
Amyloid load and stereology-based studies on 3,3′-diaminobenzidine sections were conducted in an upright Leica DMRB microscope (Leica, Wetzlar, Germany) equipped with a motorized stage and a charge-coupled device camera (model DC330; DAGE-MTI, Inc., Michigan City, IN) and coupled with the software BIOQUANT NOVA PRIME, version 6.90.10 (MBSR, Nashville, TN). Amyloid load was measured as the percentage of total surface stained by the N-terminal–specific anti-amyloid β (Aβ) antibody 10D5 (Elan Pharmaceuticals, Inc., Dublin, Ireland) in a full-thickness strip of cortex (approximately 1-cm long) using the optical threshold application of the software. The total number of amyloid plaques in a 1-cm-long strip of cortex was calculated by dividing the total number of particles higher than the threshold by the area analyzed (both parameters provided by the software) and then correcting the resultant density by the cortical thickness.
Stereology-Based Quantitation of NFTs, Astrocytes, and Microglia
Paired helical filament 1–positive NFTs, GFAP-positive astrocytes, and CD68-positive microglial cells were counted with the optic dissector technique,6 using either 100 cells or 1000 optical dissectors as the end point. The objective/dissector size used in each case was 40/150 × 150 μm for paired helical filament 1–positive neurons, 40/50 × 50 μm for GFAP-positive astrocytes, and 100/20 × 20 μm for CD68-positive microglial cells. Intraneuronal and extracellular ghost tangles were not distinguished. Because different pathological features tend to accumulate in specific layers of the cortex (ie, reactive astrocytes in layer I and NFTs in layers II and V), care was taken to cover all of the six cortical layers in the systematic random sampling to avoid selection bias. As with the amyloid plaques, the densities of NFTs, astrocytes, and microglial cells were calculated by dividing the number of cells counted in single sections by the total area of the dissectors analyzed. To avoid any overestimation of densities because of disease-related cortical atrophy, these densities were then corrected by the cortical thickness to estimate the total number of cells within a full-thickness 1-cm-long strip of cortex.
Stereology-Based Quantitation of Oligomeric Aβ-Positive and Dense-Core Plaques
We performed additional quantitative studies in a subset of 40 AD cases selected from the original AD cohort on the basis of a wide range of disease duration and in a subset of six controls without dementia (ie, those with enough dense-core plaques). These subsets were representative of their corresponding cohorts in demographic characteristics, and the AD subset was also comparable to the entire AD cohort in neuropathological quantitative measures (Table 1; see also Supplemental Table S1 and Figure S1 at http://ajp.amjpathol.org). To study the progression of compact and oligomeric species of Aβ, we quantified the number of dense-core plaques and oligomeric Aβ-positive plaques in sections doubly stained with thioflavin-S and NAB61 antibody. The NAB61 antibody was provided by Dr. Virginia Lee (University of Pennsylvania, Philadelphia) and has been previously characterized. It is a conformation-specific anti-Aβ mouse monoclonal antibody that binds to Aβ dimers, small oligomers, and higher-order Aβ assemblies and stains a subset of mature dense-core plaques.7 Virtually no thioflavin-S–negative plaque was immunoreactive for NAB61. In this study, 100 dense-core plaques per case were randomly sampled, as previously described, and their positivity for NAB61 was qualitatively assessed. The densities of dense-core plaques and NAB61-positive plaques obtained were corrected by the cortical thickness to calculate total numbers of plaques within a 1-cm-long full-thickness strip of cortex.
Spatial Relationships of Dense-Core Plaques, NFTs, Astrocytes, and Microglia
Single sections from the subset of 40 AD cases were also doubly stained with thioflavin-S and GFAP or Iba1 to investigate the spatial relationship between glial responses and dense-core plaques and NFTs along the course of the disease. Optimal fluorescent immunolabeling of activated microglia was achieved with antibody Iba1 (Wako, Osaka, Japan), another marker widely used for activated microglia. Sections were placed on the motorized stage of an upright BX51 Olympus microscope equipped with CAST stereology software. One hundred GFAP-positive astrocytes or Iba1-positive microglial cells per section were randomly selected under the ×20 or the ×40 objective, respectively, and their distance with respect to the closest dense-core plaque or NFT was measured with the appropriate tool of the software. For consistency, only cells with a visible nucleus in the DAPI staining were considered. Astrocytes and microglial cells were classified into three categories: i) close to plaques, if located ≤50 μm from the edge of a plaque (regardless of the presence of an NFT within this boundary); ii) close to NFTs, if located ≤50 μm from an NFT but far (>50 μm) from dense-core plaques; and iii) far from plaques and NFTs, if the closest plaque and NFT to the glial cell were located >50 μm. Densities of glial cells in each of these categories were obtained as previously described.
APOE Genotyping
The APOE genotype was determined in all of the study subjects by restriction fragment length polymorphism analysis, as previously described.8
Statistics
Statistics were performed, and graphs were obtained with GraphPad Prism software for Mac, version 5.0. The normality of data sets was tested with the D'Agostino-Pearson omnibus test. For correlations of cortical thickness, amyloid load, and total number of astrocytes/microglia with disease duration, two different fit models were examined using the least-squares fitting method: linear regression versus one-phase exponential association (or decay in the case of cortical thickness). The first model assumes a linear increase of the pathological features over time, whereas the second model consists of an initial increase followed by a plateau. Next, these two fit models were compared using the Akaike's Informative Criteria method with no constraints, and the model most likely to have generated the data was selected based on the magnitude of the difference between both fit models, the probabilities of the models being correct (as calculated by the statistical software), and their goodness of fit (R2). When the straight-line model was preferred, a P value indicating whether the slope of the straight line is significantly different from 0 and both the correlation coefficient (r) and the P value of Spearman's rank correlation test were also reported. Because none of the data sets was normally distributed, cross correlations between these pathological quantitative measures were investigated with the Spearman's rank correlation test. The significance level was set at a two-sided P < 0.05 in all statistical analyses.
