

Abstract
Gestational choriocarcinoma is a malignant trophoblastic tumor. The development of novel molecular-targeted therapies is needed to reduce the toxicity of current multiagent chemotherapy and to treat successfully the chemoresistant cases. The molecular mechanisms underlying choriocarcinoma tumorigenesis remain uncharacterized, however, and appropriate choriocarcinoma animal models have not yet been developed. In this study, we established a choriocarcinoma model by inoculating mice with induced-choriocarcinoma cell–1 (iC3-1) cells, generated from HTR8/SVneo human trophoblastic cells retrovirally transduced with activated H-RAS (HRASV12). The iC3-1 cells exhibited constitutive activation of the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways and developed into lethal tumors in all inoculated mice. Histopathological analysis revealed that the tumors consisted of two distinct types of cells, reminiscent of syncytiotrophoblasts and cytotrophoblasts, as seen in the human choriocarcinoma. The tumors expressed HLA-G and cytokeratin (trophoblast markers) and hCG (a choriocarcinoma marker). Comparative analysis of gene expression profiles between iC3-1 cells and parental HTR8/SVneo cells revealed that iC3-1 cells expressed matrix metalloproteinases, epithelial–mesenchymal transition-related genes, and SOX3 at higher levels than parental trophoblastic cells. Administration of SOX3-specific short-hairpin RNA decreased SOX3 expression and attenuated the tumorigenic activity of iC3-1 cells, suggesting that SOX3 overexpression might be critically involved in the pathogenesis of choriocarcinoma. Our murine model represents a potent new tool for studying the pathogenesis and treatment of choriocarcinoma.
Gestational choriocarcinoma is a malignant trophoblastic tumor that occurs in the uterus after any types of pregnancy but is most often associated with a complete hydatidiform mole.1 It is a rare disease with an estimated incidence of approximately 1 in 20,000 to 40,000 pregnancies. Geographical differences in its incidence have also been reported: 2 to 7 in 100,000 pregnancies in North America and Europe, and 5 to 202 in 100,000 pregnancies in Asia.2,3 Gestational trophoblastic tumors, including choriocarcinoma, are among the most highly responsive cancers to chemotherapy, whereas nongestational choriocarcinoma is generally more refractory to chemotherapy.4,5 Most patients with nonmetastatic or low-risk metastatic gestational choriocarcinoma are successfully treated with single-agent chemotherapy.6 Metastatic cases are also curable by multiagent chemotherapy with or without adjuvant surgical procedures or radiation therapy.7–9 However, multiagent chemotherapies, such as EMA-CO (etoposide, methotrexate, actinomycin D, vincristine, and cyclophosphamide), induce considerable toxicity-related adverse effects in patients. Furthermore, approximately 7% of low-risk [International Federation of Gynecology and Obstetrics (FIGO) stages I, score <7]10 patients and 27% of high-risk (FIGO stages II to IV, score > or = 7)10 patients will have an incomplete response to first-line single-agent or multiagent chemotherapy, and will relapse from remission.8,11 Thus, it is still necessary to develop novel, specific, and less toxic therapeutic approaches for gestational choriocarcinoma.
In addition to being rare, gestational choriocarcinoma is mainly treated with chemotherapy on the basis of human chorionic gonadotropin (hCG) levels rather than surgery, making it difficult to obtain enough clinical samples for investigating the molecular pathogenesis of this disease. Furthermore, the absence of an appropriate animal model is a critical deficit for the development of novel therapeutic approaches.
Gestational choriocarcinoma of the uterus is always associated with either normal pregnancy or conditions akin to pregnancy, such as hydatidiform mole, spontaneous abortion, or ectopic pregnancy. During pregnancy, to achieve adequate fetal–maternal exchange of molecules, extravillous trophoblast cells show considerable invasive ability into uterine decidua and into the inner third of the myometrium—a process that is highly regulated.12 Because of their invasive ability during the physiological process of pregnancy, extravillous trophoblast cells aberrantly retained in the uterus after pregnancy might be susceptible to oncogenic events leading to gestational choriocarcinoma. Thus, we hypothesized that extravillous trophoblast cells are the putative cells of origin of gestational choriocarcinoma.
The precise molecular mechanisms leading to the development of gestational choriocarcinoma remain to be elucidated. However, previous studies have reported that the expression of RAS GTPase-activating protein, a major inactivator of RAS activity, was decreased in gestational choriocarcinoma compared with normal placenta and noninvasive hydatidiform mole.13 Furthermore, activated RAS signaling can induce choriocarcinoma from murine embryonic stem cells via differentiation into trophectodermal lineages,14 suggesting that RAS signaling plays a role in choriocarcinoma tumorigenesis.
In this study, we have achieved the development of a choriocarcinoma mouse model by overexpressing oncogenic HRASV12 in the immortalized human extravillous trophoblast cell line HTR8/SVneo. Importantly, the tumors contained the two typical trophoblast cell types, syncytiotrophoblasts and cytotrophoblasts, and are therefore histologically similar to human choriocarcinoma. Gene expression study revealed that matrix metalloproteinase (MMP) family genes, epithelial–mesenchymal transition (EMT)–related genes, and SOX family genes were upregulated in induced-choriocarcinoma cell–1 (iC3-1) cells, established as induced-choriocarcinoma cells from the tumor. In particular, SOX3 down-regulation by SOX3-specific short-hairpin RNA (shRNA) markedly attenuated the tumorigenic activity of iC3-1 cells.
Materials and Methods
Cell Culture
HTR8/SVneo, a human extravillous trophoblast cell line immortalized using SV40 T antigen (SV40Tag), was kindly provided by Dr. C. H. Graham (Queen's University, Kingston, ON, Canada).15 Cells were cultured in Dulbecco's modified Eagle's medium (Gibco-BRL, Burlington, ON) supplemented with 10% heat-inactivated fetal calf serum, penicillin (100 U/mL) and streptomycin (100 U/mL) at 37°C in a 5% CO2 atmosphere. Choriocarcinoma cell lines JEG3 and JAR were obtained from American Type Culture Collection (ATCC; Rockville, MD). Both cell lines were cultured in RPMI media with 5% heat-inactivated fetal calf serum and penicillin (100 U/mL) and streptomycin (100 U/mL) at 37°C in a 5% CO2 atmosphere. The colon adenocarcinoma cell line HCT116 was also obtained from ATCC. Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal calf serum, penicillin (100 U/mL), and streptomycin (100 U/mL) at 37°C in a 5% CO2 atmosphere.
Retroviral Gene Transfer
The pMX retroviral vector and Plat-A packaging cells were generously provided by Dr. Toshio Kitamura (Institute of Medical Science, University of Tokyo, Tokyo, Japan). The HRASV12 gene (kindly provided by Dr. Pier P. Pandolfi, Harvard Medical School, Boston, MA) was cloned into the retroviral plasmid pMX-IRES-EGFP. Both pMX-HRASV12-IRES-EGFP and pMX-IRES-EGFP were transfected into Plat-A cells using FuGENE HD Reagent (Roche Applied Science, Mannheim, Germany), as previously described.16 Twenty-four hours after transfection, the supernatant was collected, designated the first virus-containing supernatant, and replaced with new medium. After another 24 hours, the new supernatant was collected and designated as the second virus-containing supernatant. To further increase infection efficiency, retroviruses in the virus-containing supernatants were concentrated by a medium-speed centrifuge (8000 × g at 4°C for 16 hours), filtered through a 45-μm pore-sized filter, and supplemented with 8 μg/mL polybrene (hexadimethrine bromide; H9268, Sigma-Aldrich, St. Louis, MO). The concentrated first virus-containing supernatant was then transferred to the cultured HTR8/SVneo cells and incubated overnight. Twenty-four hours after transduction, the virus-containing medium was replaced with the concentrated second virus-containing supernatant.