Results
Disease Duration Is a Reliable Proxy of Disease Severity
We have previously used disease duration (defined from the onset of cognitive symptoms) as a proxy of disease severity to avoid the floor effects of neuropsychological tests in patients with advanced dementia, who are typically not testable. More important, the three major pathological correlates of cognitive decline (ie, NFT burden, neuron loss, and synaptic loss) also correlated with disease duration in our previous quantitative post-mortem studies2,9,10 on the temporal neocortex. Herein, we measured the cortical thickness of the temporal neocortex specimens from the AD cohort as an index of synaptic, dendritic, and neuronal integrity. We found a significant negative correlation between cortical thickness and symptomatic disease duration, further validating the use of disease duration as a surrogate of disease severity (r = −0.3977, P < 0.0001) (Figure 1A and Table 3).
Figure 1.
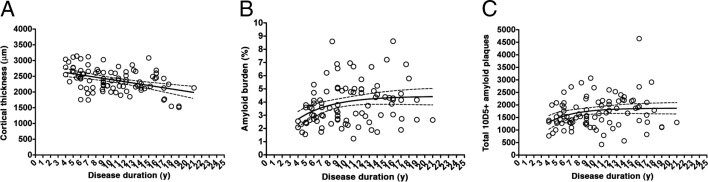
Progression of cortical atrophy and amyloid deposition in the temporal neocortex in AD (see also Table 3). A: Cortical thickness decreased linearly, along with the symptomatic disease duration, indicating that this is a reliable proxy of disease severity. Amyloid burden (B) and total number of plaques (C) increased during the first years of the clinical course of the disease but reached a plateau soon after.
Table 3.
Summary of the Results from the AD and Control Cohorts
| Variable | AD cohort (n = 91) |
AD + CTRL with plaques (n = 101) |
AD + all CTRL (n = 106) |
|||
|---|---|---|---|---|---|---|
| Linear | One-phase exponential | Linear | One-phase exponential | Linear | One-phase exponential | |
| Cortical thickness (μm) | ||||||
| ΔAICc | 1.804 | NA | NA | |||
| Probability (%) | 71.13 | 28.87 | 100 | Not converged | 100 | Not converged |
| Goodness (R2) | 0.1797 | 0.1831 | 0.2130 | NA | 0.2296 | NA |
| Slope ≠ 0? (P) | <0.0001 | NA | <0.0001 | NA | <0.0001 | NA |
| Spearman's r | −0.3977 | NA | −0.4523 | NA | −0.4649 | NA |
| Spearman′s P | <0.0001 | NA | <0.0001 | NA | <0.0001 | NA |
| Amyloid burden (%) | ||||||
| ΔAICc | 4.347 | 10.83 | 18.23 | |||
| Probability (%) | 10.22 | 89.78 | 0.44 | 99.56 | 0.01 | 99.99 |
| Goodness (R2) | 0.0657 | 0.1093 | 0.2317 | 0.3099 | 0.3117 | 0.4205 |
| Slope ≠ 0? (P) | 0.0142 | NA | <0.0001 | NA | <0.0001 | NA |
| Total amyloid plaques | ||||||
| ΔAICc | 1.761 | 11.96 | 21.72 | |||
| Probability (%) | 29.31 | 70.69 | 0.25 | 99.75 | <0.01 | >99.99 |
| Goodness (R2) | 0.0391 | 0.0575 | 0.2101 | 0.2983 | 0.2952 | 0.4258 |
| Slope ≠ 0? (P) | 0.0602 | NA | <0.0001 | NA | <0.0001 | NA |
| Total astrocytes | ||||||
| ΔAICc | 0.7545 | 3.005 | 3.953 | |||
| Probability (%) | 59.32 | 40.68 | 81.80 | 18.20 | 87.83 | 12.17 |
| Goodness (R2) | 0.1951 | 0.1884 | 0.2433 | 0.2204 | 0.2705 | 0.2428 |
| Slope ≠ 0? (P) | <0.0001 | NA | <0.0001 | NA | <0.0001 | NA |
| Spearman's r | 0.4070 | NA | 0.5037 | NA | 0.5471 | NA |
| Spearman's P | <0.0001 | NA | <0.0001 | NA | <0.0001 | NA |
| Total microglia | ||||||
| ΔAICc | 5.171 | 33.18 | 42.25 | |||
| Probability (%) | 92.99 | 7.01 | >99.99 | <0.01 | >99.99 | <0.01 |
| Goodness (R2) | 0.0960 | 0.0431 | 0.1728 | −0.1489 | 0.2109 | −0.1755 |
| Slope ≠ 0? (P) | 0.0028 | NA | <0.0001 | NA | <0.0001 | NA |
| Spearman's r | 0.3545 | NA | 0.4326 | NA | 0.4728 | NA |
| Spearman's P | 0.0006 | NA | <0.0001 | NA | <0.0001 | NA |
The probability of being correct and the goodness of fit (R2) of both the linear regression and the one-phase exponential association models (or decay, in the case of cortical thickness) are shown for the main neuropathological measures in the AD cohort alone, the AD cohort plus the controls without dementia and with plaques, and the AD cohort plus the entire control cohort. The best-fit model is boldfaced. In the linear regression model, P indicates whether the slope is significantly different from 0. When the linear regression model was the preferred-fit model, the correlation coefficient and the P value from the Spearman's rank correlation test are also shown. For the amyloid burden and the total number of amyloid plaques, the nonlinear model remains the best fit, despite the linear fit yielding a straight line with a slope significantly different from 0 (because of the anchoring effect of controls close to 0). Also, the R2 of the one-phase exponential association model is negative for some neuropathological measures, indicating that the best-fit curve fits the data even worse than a horizontal line. Statistics in Materials and Methods provides further details.
ΔAICc, magnitude of the difference between both fit models; CTRL, control without dementia; NA, not applicable.
Progression of Amyloid Deposition in the Temporal Neocortex
Next, we traced the progression of amyloid deposition and patterns of glial immunostaining throughout the clinical disease course. Amyloid burden, determined as the percentage of cortical surface immunoreactive for the anti-Aβ antibody 10D5, reached a plateau early after symptomatic onset and remained relatively stable thereafter (Figure 1B and Table 3). An analysis of total number of plaques yielded similar results (Figure 1C and Table 3). Like 10D5-immunoreactive plaques in the original AD cohort, the number of dense-core plaques determined in a subset of 40 AD cases remained relatively stable throughout the disease clinical course after an initial increase (Figure 2, A and B, and Table 4). Last, the amount of NAB61-positive oligomeric Aβ-enriched plaques also remained constant throughout the disease clinical course (Figure 2, C and D, and Table 4).
Figure 2.
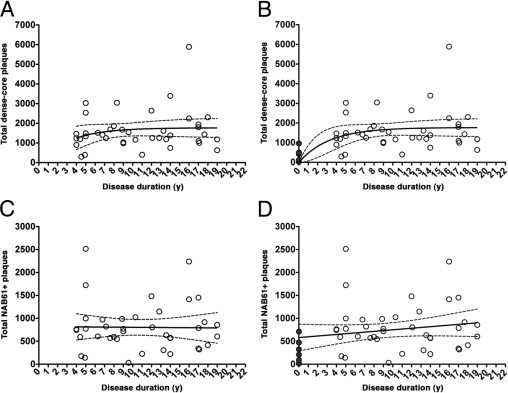
Progression of fibrillar and oligomeric Aβ burden in the temporal neocortex (see also Table 4). A and C: Only the AD subset is shown (open circles, n = 40). B and D: The highly selected subset of controls without dementia and with dense-core plaques is also included, with a disease duration of 0 years (dark gray circles, n = 6). In A and B, a small increase in the number of dense-core plaques was only detectable during the first years after the onset of cognitive symptoms. In C and D, the number of NAB61-positive oligomeric Aβ-enriched plaques remained unchanged after symptom onset.
Table 4.
Summary of the Results from the AD and Control Subsets Concerning Fibrillar and Oligomeric Aβ Burden
| Variable | AD subset (n = 40) |
AD + CTRL with dense-core plaques (n = 46) |
||
|---|---|---|---|---|
| Linear | One-phase exponential | Linear | One-phase exponential | |
| Total dense-core plaques | ||||
| ΔAICc | 0.2525 | 1.833 | ||
| Probability (%) | 46.85 | 53.15 | 28.57 | 71.43 |
| Goodness (R2) | 0.0248 | 0.0310 | 0.1297 | 0.1637 |
| Slope ≠ 0? (P) | 0.3313 | NA | 0.0140 | NA |
| Total NAB61 + plaques | ||||
| ΔAICc | NA | NA | ||
| Probability (%) | 100 | Not converged | 100 | Not converged |
| Goodness (R2) | 0.0002 | 0.0361 | ||
| Slope ≠ 0? (P) | 0.9313 | 0.2060 | ||
| Spearman's r | −0.0037 | NA | 0.2267 | NA |
| Spearman's P | 0.9816 | NA | 0.1298 | NA |
The ΔAICc represents the magnitude of the difference between the two fit models compared. The best-fit model is boldfaced. In the linear model, P indicates whether the slope is significantly different from 0. When the linear regression model was the preferred-fit model, the correlation coefficient and the P value from the Spearman's rank correlation test are also shown. The nonlinear model is the best fit for total dense-core plaques in the AD + CTRL analysis, despite the linear fit yielding a straight line with a slope significantly different from 0 (likely because of the anchoring effect caused by the controls). Statistics in Materials and Methods provides further details.
ΔAICc, magnitude of the difference between both fit models; CTRL, control without dementia; NA, not applicable.
Progression of Glial Responses and Relationship to AD Pathological Hallmarks
Despite being traditionally regarded as plaque-associated pathological features, the progression of astrocytic and microglial responses differed from that of amyloid plaques both qualitatively and quantitatively. Both reactive glial cell types increased linearly through the entire clinical course of the disease, even when the amyloid burden was no longer increasing [r = 0.4070 (P < 0.0001) and r = 0.3545 (P = 0.0006) for astrocytes and microglia, respectively]. The inclusion in these analyses of either controls without dementia who had plaques (n = 10) or all controls (n = 15) further accentuated the difference in the patterns of progression of amyloid burden and glial responses (Figure 3, A–D, and Table 3). Indeed, neither astrocytosis nor microgliosis correlated with the amyloid burden [r = −0.0963 (P = 0.3637) and r = 0.0062 (P = 0.9538), respectively] or the total number of plaques [r = −0.0774 (P = 0.4660) and r = 0.1124 (P = 0.2888), respectively] (Figure 4, A–D).
Figure 3.
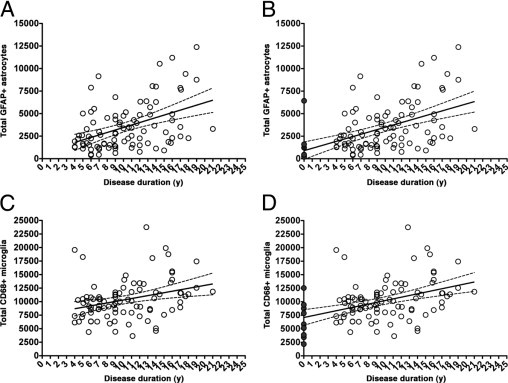
Progression of glial responses in the temporal neocortex (see also Table 3). A and C: Only the AD cohort is shown (open circles, n = 91). B and D: The controls without dementia and with plaques are also included, with a clinical disease duration of 0 years (dark gray circles, n = 10). Both astrocytosis (A and B) and microgliosis (C and D) significantly increased in a linear fashion, along with disease progression.
Figure 4.
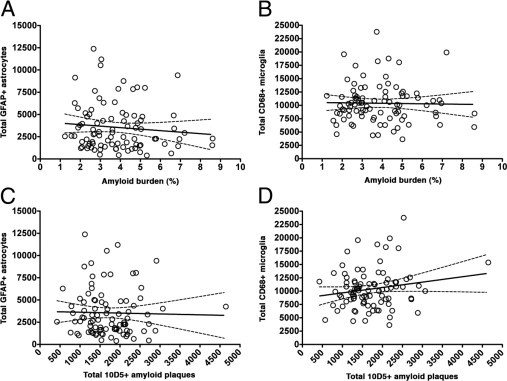
Correlations between glial responses and measures of amyloid deposition in AD. Neither astrocytosis nor microgliosis correlated with amyloid burden (A and B) or total number of plaques (C and D).
Unlike the measures of amyloid deposition, both astrocytosis and microgliosis correlated positively with the NFT burden [r = 0.3419 (P = 0.0009) and r = 0.4635 (P < 0.0001) respectively]. Also, astrocytosis, but not microgliosis, correlated negatively with cortical thickness [r = −0.4647 (P < 0.0001) and r = −0.0199 (P = 0.8517), respectively] (Figure 5, A–D), further arguing that the relationship between reactive glia and amyloid plaques might not be as straightforward as previously thought. Last, astrocytosis and microgliosis tightly covaried together (r = 0.3377, P = 0.0011) (data not shown).