Inoculation of HTR8/SVneo/EGFP and HTR8/SVneo/HRASV12
HTR8/SVneo cells infected with enhanced green fluorescent protein (EGFP)-expressing retrovirus (HTR8/SVneo/EGFP cells); those with HRASV12-expressing retrovirus (HTR8/SVneo/HRASV12 cells) were grown to 80% confluence, detached with trypsin/EDTA, and resuspended in medium. EGFP-positive infected cells were sorted by FACS Vantage SE (BD, Franklin Lakes, NJ), and 5 × 106 cells in a volume of 0.2 mL were injected into the subcutaneous or intraperitoneal space of 8-week-old BALB/c slc-nu/nu female nude mice (Sankyo Labo Service Corporation, Tokyo, Japan). Each inoculation was performed in five mice. All animal care and procedures were performed in accordance with the guidelines of Keio University. Nude mice were maintained under specific pathogen-free conditions at constant temperature and humidity. Every third day, the tumor size was measured using calipers.
Senescence-Associated β-Galactosidase
Senescence-associated β-galactosidase (SA β-gal) activity was evaluated using the Senescence Cells Histochemical Staining Kit (CS0030; Sigma-Aldrich) according to the manufacturer's instructions.
Cytology, Histology, and Immunohistochemistry
Cytological specimens were obtained from formed tumors by cotton swabbing and were subjected to Papanicolaou staining. For immunohistochemistry, formalin-fixed, paraffin-embedded, 4-μm-thick serial tissue sections were immunostained using the avidin–biotin peroxidase complex method (Vectastain ABC Elite kit, Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. Sections were stained with antibodies against HLA-G (ab7759, Abcam, Cambridge, UK), Cytokeratin (ab27988, Abcam), hCG (N1534, DAKO, Glostrup, Denmark), hPL (A0137, DAKO), E-cadherin (334000, ZYMED, San Francisco, CA) and Vimentin (N1521, DAKO), and were counterstained with hematoxylin to visualize the cell nuclei.
Establishment and Implantation of iC3-1 Cells
The cells used to establish the induced-choriocarcinoma cell line, designated iC3-1, were collected by mixing trypsin with tissue from a single HTR8/SVneo/HRASV12-derived tumor. The tumor was dissected from mice under anesthesia, minced into fine fragments with trypsin, and transferred to a culture dish after passing through a 45-μm filter. Culture conditions were the same as those for the HTR8/SVneo cells. Each inoculation of iC3-1 into nude mice was performed into five mice as described above.
Immunoblotting
Lysates from HTR8/SVneo/EGFP and iC3-1 cells were electrophoresed and transferred to a polyvinylidene fluoride membrane (Immobilon-P; Millipore, Bedford, MA). The membranes were incubated with primary antibodies at 4°C overnight and with secondary antibodies at room temperature for 1 hour. Peroxidase activity was detected by Western Lightning chemiluminescence reagents (Perkin Elmer, Boston, MA). Primary antibody sources and concentrations used are as follows: H-RAS, 1 μg/mL (sc-520; Santa Cruz Biotechnology, Santa Cruz, CA); p44/42 MAPK, 19 ng/mL (#9102; Cell Signaling Technology, Danvers, MA); Phospho-p44/42 MAPK, 123 ng/mL (#9101; Cell Signaling Technology); Akt, 38 ng/mL (#9272; Cell Signaling Technology), Phospho-Akt, 37.5 ng/mL (#4060; Cell Signaling Technology), and α-TUBULIN, 5 μg/mL (T5168; Sigma-Aldrich).
Microarray Analysis Comparing iC3-1 and HTR8/SVneo/EGFP
Gene expression profiles comparing iC3-1 and HTR8/SVneo/EGFP were performed using the Human Whole Genome Oligo Microarrays Kit (G4112F; Agilent, Santa Clara, CA) according to the manufacturer's instructions. In brief, total RNA from iC3-1 and HTR8/SVneo/EGFP was extracted using the Isogen (Nippon Gene, Tokyo, Japan) kit, according to the manufacturer's instructions. After resuspension in RNase-free water, 400 ng total RNA was amplified and labeled using the Agilent Low RNA Input Fluorescent Linear Amplification Kit, according to the manufacturer's protocol. Tumor RNA was labeled with Cy5, and Universal Human Reference RNA (Stratagene) was labeled with Cy3. Hybridization of tumor and reference RNA was performed following the Agilent users' manual protocol for oligonucleotide microarray hybridization. A total of 750 ng labeled cRNA per channel was mixed and hybridized onto an array from the 44k 60-mer Human Whole Genome Oligo Microarrays kit (G4112F) at 60°C and 4 rpm for 17 hours. The arrays were scanned using the Agilent dual-laser DNA microarray scanner (G2565A), and data were extracted using Agilent Feature Extraction Software.
Quantitative and Semiquantitative RT-PCR Analysis
Total RNA was extracted from the sorted tumor cells using Isogen (Nippon Gene, Tokyo, Japan) and was reverse-transcribed using a Prime Script RT-PCR kit (Takara Bio, Shiga, Japan). Quantitative RT-PCR was performed using the Thermal Cycler Dice Real Time System (Takara Bio) using SYBR Premix Ex Taq (Takara Bio). The PCR conditions were as follows: 95°C for 15 minutes, 35 cycles of 95°C for 15 seconds and 64°C for 30 s, and a single cycle of 95°C for 15 s, 60°C for 30 seconds, and 95°C for 15 seconds. The PCR amplification was followed by dissociation-curve analysis to confirm specificity. Relative mRNA levels were calculated by normalization of the cycle threshold (Ct) values of the target gene to those of the reference gene (ACTB). Data are presented as mean ± SD of triplicates. The primer sets for the RT-PCR are listed in Table 1.
Table 1.