Figure 5.
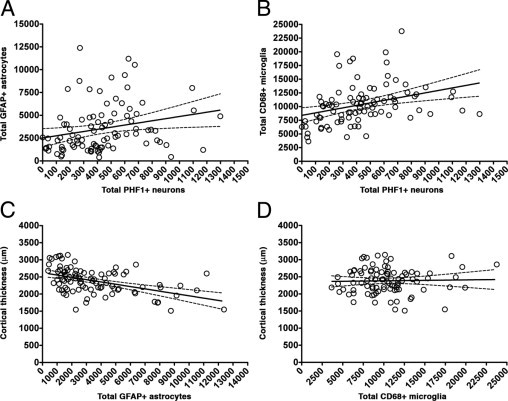
Correlations between glial responses and markers of neurodegeneration in AD. Astrocytosis (A) and microgliosis (B) correlated positively with the burden of NFTs. Patients with higher astrocytosis tended to have more cortical atrophy (C), but no association was observed between the extent of microgliosis and cortical thickness (D). PHF, paired helical filament.
The APOE genotype did not affect the magnitude of astrocytosis or microgliosis and did not significantly influence the progression of glial responses (see Supplemental Figure S2 at http://ajp.amjpathol.org).
Spatial Relationships between Glial Responses and Amyloid Plaques/NFTs
It is well established that activated glial cells are associated with individual senile plaques. In an attempt to better understand the previous results, we examined reactive glia in the microenvironment near plaques and tangles. We compared the progression of plaque-associated reactive glial cells with NFT-associated reactive glial cells and reactive glial cells in the neuropil not close to either a plaque or a tangle in the subset of 40 AD cases using the stereology-based procedures described in Materials and Methods. We observed a significant increase in the density of reactive astrocytes and, to a lesser extent, activated microglia in the proximity of dense-core plaques, along with disease progression [r = 0.6275 (P < 0.0001) and r = 0.3073 (P = 0.0538), respectively] (Figure 6, A, D, G, and J). We also observed a significant, although weaker, linear increase of astrocytosis and, particularly, microgliosis in the vicinity of NFTs as the disease progresses [r = 0.3125 (P = 0.0497) and r = 0.3586 (P = 0.0231), respectively] (Figure 6, B, E, H, and K), suggesting that a causal link underlies the positive correlations between accumulation of reactive glia and NFT burden previously shown. By contrast, the density of astrocytes and microglial cells not associated with either dense-core plaques or NFTs (ie, free in the neuropil) did not significantly correlate with the progression of the disease [r = 0.1883 (P = 0.2447) and r = 0.0921 (P = 0.5720), respectively] (Figure 6, C, F, I, and L).
Figure 6.
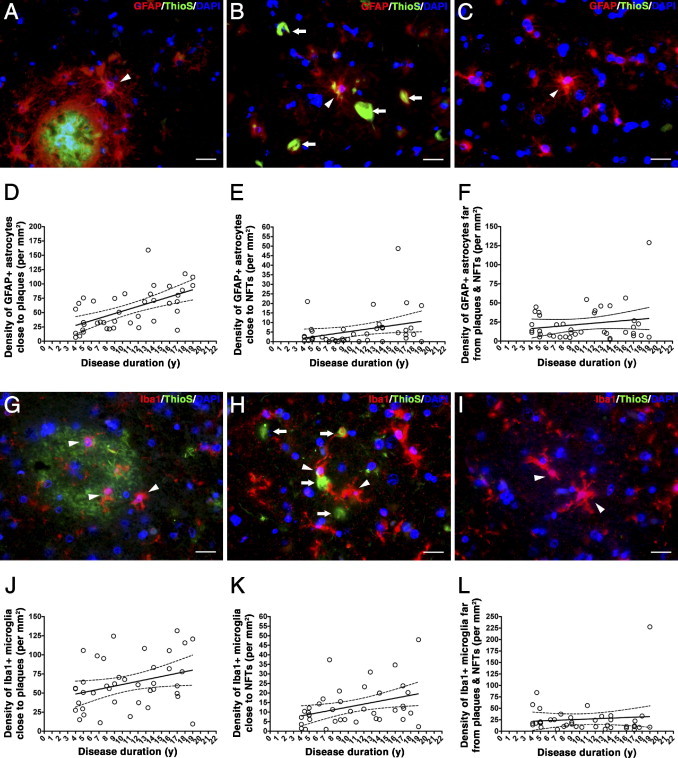
Spatial relationship of glial responses and dense-core plaques and NFTs. A–C: Photomicrographs of triple staining for GFAP (red), thioflavin-S (ThioS; green), and DAPI (blue) in an AD case. GFAP-positive reactive astrocytes (arrowhead) cluster in the vicinity (≤50 μm) of dense-core plaques (A), but they can also be seen in close association to NFTs (B, arrows) and free in the neuropil (C). Scale bar = 20 μm. The density of reactive astrocytes increased linearly in the proximity of dense-core plaques (D) and close to NFTs (E); by contrast, reactive astrocytes did not increase far (>50 μm) from dense-core plaques and NFTs (F). G–I: Photomicrographs of triple staining for Iba1 (red), ThioS (green), and DAPI (blue) in an AD case. Iba1-positive activated microglial cells (arrowheads) cluster in the vicinity (≤50 μm) of dense-core plaques (G), but they can also be found near NFTs (H, arrows) and free in the neuropil (I). Scale bar = 20 μm. Microgliosis increased linearly close to both plaques (J) and NFTs (K), but no significant increase in the number of activated microglial cells was observed far (>50 μm) from dense-core plaques and NFTs (L).
Discussion
Much evidence from previous clinicopathological studies9–15 has established that the NFT burden and both neuronal and synaptic loss, but not amyloid plaque burden, correlate with the severity of cognitive impairment in AD. However, despite invariably accompanying these classic hallmarks, particularly amyloid plaques, these previous reports have not addressed the progression of glial responses in AD. Although post-mortem studies are inherently cross-sectional and, therefore, any longitudinal extrapolation should be interpreted with caution, quantitative unbiased stereology-based analysis in a large cohort of subjects with a broad range of disease duration enabled us to track the progression of amyloid deposition, NFTs, and glial responses within the AD temporal neocortex and to investigate their relationships. Our findings can be summarized as follows: i) astrocytosis and microgliosis occur both around dense-core plaques and in the proximity of NFTs; ii) astrocytosis and microgliosis increase linearly with disease duration, despite amyloid burden reaching a plateau early in the clinical course of the disease; iii) astrocytosis and microgliosis covary with the burden of NFTs through the entire clinical course of the disease. Taken together, the number of reactive astrocytes and activated microglial cells tracks better with tangles than with plaques (Figure 7).