Primers Used for RT-PCR and Quantitative RT-PCR
| Gene symbol | Primer direction | Primer sequence |
|---|---|---|
| RT-PCR | ||
| HLA-G | Sense | 5′-CTGACCCTGACCGAGACCT-3′ |
| Antisense | 5′-CTCGCTCTGGTTGTAGTAGCC-3′ | |
| KRT | Sense | 5′-TGGTCACCACACAGTCTGCT-3′ |
| Antisense | 5′-CCAAGGCATCACCAAGATTA-3′ | |
| GAPDH | Sense | 5′-CCTCCCGCTTCGCTCTCT-3′ |
| Antisense | 5′-TGGCGACGCAAAAGAAGAT-3′ | |
| CGB | Sense | 5′-TCGGGTCACGGCCTCCT-3′ |
| Antisense | 5′-CAGCACGCGGGTCATGGT-3′ | |
| Quantitative RT-PCR | ||
| CDH1 | Forward | 5′-ATTCTGATTCTGCTGCTCTTG-3′ |
| (E-CADHERIN) | Reverse | 5′-AGTAGTCATAGTCCTGGTCTT-3′ |
| VIM | Forward | 5′-TTCCAAACTTTTCCTCCCTGAACC-3′ |
| (VIMENTIN) | Reverse | 5′-TCAAGGTCATCGTGATGCTGAG-3′ |
| CDH2 | Forward | 5′-CTCCTATGAGTGGAACAGGAACG-3′ |
| (N-CADHERIN) | Reverse | 5′-TTGGATCAATGTCATAATCAAGTGCTGTA-3′ |
| TWIST1 | Forward | 5′-CGGGAGTCCGCAGTCTTA-3′ |
| (TWIST) | Reverse | 5′-TGAATCTTGCTCAGCTTGTC-3′ |
| SNAI1 | Forward | 5′-GAGGCGGTGGCAGACTAG-3′ |
| (SNAIL) | Reverse | 5′-GACACATCGGTCAGACCAG-3′ |
| SNAI2 | Forward | 5′-CATGCCTGTCATACCACAAC-3′ |
| (SLUG) | Reverse | 5′-GGTGTCAGATGGAGGAGGG-3′ |
| SOX1 | Forward | 5′-GCCCAGGAGAACCCCAAG-3′ |
| Reverse | 5′-CGTCTTGGTCTTGCGGC-3′ | |
| SOX2 | Forward | 5′-CAGACTTCACATGTCCCAGC-3′ |
| Reverse | 5′-GGCAGTGTGCCGTTAATGG-3′ | |
| SOX3 | Forward | 5′-CCAGTCGTGTCGCGTCTGT-3′ |
| Reverse | 5′-GCACACCTGGCTATAAATTAACATTG-3′ | |
| ACTB | Forward | 5′-GGGTCTGGACCTGGCTGGCCGGGACTCG-3′ |
| (β-ACTIN) | Reverse | 5′-GGGCCGCCGATCCACACGGAGTACTTGC-3′ |
RNAi for SOX3
To obtain stable shRNA oligo-mediated gene silencing, the cells were transfected using MISSION Lentiviral Transduction Particles (Sigma-Aldrich), MISSION pLKO.1-puro Control Transduction Particles (Sigma-Aldrich), and MISSION shSOX3 Lentiviral Transduction Particles (TRCN0000016073, Sigma-Aldrich). Transduction into iC3-1 cells was performed according to the manufacturer's instructions. Briefly, iC3-1 cells were harvested at 50% confluency 24 hours before transduction. After the addition of 8 μg/mL hexadimethrine bromide, the appropriate amount of viral particles at a suitable multiplicity of infection (MOI) was added, the plate was swirled gently to mix, and the cell–viral particle mixture was incubated at 37°C overnight. The viral particle–containing medium was removed and replaced with fresh culture medium the next day. The following day, this medium was replaced with medium containing 1 μg/mL puromycin for the selection of transduced cells to optimize stable expression. The puromycin-containing medium was replaced every 3 days until resistant colonies could be identified 12 days after selection. The cells cultured under these conditions were tested for SOX3 knockdown by quantitative RT-PCR. Two individual experiments were performed, each in triplicate.
Statistical Analysis
Data are presented as mean ± SD, and were analyzed using the unpaired Student's t-test. Survival curves were analyzed by the log-rank test. A value of P < 0.05 was considered statistically significant.
Results
HRASV12-Transformed HTR8/SVneo Trophoblast Cells
Primary normal trophoblast cells are known to easily differentiate and, therefore, are difficult to keep in an immature state in vitro.17,18 As the target cells for the transformation, HTR8/SVneo cells that were originally generated from human first-trimester placental tissue and immortalized by SV40Tag transduction were chosen.15 Previous studies have suggested that activated RAS signaling may play a role in choriocarcinoma tumorigenesis.13,14 Therefore, we used the oncogenic H-RAS (HRASV12) for the induction of choriocarcinoma from immortalized human trophoblast cells.19 We introduced oncogenic HRASV12 into HTR8/SVneo cells by retroviral gene transfer, and sorted the infected cells using EGFP. Parental HTR8/SVneo cells and GFP-transduced HTR8/SVneo/EGFP cells exhibited epithelial-like morphology, whereas HTR8/SVneo/HRASV12 cells showed a mixture of smaller cells with spindle-like shapes and larger cells with flattened shapes (Figure 1 A). We confirmed that HTR8/SVneo/HRASV12 cells express both EGFP and HRASV12 (Figure 1B). Notably, HTR8/SVneo/HRASV12 cells exhibited slower proliferation than control cells (Figure 1C). Previous reports have indicated that the overexpression of oncogenic RAS induces premature senescence in normal cells in vitro, although it confers tumorigenic activity to normal cells in vivo.20,21 Indeed, the percentage of senescence-associated β-galactosidase–positive cells was higher in HTR8/SVneo/HRASV12 than in HTR8/SVneo/EGFP (Figure 1, D and E).
Figure 1.
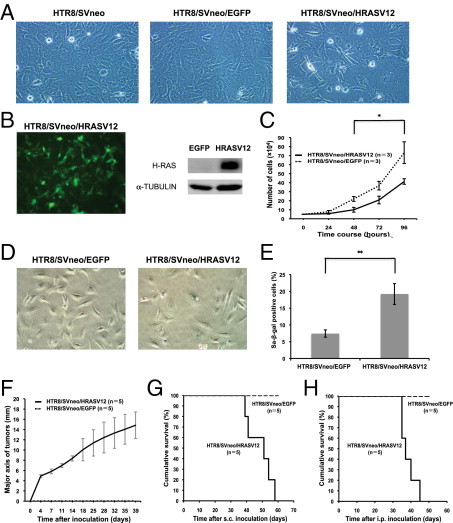
Cell character and tumorigenic activity of HRASV12-transformed HTR8/SVneo trophoblast cells. A: Cell morphology of HTR8/SVneo (left, ×100 magnification), HTR8/SVneo/EGFP (middle, ×100 magnification), and HTR8/SVneo/HRASV12 (right, ×100 magnification). B: Confirmation of the forced expression of EGFP (left, ×100 magnification) and expression of HRASV12 by Western blot in HTR8/SVneo/HRASV12 (HRASV12) compared with HTR8/SVneo/EGFP (EGFP) (right). C: Cell growth of HTR8/SVneo/EGFP and HTR8/SVneo/HRASV12 (*P < 0.05). D: Cellular senescence was evaluated by senescence-associated β-galactosidase (SA β-gal) staining (left: HTR8/SVneo/EGFP; right: HTR8/SVneo/HRASV12). E: In each of five different fields, cells (>100) were scored to determine the percentage of SA β-gal–positive cells (**P < 0.01). F: Measurement of the major axis of tumors derived from HTR8/SVneo/HRASV12 by subcutaneous injection into nude mice (mm). HTR8/SVneo/EGFP did not form any tumors. G, H: Survival curves for the nude mice injected with HTR8/SVneo/HRASV12 cells or HTR8/SVneo/EGFP cells subcutaneously (G) or intraperitoneally (H).
To evaluate in vivo tumorigenic ability, we subcutaneously and intraperitoneally injected the FACS-sorted transfected cells into nude mice. Trophoblast cells immortalized by SV40Tag, such as HTR8/SVneo cells, were reported to be nontumorigenic.22 In fact, subcutaneously or intraperitoneally injected HTR8/SVneo/EGFP cells did not develop tumors over the course of 120 days of observation (n = 5) (Figure 1, F–H). In contrast, HTR8/SVneo/HRASV12-derived tumors grew rapidly, and inoculated mice died at approximately 51 days (median; subcutaneous injection group; n = 5) and 37 days (median; intraperitoneal injection group; n = 5) after inoculation (Figure 1, F–H).
Collectively, the overexpression of HRASV12-transformed HTR8/SVneo trophoblast cells and conferred tumorigenic activity on them in vivo.