Figure 7.
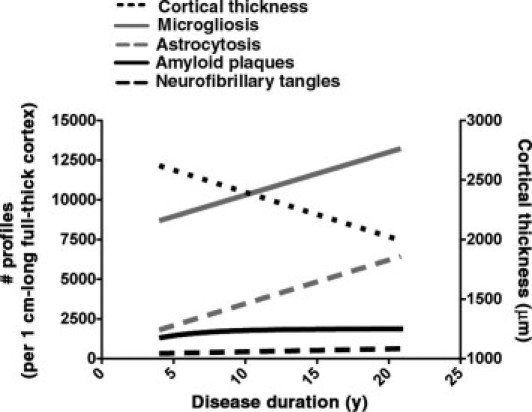
A model of the progression of AD-related pathological features in the temporal neocortex based on the present results. The regression lines for each of the pathological measures were plotted within a single graph. For clarity purposes, the regression line for cortical thickness was linked to the y axis with a different scale (right). Amyloid deposition reaches a plateau early after the onset of cognitive symptoms, whereas reactive astrocytes, activated microglial cells, and NFTs keep accumulating as the cortical mantle atrophies through the disease course. Remarkably, the slopes indicate that, within a reference area of 1-cm-long full-thickness temporal neocortex, activated microglia and reactive astrocytes accumulate at a strikingly similar average rate of 270 and 275 cells per year, respectively, which is >10-fold higher than the rate of neurofibrillary degeneration (average, 18 tangles per year). Last, cortical thickness decreased at an average rate of 37 μm/year.
Dynamics of Amyloid Deposition in the Temporal Neocortex
Previous post-mortem studies2,9–13 have established that the amyloid burden remains relatively stable after the first stages of AD dementia and, thus, does not correlate with the severity of dementia. The inclusion of controls without dementia and with AD pathological changes and, particularly, of patients with AD who died shortly after the clinical onset enabled us to trace the natural history of brain β-amyloidosis as a continuum between normal aging and AD. Taken together, our analyses of amyloid deposition support a saturation model in which most amyloid accumulation occurs in the earliest phases, after which there is little or no further accumulation of total amyloid burden in a particular brain region (Figure 7). The pace at which this pattern of deposition and plateau occurs may well play out at different rates or times, depending on the brain region, as suggested by the hierarchical distribution of amyloid deposits previously described.9,16–18 The type of amyloid plaque did not vary substantially once plaques were deposited. The natural history of fibrillar (thioflavin-S–positive) amyloid deposition paralleled that of total (10D5-immunoreactive) amyloid deposition, also in keeping with our previous results.2 Interestingly, NAB61-positive oligomeric Aβ was readily observed in association with the dense-core plaques throughout the disease course.
Implications for the Role of Reactive Glial Cells in AD
Because the increase in reactive glial cells cannot be attributed to a progressive buildup of plaque burden, we reasoned that it might be explained by either the same original plaques accumulating more reactive glial cells as the disease advances or the reactive glial cells spreading throughout the neuropil, perhaps contributing to neuronal dysfunction and neurodegeneration as additional neuronal lesions and synaptic loss accrue. Both explanations are supported by our results. On one hand, we observed a progressive increase in the density of reactive astrocytes and activated microglial cells in the proximity (≤50 μm) of dense-core plaques, indicating that Aβ plaques are permanently recruiting and activating glial cells. On the other hand, both astrocytosis and microgliosis covaried with the number of NFTs and increased in their proximity, and astrocytosis also correlated negatively with cortical thickness, suggesting a partial emancipation of glial cells from amyloid plaques. Whether the increasing number of reactive glial cells is responding to the ongoing neurodegenerative process (and, thus, a surrogate marker of neurodegeneration) or actively contributes to neurodegeneration cannot be determined by cross-sectional human neuropathological studies. However, along this line, astrocytosis was the best correlate of neuron loss and cognitive deficits in an Aβ precursor protein/τ double-transgenic mouse model that closely recapitulates the pathological features of human AD.19 In addition, recent experimental studies with in vitro and mouse models of AD have linked both astrocytes and microglia to neurodegeneration20–25 and have shown that activated microglia can lead to τ hyperphosphorylation and aggregation.26,27
The idea of reactive glia as a hostile environment for neurons in the context of neurodegeneration has been recently proposed by elegant studies28,29 on amyotrophic lateral sclerosis and tauopathy mouse models. If this idea proves to apply to AD, an increasing number of reactive glial cells around dense-core plaques might well contribute to their local toxicity by releasing soluble biologically active toxic molecules, such as pro-inflammatory cytokines and reactive oxygen species. These toxic biomolecules, together with soluble Aβ oligomers existing around the plaques, might account for the plaque-associated neurodegenerative changes, including neuritic dystrophies, an increased neurite curvature ratio, and synaptic and neuron loss, which might be slowly progressive.