HTR8/SVneo/HRASV12-Derived Tumors Mimic Human Choriocarcinoma
We microscopically examined the established tumors to determine their histological type. The tumors established by subcutaneous injection were hemorrhagic and centrally necrotic (Figure 2A). The cytology of cells obtained from the tumor stump showed typical abnormal Langhans-type trophoblast cells, with extensive large cells and hyperstained nuclei (Figure 2B). Importantly, the tumors consisted of an admixture of two distinct types of cells, reminiscent of multinucleated syncytiotrophoblasts and mononuclear cytotrophoblasts (Figure 2, C and I). Most of the tumor cells were GFP positive and rarely contained tumor stroma or vessels (Figure 2D). The viable tumor cells were located at the periphery of hemorrhagic foci (Figure 2E). Tumors were also positive for the trophoblast markers human leukocyte antigen–G (HLA-G) and cytokeratin (Figure 2F; see also Supplemental Figure S1, A and C at http://ajp.amjpathol.org).23,24 It is sometimes difficult to distinguish choriocarcinoma from other trophoblastic tumors, such as placental site trophoblastic tumor (PSTT) in the clinical setting. To further define the histological type of the established tumors, we evaluated the expression of hCG and human placental lactogen (hPL), which are the differential diagnostic markers for choriocarcinoma and PSTT, respectively.25 Notably, the tumor cells exhibited immunoreactivity to hCG, but they did not express hPL (Figure 2G; see also Supplemental Figure S1, B, C, and D at http://ajp.amjpathol.org). These cytological and histopathological findings were compatible with human choriocarcinoma.
Figure 2.
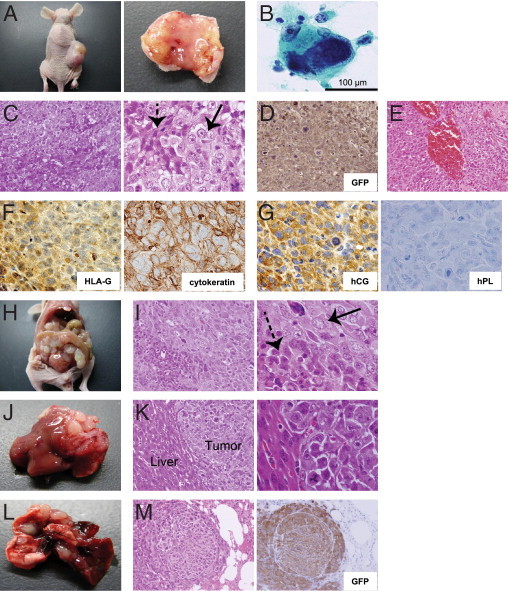
Histopathological findings of HTR8/SVneo/HRASV12-derived tumors mimicking human choriocarcinoma. A: Tumor formation derived from HTR8/SVneo/HRASV12 cells by subcutaneous injection into a nude mouse (left). The tumors were hemorrhagic and centrally necrotic (right). B: A tumor stamp specimen visualized with Papanicolaou staining reveals a marked large cell with hyperstained nucleus. C: H&E staining of tumors derived from HTR8/SVneo/HRASV12 cells by subcutaneous injections. The characteristic two-cell pattern, consisting of multinucleated syncytiotrophoblasts (dotted arrow) and mononuclear cytotrophoblasts (arrow), is shown (left, ×200 magnification; right, ×600 magnification). D: Most of the tumor cells were GFP-positive and rarely contained tumor stroma or vessels (GFP immunohistochemistry, ×200 magnification). E: Viable tumor cells were located at the periphery of hemorrhagic foci (H&E staining, ×200 magnification). F: Immunohistochemistry revealed that tumors expressed HLA-G (left, ×600 magnification) and cytokeratin (right, ×600 magnification). G: Immunohistochemistry revealed that tumors expressed hCG (left, ×600 magnification), but not hPL (right, ×600 magnification). H: Tumor formation from intraperitoneally injected cells. I: Intraperitoneally injected cells also exhibited the characteristic two-cell pattern consisting of multinucleated syncytiotrophoblasts (dotted arrow) and mononuclear cytotrophoblasts (arrow) (left, ×200 magnification; right, ×600 magnification). J: Tumors generated from intraperitoneally injected cells invading the liver. K: H&E staining of the tumor depicted in J (left, ×200 magnification; right, ×600 magnification). L: Tumor development in the lung by intravenous injection of HTR8/SVneo/HRASV12 cells. M: H&E staining of cells from the tumor depicted in L (left, ×200 magnification; right, GFP immunohistochemistry, ×200 magnification).
The intraperitoneally injected cells also developed markedly invasive tumors (Figure 2, H–K). Furthermore, we confirmed tumor formation in the lung by injection from the tail vein (Figure 2, L and M).
Collectively, these findings suggested that HTR8/SVneo/HRASV12 cells developed into tumors with characteristics similar to those of human choriocarcinoma in vivo.
Establishment of iC3-1 Cell Line
In addition to tumorigenic cells, the HTR8/SVneo/HRASV12 cells included a cell population undergoing senescence in vitro (Figure 1, D and E). Therefore, we established a subline, termed induced-choriocarcinoma cell–1 (iC3-1), from an HTR8/SVneo/HRASV12-derived subcutaneous tumor to endow the cells with tumor-initiating activity. The iC3-1 cells were significantly smaller than HTR8/SVneo/EGFP cells (Figure 3A). RT-PCR analysis revealed that both iC3-1 cells and HTR8/SVneo/EGFP cells expressed HLA-G and KRT as trophoblast markers, and CGB as choriocarcinoma marker (Figure 3B), suggesting that they originated from trophoblast cells and mimicked choriocarcinoma. Another choriocarcinoma cell line, JEG3, also expressed these three genes, whereas the HLA-G–negative choriocarcinoma cell line JAR26 did not express HLA-G, and colon cancer cell line HCT116 cells did not express CGB (Figure 3B). We observed enhanced activation of both mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways in iC3-1 compared with HTR8/SVneo/EGFP cells (Figure 3C). To examine the tumorigenic potential of iC3-1 cells in vivo, we inoculated them subcutaneously or intraperitoneally into nude mice. The iC3-1 cells rapidly generated lethal tumors, and the inoculated mice died at approximately 48 days (median; subcutaneous injection group; n = 5) and 29 days (median; intraperitoneal injection group; n = 5) after the inoculation (Figure 3, D and E). Histopathological examination revealed that the tumors contained two types of cells, reminiscent of multinucleated syncytiotrophoblasts and mononuclear cytotrophoblasts (Figure 3F) and were positive for HLA-G, cytokeratin, and hCG as is seen in human choriocarcinoma (Figure 3G; see also Supplemental Figure S1, E–G at http://ajp.amjpathol.org).
Figure 3.