Diagnostic and Therapeutic Implications
Follow-up PET studies using the fibrillar amyloid-specific radiotracer Pittsburgh compound B (PiB) have yielded conflicting results regarding the progression of amyloid deposition in vivo in patients with AD-type dementia, with some studies30,31 reporting little or no increase in PiB uptake over time and other studies32,33 describing a significant increase in patients with dementia. Technical issues inherent to PET, such as test-retest reliability or a dynamic range of detection, might account for these divergent findings to some extent. Our post-mortem analyses argue against a marked increase in PiB uptake in a region with established amyloid deposition (eg, the temporal neocortex) once the dementia syndrome is overt. However, PiB might be useful to track the spreading of amyloid deposition to additional cortical areas in vivo in elderly controls without dementia, patients with mild cognitive impairment, and perhaps patients with mild dementia. It is also possible that regions of interest with later amyloid deposition, such as the occipital cortex, might still show increasing PiB uptake in patients with established dementia.30 Longer serial PET studies,34 combined with serial cognitive assessments in larger samples of elderly controls and patients with mild cognitive impairment and AD, will determine whether PiB PET is useful as a surrogate biomarker of progression at each stage of the disease. The current post-mortem quantitative analysis suggests that imaging some sort of glial marker might also be a useful approach to track the progression of AD in vivo. A positive glial PET might also add diagnostic and prognostic specificity to a positive amyloid PET study at the preclinical and mild cognitive impairment phases of the disease. Our results predict that the pattern of glial radiotracer uptake would closely match those of PiB uptake and cortical atrophy. However, the first PET studies using [11C]-(R)-PK11195, a radiotracer for the peripheral benzodiazepine receptor expressed by activated microglia, have reported contradictory results: although the original study35 described an abnormal uptake in vulnerable cortical regions of patients with mild AD but not in controls without dementia, more recent studies36–38 have reported a great overlap between both groups and no correlation between [11C]-(R)-PK11195 uptake and severity of cognitive decline. Novel radioligands targeting the same receptor in microglia have recently proven to track considerably better with τ-induced neurodegeneration in mice overexpressing mutant human τ than with amyloid burden in animals modeling Aβ plaque deposition.39
If glial responses become partially independent from amyloid plaques and their contribution to neurodegeneration is relevant, then removal of amyloid plaques with anti-Aβ-directed therapies, such as passive or active immunization, might not be sufficient to block this neurotoxicity. In this scenario, the transient microglial response triggered by anti-Aβ immunization might have deleterious effects on the neuropil, even though there is an overall decrease in gliosis on clearance of amyloid plaques.40,41 Glia-mediated inflammation has already been considered as a therapeutic target. NSAIDs became promising agents after the first large epidemiological studies42,43 revealed that a long-term treatment substantially reduced the risk of AD. Preclinical research44 has demonstrated that NSAIDs can decrease Aβ42 levels by modulating the activity of the γ-secretase complex, independently from their inhibitory effect on the pro-inflammatory cyclooxygenase and peroxisome proliferator-activated receptor-γ pathways. However, the clinical trials44 with NSAIDs completed thus far have failed to delay the progression of cognitive decline in AD or the conversion from mild cognitive impairment to AD, and recent epidemiological studies have suggested that NSAIDs might have no effect on the risk of AD45 or might even increase it.46 Moreover, two neuropathological studies45,47 found an increased burden of AD pathological features, particularly amyloid plaques, in NSAID users compared with nonusers. Other molecular targets involved in the first steps of glial activation, rather than in the production of singular pro-inflammatory cytokines, remain to be explored. Recently, the activation phenotype of both astrocytes and microglial cells was reported to be largely mediated through the calcineurin–nuclear factor of activated T cells pathway.48,49 Fibrillar and particularly oligomeric Aβ can aberrantly activate this molecular signaling pathway in glial cells48,50 and neurons, causing the morphological triad of dystrophic neurites, dendritic simplification, and loss of dendritic spines, subsequently leading to cognitive impairment.51–53 More important, Food and Drug Administration–approved and available calcineurin inhibitors, such as cyclosporine and FK-506 (tacrolimus), can revert these Aβ-related pathological changes both in vitro and in vivo and ameliorate both the neuropathological features and the memory deficits of AD mouse models.26,51–53
In summary, our findings indicate a major role of gliosis in the pathophysiological features of both amyloid plaques and NFTs, suggesting that glial cells might be proactive players linking Aβ with downstream neurodegenerative events beyond their role in the initial steps of the amyloid cascade. The implications of these results for the development of imaging biomarkers and disease-modifying drugs encourage further research.
Acknowledgments
We thank the patients and caregivers involved in research at Massachusetts General Hospital, Dr. Virgina Lee (University of Pennsylvania, Philadelphia) for providing the NAB61 anti-oligomeric Aβ-specific antibody, and Elan Pharmaceuticals, Inc. for supplying the 10D5 anti-Aβ mouse monoclonal antibody.
Footnotes
Supported by grants from the NIH (P50AG05134 and AG08487) and a research fellowship from the Foundation Alfonso Martín Escudero, Madrid, Spain (A.S.-P.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.05.047.
Supplementary data
The subset of patients with AD used to investigate the spatial relationships between glial responses and plaques and NFTs is representative of the entire AD cohort. The following are shown: progression of cortical thickness (A), amyloid burden (B), total number of plaques (C), astrocytosis (D), and microgliosis (E) in the subset of 40 AD cases. Figures 1, 2, and 3 and Table 3 have comparisons.
There was no significant effect of APOEε4 on the progression of glial responses. Astrocytosis (A) and microgliosis (B) were more severe in patients with AD than in age-matched controls without dementia, but no significant difference was observed between APOEε4 carriers (n = 59) and noncarriers (n = 32) within the AD group (one-way analysis of variance Kruskal-Wallis test: P < 0.0001 and P = 0.0004, respectively; Dunn's multiple comparison test: **P < 0.001, ***P < 0.0001). The progression of astrocytosis (C) and microgliosis (D) did not substantially differ between APOEε4 carriers and noncarriers.