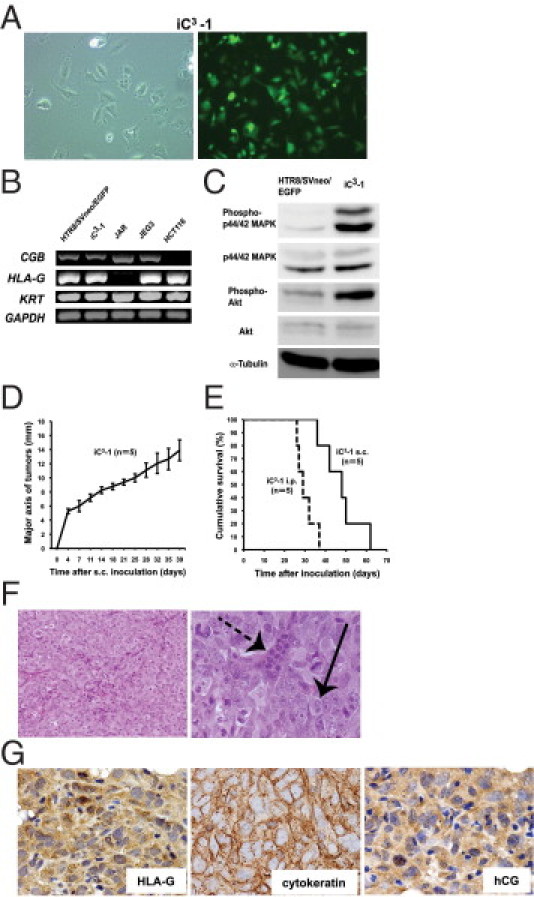
The characteristics and tumorigenic activity of iC3-1. A: Morphology of iC3-1 cells (left, ×100 magnification) and confirmation of the forced expression of EGFP (right, ×100 magnification). B: RT-PCR analysis of CGB, HLA-G, and KRT. GAPDH was used as internal control. C: Western blot analysis of Phospho-p44/42 MAPK, p44/42 MAPK, Phospho-Akt and Akt. α-TUBULIN was used as a loading control. D: Measurement of the major axis of tumors derived from iC3-1 by subcutaneous injection into nude mice (mm). E: Survival curves for the nude mice injected with iC3-1 subcutaneously or intraperitoneally. F: iC3-1 cells also formed tumors exhibiting the characteristic two-cell pattern consisting of multinucleated syncytiotrophoblasts (dotted arrow) and mononuclear cytotrophoblasts (arrow) (left, ×200 magnification; right, ×600 magnification). G: Immunohistochemistry revealed that tumors formed by iC3-1 cells also expressed HLA-G (left, ×600 magnification) and cytokeratin (middle, ×600 magnification) hCG (right, ×600 magnification).
Involvement of Extracellular Matrix Remodeling and Epithelial–Mesenchymal Transition in iC3-1 Cells
To gain insight into the critical events for the generation of choriocarcinoma, we compared the gene expression profiles of iC3-1 and HTR8/SVneo/EGFP cells. Among the 44,000 genes examined, 3094 genes were upregulated and 3126 genes were down-regulated in iC3-1 cells compared with HTR8/SVneo/EGFP cells. Among these, the most upregulated gene, IL13RA2, was previously reported as a novel epithelial–mesenchymal transition (EMT)–associated gene (Table 2).27 In contrast, the most down-regulated gene, PEG1/MEST (paternally expressed gene 1/mesoderm-specific transcript), was reported to be involved in malignant transformation (Table 2).28–30 The physiological invasion into myometrium by extravillous trophoblast cells is known to be supported by the extracellular matrix (ECM) and matrix metalloproteinases (MMPs), and negatively regulated by tissue inhibitor of metalloproteinase (TIMP).31,32 In iC3-1 cells, MMPs were significantly upregulated and TIMP2 was significantly down-regulated compared with nontumorigenic HTR8/SVneo/EGFP cells (Table 2).
Table 2.
Genes Identified through Gene Expression Profile Analysis of iC3-1 and HTR8/SVneo/EGFP Cells
| No. | GenBank no. | Gene symbol | Fold change | P value |
|---|---|---|---|---|
| Top 10 upregulated genes, of a total of 3094 upregulated genes | ||||
| 1 | NM_000640 | IL13RA2 | 1880.834 | 8.75 × 10-05 |
| 2 | AF020589 | COX6A1 | 1209.975 | 3.93 × 10-06 |
| 3 | NM_005328 | HAS2 | 664.8997 | 1.22 × 10-04 |
| 4 | NM_004049 | BCL2A1 | 620.046 | 1.21 × 10-04 |
| 5 | M14087 | UNKNOWN | 443.7551 | 1.32 × 10-05 |
| 6 | NM_007036 | ESM1 | 285.2013 | 2.87 × 10-05 |
| 7 | NM_181755 | HSD11B1 | 260.9386 | 1.61 × 10-04 |
| 8 | NM_003483 | HMGA2 | 232.0927 | 1.70 × 10-04 |
| 9 | NM_001704 | BAI3 | 210.0257 | 1.75 × 10-04 |
| 10 | NM_024717 | MCTP1 | 173.3222 | 1.88 × 10-04 |
| Top 10 downregulated genes, of a total of 3126 downregulated genes | ||||
| 1 | NM_002402 | MEST | 0.010001 | 4.16 × 10-05 |
| 2 | NM_004576 | PPP2R2B | 0.010003 | 2.89 × 10-04 |
| 3 | NM_002543 | OLR1 | 0.010007 | 5.71 × 10-03 |
| 4 | NM_005264 | GFRA1 | 0.010012 | 1.11 × 10-02 |
| 5 | NM_020311 | CXCR7 | 0.010015 | 4.50 × 10-03 |
| 6 | NM_000609 | CXCL12 | 0.010016 | 2.13 × 10-03 |
| 7 | M31157 | UNKNOWN | 0.010028 | 7.57 × 10-04 |
| 8 | AK025758 | UNKNOWN | 0.010245 | 8.12 × 10-04 |
| 9 | XM_211749 | UNKNOWN | 0.010967 | 1.63 × 10-02 |
| 10 | NM_002825 | PTN | 0.0111 | 1.73 × 10-03 |
| MMP genes found to be upregulated or downregulated | ||||
| NM_002421 | MMP1 | 92.33915 | 1.08 × 10-04 | |
| NM_002422 | MMP3 | 44.93045 | 3.43 × 10-04 | |
| NM_002425 | MMP10 | 7.441263 | 1.28 × 10-03 | |
| NM_002426 | MMP12 | 4.228032 | 1.40 × 10-02 | |
| NM_004530 | MMP2 | 3.457115 | 3.58 × 10-04 | |
| NM_004995 | MMP14 | 3.060105 | 4.00 × 10-03 | |
| AK 057217 | TIMP2 | 0.442898 | 8.40 × 10-04 | |
| SOX genes found to be upregulated or downregulated | ||||
| NM_005634 | SOX3 | 79.56935 | 2.60 × 10-04 | |
| NM_005986 | SOX1 | 3.31906 | 3.02 × 10-02 | |
| NM_003107 | SOX4 | 0.424586 | 4.82 × 10-03 | |
| NM_014587 | SOX8 | 0.409181 | 6.17 × 10-03 | |
| NM_152989 | SOX5 | 0.315828 | 1.31 × 10-02 | |
| NM_000346 | SOX9 | 0.261543 | 1.06 × 10-02 | |
EMT is considered a critical event observed when epithelial-type cancers become invasive and metastatic.33 Microarray analysis revealed that TWIST, a regulator of EMT,34 was upregulated 2.65-fold in iC3-1cells compared with HTR8/SVneo/EGFP cells. Furthermore, IL13RA2 and HAS2, which were recently suggested to be involved in EMT in human prostate cancer27 and during cardiac development,35 respectively, were also upregulated (Table 2). Whereas HTR8/SVneo/EGFP cells exhibited epithelial-like morphology, iC3-1cells displayed a mesenchymal/fibroblastic morphology with decreased cell–cell junctions that were indicative of an invasive phenotype (Figure 4A). Consistent with this morphological difference, quantitative RT-PCR revealed that the expression of CDH1 (E-CADHERIN) was down-regulated and that of VIM (VIMENTIN), CDH2 (N-CADHERIN), TWIST1 (TWIST), SNAI1 (SNAIL), and SNAI2 (SLUG) was upregulated in iC3-1 cells as compared with expression of these genes in HTR8/SVneo/EGFP cells (Figure 4B). Furthermore, the tumors showed low immunoreactivity with E-CADHERIN, whereas they were strongly positive for VIMENTIN (Figure 4C; see also Supplemental Figure S1, E and H at http://ajp.amjpathol.org).