References
- 1.Itagaki S., McGeer P.L., Akiyama H., Zhu S., Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–182. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 2.Ingelsson M., Fukumoto H., Newell K.L., Growdon J.H., Hedley-Whyte E.T., Frosch M.P., Albert M.S., Hyman B.T., Irizarry M.C. Early Aβ accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 3.McKhann G., Drachman D., Folstein M., Katzman R., Price D., Stadlan E.M. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of the Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 4.The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease: Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. Neurobiol Aging. 1997;18(Suppl):S1–S2. [PubMed] [Google Scholar]
- 5.Freeman S.H., Kandel R., Cruz L., Rozkalne A., Newell K., Frosch M.P., Hedley-Whyte E.T., Locascio J.J., Lipsitz L.A., Hyman B.T. Preservation of neuronal number despite age-related cortical brain atrophy in elderly subjects without Alzheimer disease. J Neuropathol Exp Neurol. 2008;67:1205–1212. doi: 10.1097/NEN.0b013e31818fc72f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hyman B.T., Gómez-Isla T., Irizarry M.C. Stereology: a practical primer for neuropathology. J Neuropathol Exp Neurol. 1998;57:305–310. doi: 10.1097/00005072-199804000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Lee E.B., Leng L.Z., Zhang B., Kwong L., Trojanowski J.Q., Abel T., Lee V.M. Targeting amyloid-β peptide (Aβ) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Aβ precursor protein (APP) transgenic mice. J Biol Chem. 2006;281:4292–4299. doi: 10.1074/jbc.M511018200. [DOI] [PubMed] [Google Scholar]
- 8.Ingelsson M., Shin Y., Irizarry M.C., Hyman B.T. Genotyping of apolipoprotein E: comparative evaluation of different protocols. Curr Protoc Hum Gen. 2003;38:1–13. doi: 10.1002/0471142905.hg0914s38. [DOI] [PubMed] [Google Scholar]
- 9.Arriagada P.V., Growdon J.H., Hedley-Whyte E.T., Hyman B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 10.Gómez-Isla T., Hollister R., West H., Mui S., Growdon J.H., Petersen R.C., Parisi J.E., Hyman B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 11.Hyman B.T., Marzloff K., Arriagada P.V. The lack of accumulation of senile plaques or amyloid burden in Alzheimer's disease suggests a dynamic balance between amyloid deposition and resolution. J Neuropathol Exp Neurol. 1993;52:594–600. doi: 10.1097/00005072-199311000-00006. [DOI] [PubMed] [Google Scholar]
- 12.Bierer L.M., Hof P.R., Purohit D.P., Carlin L., Schmeidler J., Davis K.L., Perl D.P. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Arch Neurol. 1995;52:81–88. doi: 10.1001/archneur.1995.00540250089017. [DOI] [PubMed] [Google Scholar]
- 13.Giannakopoulos P., Herrmann F.R., Bussière T., Bouras C., Kövari E., Perl D.P., Morrison J.H., Gold G., Hof P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 14.DeKosky S.T., Scheff S.W. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 15.Terry R.D., Masliah E., Salmon D.P., Butters N., DeTeresa R., Hill R., Hansen L.A., Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 16.Braak H., Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 17.Arriagada P.V., Marzloff K., Hyman B.T. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer's disease. Neurology. 1992;42:1681–1688. doi: 10.1212/wnl.42.9.1681. [DOI] [PubMed] [Google Scholar]
- 18.Thal D.R., Rüb U., Orantes M., Braak H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology. 2001;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 19.DaRoucha-Souto B., Scotton T.C., Coma M., Serrano-Pozo A., Hashimoto T., Serenó L., Rodríguez M., Sánchez M.B., Hyman B.T., Gómez-Isla T. Brain oligomeric Aβ but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in APP/tau-transgenic mice. J Neuropathol Exp Neurol. 2011;70:360–376. doi: 10.1097/NEN.0b013e318217a118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fonseca M.I., Zhou J., Botto M., Tenner A.J. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer's disease. J Neurosci. 2004;24:6457–6465. doi: 10.1523/JNEUROSCI.0901-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ortinski P.I., Dong J., Mungenast A., Yue C., Takano H., Watson D.J., Haydon P.G., Coulter D.A. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci. 2010;13:584–591. doi: 10.1038/nn.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allaman I., Gavillet M., Bélanger M., Laroche T., Viertl D., Lashuel H.A., Magistretti P.J. Amyloid-β aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J Neurosci. 2010;30:3326–3338. doi: 10.1523/JNEUROSCI.5098-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jana A., Pahan K. Fibrillar amyloid-β-activated human astroglia kill primary human neurons via neutral sphingomyelinase: implications for Alzheimer's disease. J Neurosci. 2010;30:12676–12689. doi: 10.1523/JNEUROSCI.1243-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giulian D., Haverkamp L.J., Yu J.H., Karshin W., Tom D., Li J., Kirkpatrick J., Kuo L.M., Roher A.E. Specific domains of β-amyloid from Alzheimer plaque elicit neuron killing in human microglia. J Neurosci. 1996;16:6021–6037. doi: 10.1523/JNEUROSCI.16-19-06021.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuhrmann M., Bittner T., Jung C.K., Burgold S., Page R.M., Mitteregger G., Haass C., LaFerla F.M., Kretzschmar H., Herms J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci. 2010;13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshiyama Y., Higuchi M., Zhang B., Huang S.M., Iwata N., Saido T.C., Maeda J., Suhara T., Trojanowski J.Q., Lee V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 27.Bhaskar K., Konerth M., Kokiko-Cochran O.N., Cardona A., Ransohoff R.M., Lamb B.T. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68:19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamanaka K., Chun S.J., Boillee S., Fujimori-Tonou N., Yamashita H., Gutmann D.H., Takahashi R., Misawa H., Cleveland D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hampton D.W., Webber D.J., Bilican B., Goedert M., Spillantini M.G., Chandran S. Cell-mediated neuroprotection in a mouse model of human tauopathy. J Neurosci. 2010;30:9973–9983. doi: 10.1523/JNEUROSCI.0834-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engler H., Forsberg A., Almkvist O., Blomquist G., Larsson E., Savitcheva I., Wall A., Ringheim A., Längström B., Nordberg A. Two-year follow-up of amyloid deposition in patients with Alzheimer's disease. Brain. 2006;129:2856–2866. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- 31.Scheinin N.M., Aalto S., Koikkalainen J., Lötjönen J., Karrasch M., Kemppainen N., Vlitanen M., Nägren K., Helin S., Scheinin M., Rinne J.O. Follow-up of [11C]PIB uptake and brain volume in patients with Alzheimer disease and controls. Neurology. 2009;73:1186–1192. doi: 10.1212/WNL.0b013e3181bacf1b. [DOI] [PubMed] [Google Scholar]