Figure 4.
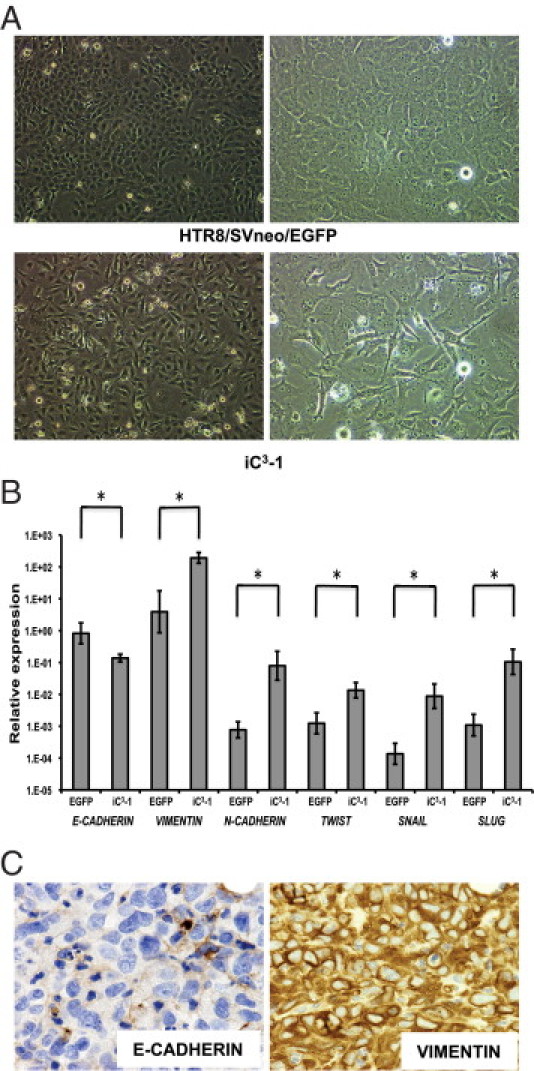
Epithelial–mesenchymal transition contributed to the tumorigenic activity of iC3-1 cells. A: Morphology of HTR8/SVneo/EGFP (upper) and iC3-1 (lower) (left, ×40 magnification: right, ×100 magnification). B: Quantitative RT-PCR analysis of EMT-related genes in iC3-1 compared with HTR8/SVneo/EGFP (*P < 0.05). C: Immunohistochemical staining of E-CADHERIN (left, ×600 magnification) and VIMENTIN (right, ×600 magnification) in iC3-1–derived tumors.
Collectively, these findings indicated that iC3-1 cells display characteristics of ECM remodeling and EMT, which may play roles in invasive and metastatic phenotypes.
Contribution of SOX3 to Choriocarcinoma Tumorigenesis
The SOX (SRY-related HMG box) gene family has been identified through the homology of the high mobility group (HMG) domain to the sex-determining lesion Y (SRY).36 Almost 30 SOX genes have been identified, including 20 that are present in both the mouse and the human genome.37 Expression of several SOX genes has been reported to be up- or down-regulated in many cancers.38 Notably, the gene expression profiling revealed that the expression of SOX3 was markedly upregulated in iC3-1 cells compared with HTR8/SVneo/EGFP cells (Table 2). SOX3 was reported to induce oncogenic transformation of chicken embryonic fibroblasts39 and to be identified as proto-oncogenes in murine T-cell lymphomas,40 suggesting that SOX3 may play a role in tumorigenesis. The role of the SOX family in the pathogenesis of choriocarcinoma has been scarcely clarified except for the frequent hypermethylation of SOX2.41 Thus, we focused on and investigated SOX3 in our tumor model.
Quantitative RT-PCR analysis revealed that the expression of SOX3 gene was upregulated in iC3-1 cells compared with that in HTR8/SVneo/EGFP or choriocarcinoma cell lines (Figure 5A). As the upregulation of SOX3 was detected both by microarray and quantitative RT-PCR analysis, we knocked down SOX3 expression by shRNA to investigate its contribution to choriocarcinoma tumorigenesis in vivo (Figure 5B). Cell growth of iC3-1/sh SOX3 cells in vitro was slower than that of iC3-1/sh control cells (Figure 5C). Subcutaneous injection of iC3-1/sh control cells developed tumors in all cases (n = 5/5), and these mice died at approximately 47 days after the injection (Figure 5, D and E). However, subcutaneous injection of iC3-1/sh SOX3 cells resulted in the suppression of tumor growth as compared with that in the control group, and the inoculated mice died at approximately 81 days (Figure 5, D and E). These findings suggest that SOX3 upregulation might be involved in choriocarcinoma tumorigenic activity.
Figure 5.
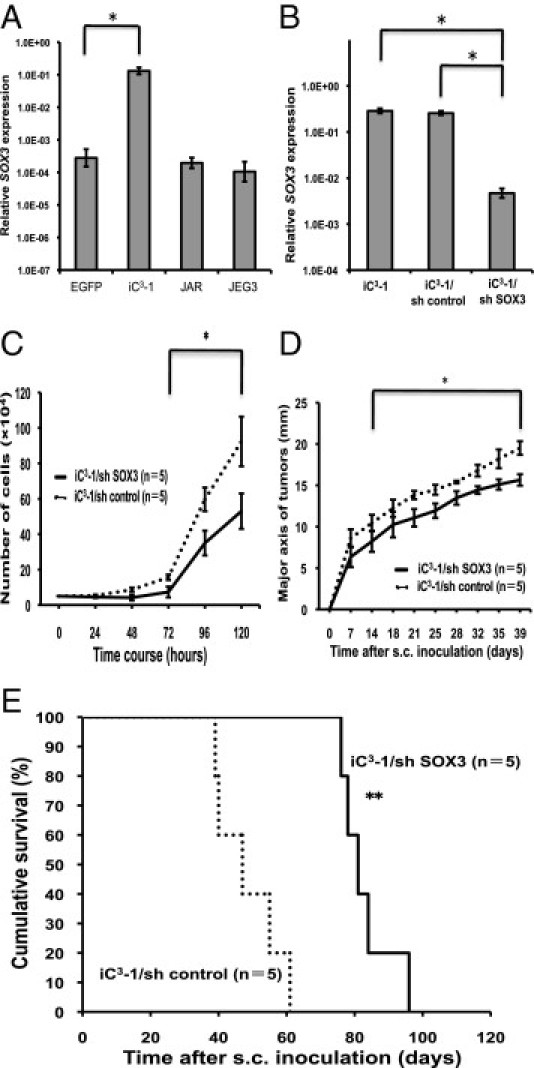
Knockdown of SOX3 attenuated the tumorigenic activity of iC3-1 cells. A: Quantitative RT-PCR showing significant upregulation of SOX3 expression in iC3-1 cells as compared with HTR8/SVneo/EGFP cells (*P < 0.05) and choriocarcinoma cell lines (JAR and JEG3). B: Quantitative RT-PCR showing significant reduction in SOX3 expression in iC3-1/sh SOX3 cells as compared with iC3-1/sh control cells (*P < 0.05). C: Proliferation of iC3-1/sh SOX3 and iC3-1/sh control cells (*P < 0.05). D: Measurement of the major axis of tumors derived from iC3-1/sh SOX3 and iC3-1/sh control cells by subcutaneous injection into nude mice (mm) (*P < 0.05). E: Survival curves for the nude mice subcutaneously injected with iC3-1/sh SOX3 and iC3-1/sh control cells (**P < 0.01).