- 32.Jack C.R., Jr, Lowe V.J., Weigand S.D., Wiste H.J., Senjem M.L., Knopman D.S., Shiung M.M., Gunter J.L., Boeve B.F., Kemp B.J., Weiner M., Petersen R.C. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rinne J.O., Brooks D.J., Rossor M.N., Fox N.C., Bullock R., Klunk W.E., Mathis C.A., Blennow K., Barakos J., Okello A.A., Rodriguez Martinez de Liano S., Liu E., Koller M., Gregg K.M., Schenk D., Black R., Grundman M. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer's disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9:363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 34.Jack C.R., Jr, Knopman D.S., Jagust W.J., Shaw L.M., Aisen P.S., Weiner M.W., Petersen R.C., Trojanowski J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cagnin A., Brooks D.J., Kennedy A.M., Gunn R.N., Myers R., Turkheimer F.E., Jones T., Banati R.B. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- 36.Edison P., Archer H.A., Gerhard A., Hinz R., Pavese N., Turkheimer F.E., Hammers A., Tai Y.F., Fox N., Kennedy A., Rossor M., Brooks D.J. Microglia, amyloid, and cognition in Alzheimer's disease: an [11C](R)PK111595-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32:412–419. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 37.Wiley C.A., Lopresti B.J., Venneti S., Price J., Klunk W.E., DeKosky S.T., Mathis C.A. Carbon 11-labeled Pittsburgh Compound B and carbon 11-labeled (R)-PK11195 positron emission tomographic imaging in Alzheimer disease. Arch Neurol. 2009;66:60–67. doi: 10.1001/archneurol.2008.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okello A., Edison P., Archer H.A., Turkheimer F.E., Kennedy J., Bullock R., Walker Z., Kennedy A., Fox N., Rossor M., Brooks D.J. Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology. 2009;72:56–62. doi: 10.1212/01.wnl.0000338622.27876.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maeda J., Zhang M.R., Okauchi T., Ji B., Ono M., Hattori S., Kumata K., Iwata N., Saido T.C., Trojanowski J.Q., Lee V.M.-Y., Staufenbiel M., Tomiyama T., Mori H., Fukumura T., Suhara T., Higuchi M. In vivo positron emission tomographic imaging of glial responses to amyloid-β and tau pathologies in mouse models of Alzheimer's disease and related disorders. J Neurosci. 2011;31:4720–4730. doi: 10.1523/JNEUROSCI.3076-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilcock D.M., DiCarlo G., Henderson D., Jackson J., Clarke K., Ugen K.E., Gordon M.N., Morgan D. Intracranially administered anti-Aβ antibodies reduce β-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koenigsknecht-Talboo J., Meyer-Luehmann M., Parsadanian M., García-Alloza M., Finn M.B., Hyman B.T., Bacskai B.J., Holtzman D.M. Rapid microglial response around amyloid pathology after systemic anti-Aβ antibody administration in PDAPP mice. J Neurosci. 2008;28:14156–14164. doi: 10.1523/JNEUROSCI.4147-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Etminan M., Gill S., Samii A. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer's disease: a systematic review and meta-analysis of observational studies. BMJ. 2003;327:128–132. doi: 10.1136/bmj.327.7407.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayden K.M., Zandi P.P., Kachaturian A.S., Szekely C.A., Fotuhi M., Norton M.C., Tschanz J.T., Pieper C.F., Corcoran C., Lyketsos C.G., Breitner J.C., Welsh-Bohmer K.A., Cache County Investigators Does NSAID use modify cognitive trajectories in the elderly?: the Cache County Study. Neurology. 2007;69:275–282. doi: 10.1212/01.wnl.0000265223.25679.2a. [DOI] [PubMed] [Google Scholar]
- 44.Sastre M., Gentleman S.M. NSAIDs: how they work and their prospects as therapeutics in Alzheimer's disease. Front Aging Neurosci. 2010;2:1–6. doi: 10.3389/fnagi.2010.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arvanitakis Z., Grodstein F., Bienias J.L., Schneider J.A., Wilson R.S., Kelly J.F., Evans D.A., Bennett D.A. Relation of NSAIDs to incident AD, change in cognitive function, and AD pathology. Neurology. 2008;70:2219–2225. doi: 10.1212/01.wnl.0000313813.48505.86. [DOI] [PubMed] [Google Scholar]
- 46.Breitner J.C., Haneuse S.J., Walker R., Dublin S., Crane P.K., Gray S.L., Larson E.B. Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology. 2009;72:1899–1905. doi: 10.1212/WNL.0b013e3181a18691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sonnen J.A., Larson E.B., Walker R.L., Haneuse S., Crane P.K., Gray S.L., Breitner J.C., Montine T.J. Nonsteroidal anti-inflammatory drugs are associated with increased neuritic plaques. Neurology. 2010;75:1203–1210. doi: 10.1212/WNL.0b013e3181f52db1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Norris C.M., Kadish I., Blalock E.M., Chen K.C., Thibault V., Porter N.M., Landfield P.W., Kraner S.D. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer's models. J Neurosci. 2005;25:4649–4658. doi: 10.1523/JNEUROSCI.0365-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nagamoto-Combs K., Combs C.K. Microgial phenotype is regulated by activity of the transcription factor NFAT (nuclear factor of activated T cells) J Neurosci. 2010;30:9641–9646. doi: 10.1523/JNEUROSCI.0828-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abdul H.M., Sama M.A., Furman J.L., Mathis D.M., Beckett T.L., Weidner A.M., Patel E.S., Baig I., Murphy M.P., LeVine H., 3rd, Kraner S.D., Norris C.M. Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci. 2009;29:12957–12969. doi: 10.1523/JNEUROSCI.1064-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu H.Y., Hudry E., Hashimoto T., Kuchibhotla K., Rozkalne A., Fan Z., Spires-Jones T., Xie H., Arbel-Ornath M., Grosskreutz C.L., Bacskai B.J., Hyman B.T. Amyloid-β induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dineley K.T., Kayed R., Neugebauer V., Fu Y., Zhang W., Reese L.C., Taglialatela G. Amyloid-beta oligomers impair fear conditioned memory in a calcineurin-dependent fashion in mice. J Neurosci Res. 2010;88:2923–2932. doi: 10.1002/jnr.22445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rozkalne A., Hyman B.T., Spires-Jones T.L. Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol Dis. 2011;41:650–654. doi: 10.1016/j.nbd.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The subset of patients with AD used to investigate the spatial relationships between glial responses and plaques and NFTs is representative of the entire AD cohort. The following are shown: progression of cortical thickness (A), amyloid burden (B), total number of plaques (C), astrocytosis (D), and microgliosis (E) in the subset of 40 AD cases. Figures 1, 2, and 3 and Table 3 have comparisons.
There was no significant effect of APOEε4 on the progression of glial responses. Astrocytosis (A) and microgliosis (B) were more severe in patients with AD than in age-matched controls without dementia, but no significant difference was observed between APOEε4 carriers (n = 59) and noncarriers (n = 32) within the AD group (one-way analysis of variance Kruskal-Wallis test: P < 0.0001 and P = 0.0004, respectively; Dunn's multiple comparison test: **P < 0.001, ***P < 0.0001). The progression of astrocytosis (C) and microgliosis (D) did not substantially differ between APOEε4 carriers and noncarriers.