Discussion
The mechanisms underlying gestational choriocarcinoma tumorigenesis have not yet been clarified. Investigation of gestational choriocarcinoma is difficult for several reasons, including an insufficient number of surgical specimens because of a preference for chemotherapy as treatment rather than surgery, as well as the general low incidence of this disease. Another source of difficulty is the lack of established animal models for choriocarcinoma. BeWo, JAR, and JEG3 are commonly used choriocarcinoma cell lines derived from human spontaneous choriocarcinoma. However, BeWo cells are nontumorigenic,22 and JAR and JEG3 cells do not generate tumors that are pathologically similar to human choriocarcinoma.22
Here, we have achieved the generation of a choriocarcinoma mouse model by overexpressing oncogenic HRASV12 in the human immortalized normal trophoblastic cell line, HTR8/SVneo. The established tumors exhibited the characteristic two-cell pattern seen in choriocarcinoma, consisting of syncytiotrophoblasts and cytotrophoblasts. In addition, they were positive for HLA-G, cytokeratin, and hCG immunoreactivity, but not for hPL immunoreactivity, which is similar to the profile of human choriocarcinoma.
In this study, we focused on human extravillous trophoblast cells as the cells of origin for human choriocarcinoma. Gestational choriocarcinoma of the uterus always occurs after pregnancy; thus, we speculate that the cell components that participate in pregnancy are involved in the tumorigenesis of gestational choriocarcinoma. During pregnancy, extravillous trophoblast cells show considerable invasive ability into the uterine decidua and the inner third of the myometrium.12 We could induce choriocarcinoma-like tumors from HTR8/SVneo immortalized extravillous trophoblast cells by transfection with oncogenic HRASV12, suggesting that extravillous trophoblast cells can serve as the cells of origin of choriocarcinoma. Furthermore, HRASV12 activates both the MAPK and PI3K pathways in HTR8/SVneo cells (Figure 3C). Therefore, the activation of MAPK and PI3K pathways may play a role in the induction of choriocarcinoma from immortalized normal extravillous trophoblast cells.
Cytotrophoblasts, syncytiotrophoblasts, and extravillous trophoblasts have been identified as primary types of human placental villous cells.42 Both syncytiotrophoblasts and extravillous trophoblasts are reported to be differentiated from cytotrophoblasts,42 and the widely used HTR8/SVneo line is derived from extravillous trophoblast cells.43–45 Intriguingly, the established tumors derived from HTR8/SVneo/HRASV12 contained the typical pattern of two distinctive cell types that are similar to cytotrophoblasts and syncytiotrophoblasts. Although further analysis will be needed to clarify the precise mechanisms underlying this, we speculate two possibilities: HTR8/SVneo cells may acquire bidirectional potential by HRASV12-derived transformation for differentiation or dedifferentiation into syncytiotrophoblast or cytotrophoblast cells respectively; and HTR8/SVneo cells might be reprogrammed during transformation by HRASV12 into a more immature state, eg, trophoblast stem-like cells, enabling them to differentiate into distinct cell lineages. The second idea is partially supported by our microarray data showing that CDX2, which is known as a trophoblast stem cell marker, was upregulated 3.04-fold in iC3-1 cells compared with HTR8/SVneo/EGFP cells.
Gestational choriocarcinoma generates epithelial-type tumors. However, given that it frequently metastasizes to other organs at an early stage, EMT might be induced in a subpopulation of choriocarcinoma tumor cells. This notion is supported by a previous report revealing that the expression of E-CADHERIN, an epithelial marker, was decreased in gestational choriocarcinoma compared with normal placental tissues.46 The present study showed that iC3-1 cells are highly invasive with activation of MAPK and PI3K pathways (Figures 2, J–M and 3C), and that EMT and ECM remodeling-associated genes were upregulated in iC3-1 (Figure 4B and Table 2). Therefore, iC3-1–derived tumors might represent a model mimicking the highly invasive type of choriocarcinoma that undergoes EMT.
PEG1/MEST was the most down-regulated gene in the microarray analysis in the present study. However, this gene is known to be upregulated in other tumors, and it also shows loss of imprinting.28–30 We performed imprinting analysis by RT-PCR to examine whether iC3-1 cells show loss of imprinting, as previously described.28 Interestingly, iC3-1 did not show loss of imprinting (data not shown), indicating that PEG1/MEST might play a role in choriocarcinoma that is different from that in other tumors.
SOX genes are known to play key roles in embryonic development as well as in sex determination,47 but they have been recently reported to also be involved in tumorigenesis.38 In this study, microarray analysis showed that SOX3 was the most and SOX1 was also significantly upregulated among the SOX genes in iC3-1 compared with HTR8/SVneo/EGFP (Table 2). SOX3 and SOX1 belong to the SOX B1 subgroup, which also includes SOX2, a gene known to be dysregulated in many cancers.48–52 Therefore, we confirmed the expression of the SOX B1 genes using quantitative RT-PCR. SOX3 and SOX1 were again found to be significantly upregulated, whereas SOX2 did not show a remarkable change in iC3-1 cells compared with HTR8/SVneo/EGFP cells (Figure 5A; see also Supplemental Figure S2, A and B, at http://ajp.amjpathol.org). SOX3 plays important roles in neuronal development, and its dysregulation is related to mental retardation53; in addition, it has been suggested that SOX3 may play a role in tumorigenesis.39,40 Thus, we explored the important possibility that the upregulation of SOX3 may be functionally involved in the tumorigenic activity of iC3-1 cells (Figure 5, C–E). Although further evaluations are needed, the present results indicate that SOX3 might also play a crucial role in choriocarcinoma.
We consider it ideal to use primary trophoblasts as cells of origin for establishing a choriocarcinoma model. However, it is known to be difficult to generate cancer cells directly from human primary cells54 whereas rodent primary cells were efficiently converted into tumorigenic cells by the coexpression of cooperating oncogenes. Immortalization was shown to be an essential step for human primary cells to become tumor cells.55,56 Therefore, we used HTR8/SVneo immortalized trophoblast cells for generating a choriocarcinoma model.
In the gene expression study, we compared iC3-1 with HTR8/SVneo, regarded as a control extravillous trophoblast cell. Although trophoblastic cell lines have been widely used as substitutes to study trophoblast function, it has been reported that the gene expression profile of primary trophoblast cells was considerably different from that of immortalized trophoblast cell lines, including HTR8/SVneo.57 Therefore, further analysis is required for identification of genes specifically altered in the induced choriocarcinoma in comparison to primary trophoblast cells.
This study identifies a possible cell of origin for choriocarcinoma, the required oncogenic event leading to tumorigenesis. We expect that our novel choriocarcinoma model will be useful for studying the pathogenesis of choriocarcinoma and for developing new therapeutic drugs.
Acknowledgments
We thank Charles H. Graham (Queen's University, Kingston, ON, Canada) for kindly donating the HTR8/SVneo cells; Pier P. Pandolfi (Harvard Medical School, Boston, MA) for the HRasV12 gene; Toshio Kitamura (University of Tokyo, Tokyo, Japan) for the Plat-A cells and the pMX-IRES-GFP retroviral vector; Tadashi Suzuki and Yumi Matsuzaki (Keio University, Tokyo, Japan) for excellent technical help in cell sorting; Ikuyo Ishimatsu for technical assistance in the histological analyses; Kuniko Arai for secretarial assistance; and Akiyoshi Noguchi for fundamental support.
Footnotes
Supported by a grant-in-aid to Keio University from the Global COE Program of the Ministry of Education, Culture, Sports, Science and Technology, Japan (Y.K.), and by grants from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (T.S. and H.S.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.05.019.
Supplementary data
Histopathological findings of serial sections of tumors that were derived from HTR8/SVneo/HRASV12 and iC3-1 cells compared with isotype controls. A: Immunohistochemistry as isotype controls (mouse IgG) in HTR8/SVneo/HRASV12–derived tumors (left, ×200 magnification; right, ×600 magnification). B: Immunohistochemistry as isotype controls (rabbit IgG) in HTR8/SVneo/HRASV12–derived tumors (left, ×200 magnification; right, ×600 magnification). C: Immunohistochemistry revealed that HTR8/SVneo/HRASV12-derived tumors expressed HLA-G (left, ×200 magnification), cytokeratin (middle, ×200 magnification) and hCG (right, ×200 magnification). The identical areas of a tumor are shown in A–C. D: Immunohistochemistry as a positive control for hPL staining in human placenta tissue (×200 magnification). E: Immunohistochemistry as isotype controls (mouse IgG) in iC3-1–derived tumors (left, ×200 magnification; right, ×600 magnification). F: Immunohistochemistry as isotype controls (rabbit IgG) in iC3-1–derived tumors (left, ×200 magnification; right, ×600 magnification). G: Immunohistochemistry revealed that iC3-1–derived tumors expressed HLA-G (left, ×200 magnification), cytokeratin (middle, ×200 magnification), and hCG (right, ×200 magnification). H: Immunohistochemical staining of E-CADHERIN (left, ×200 magnification) and VIMENTIN (right, ×200 magnification) in iC3-1–derived tumors. The identical areas of a tumor are shown in E–H.
SOXB1 expression in iC3-1 cells. A: Quantitative RT-PCR showing significant upregulation of SOX1 expression in iC3-1 cells as compared with HTR8/SVneo/EGFP cells (*P < 0.05) and two choriocarcinoma cell lines (JAR and JEG3). B: Quantitative RT-PCR showing SOX2 expression in iC3-1 cells as compared with HTR8/SVneo/EGFP cells and two choriocarcinoma cell lines (JAR and JEG3).
References
- 1.Seckl M.J., Fisher R.A., Salerno G., Rees H., Paradinas F.J., Foskett M., Newlands E.S. Choriocarcinoma and partial hydatidiform moles. Lancet. 2000;356:36–39. doi: 10.1016/S0140-6736(00)02432-6. [DOI] [PubMed] [Google Scholar]
- 2.Smith H.O. Gestational trophoblastic disease epidemiology and trends. Clin Obstet Gynecol. 2003;46:541–556. doi: 10.1097/00003081-200309000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Altieri A., Franceschi S., Ferlay J., Smith J., La Vecchia C. Epidemiology and aetiology of gestational trophoblastic diseases. Lancet Oncol. 2003;4:670–678. doi: 10.1016/s1470-2045(03)01245-2. [DOI] [PubMed] [Google Scholar]
- 4.Lurain J.R. Treatment of gestational trophoblastic tumors. Curr Treat Options Oncol. 2002;3:113–124. doi: 10.1007/s11864-002-0057-5. [DOI] [PubMed] [Google Scholar]
- 5.Fisher R.A., Savage P.M., MacDermott C., Hook J., Sebire N.J., Lindsay I., Seckl M.J. The impact of molecular genetic diagnosis on the management of women with hCG-producing malignancies. Gynecol Oncol. 2007;107:413–419. doi: 10.1016/j.ygyno.2007.07.081. [DOI] [PubMed] [Google Scholar]
- 6.Foulmann K., Guastalla J.P., Caminet N., Trillet-Lenoir V., Raudrant D., Golfier F., Schott A.M. What is the best protocol of single-agent methotrexate chemotherapy in non metastatic or low-risk metastatic gestational trophoblastic tumors?: A review of the evidence. Gynecol Oncol. 2006;102:103–110. doi: 10.1016/j.ygyno.2006.02.038. [DOI] [PubMed] [Google Scholar]
- 7.Cagayan M.S., Lu-Lasala L.R. Management of gestational trophoblastic neoplasia with metastasis to the central nervous system: a 12-year review at the Philippine General Hospital. J Reprod Med. 2006;51:785–792. [PubMed] [Google Scholar]
- 8.Lurain J.R., Singh D.K., Schink J.C. Primary treatment of metastatic high-risk gestational trophoblastic neoplasia with EMA-CO chemotherapy. J Reprod Med. 2006;51:767–772. [PubMed] [Google Scholar]
- 9.Soper J.T. Role of surgery and radiation therapy in the management of gestational trophoblastic disease. Best Pract Res Clin Obstet Gynaecol. 2003;17:943–957. doi: 10.1016/s1521-6934(03)00091-9. [DOI] [PubMed] [Google Scholar]
- 10.Ngan H.Y., Bender H., Benedet J.L., Jones H., Montruccoli G.C., Pecorelli S., FIGO Committee on Gynecologic Oncology Gestational trophoblastic neoplasia, FIGO 2000 staging and classification. Int J Gynaecol Obstet. 2003;83(Suppl 1):175–177. doi: 10.1016/s0020-7292(03)90120-2. [DOI] [PubMed] [Google Scholar]
- 11.Growdon W.B., Wolfberg A.J., Goldstein D.P., Feltmate C.M., Chinchilla M.E., Lieberman E.S., Berkowitz R.S. Low-risk gestational trophoblastic neoplasia and methotrexate resistance: predictors of response to treatment with actinomycin D and need for combination chemotherapy. J Reprod Med. 2010;55:279–284. [PubMed] [Google Scholar]
- 12.Huppertz B., Hemmings D., Renaud S.J., Bulmer J.N., Dash P., Chamley L.W. Extravillous trophoblast apoptosis—a workshop report. Placenta. 2005;26(Suppl A):S46–S48. doi: 10.1016/j.placenta.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Ståhle-Bäckdhal M., Inoue M., Zedenius J., Sandstedt B., DeMarco L., Flam F., Silfverswärd C., Andrade J., Friedman E. Decreased expression of Ras GTPase activating protein in human trophoblastic tumors. Am J Pathol. 1995;146:1073–1078. [PMC free article] [PubMed] [Google Scholar]
- 14.Lu C.W., Yabuuchi A., Chen L., Viswanathan S., Kim K., Daley G.Q. Ras-MAPK signaling promotes trophoectoderm formation from embryonic stem cells and mouse embryos. Nat Genet. 2008;40:921–926. doi: 10.1038/ng.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graham C.H., Hawley T.S., Hawley R.G., MacDougall J.R., Kerbel R.S., Khoo N., Lala P.K. Establishment and characterization of human trophoblast cells with extended life span. Exp Cell Res. 1993;206:204–211. doi: 10.1006/excr.1993.1139. [DOI] [PubMed] [Google Scholar]
- 16.Morita S., Kojima T., Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–1066. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 17.Yagel S., Casper R.F., Powell W., Parhar R.S., Lala P.K. Characterization of pure human first-trimester cytotrophoblast cells in long-term culture: growth pattern, markers and hormone production. Am J Obstet Gynecol. 1989;160:938–945. doi: 10.1016/0002-9378(89)90314-1. [DOI] [PubMed] [Google Scholar]
- 18.Irving J.A., Lysiak J.J., Graham C.G., Hearns S., Han V.K.M., Lala P.K. Characterization of trophoblast cells migrating from first trimester human chorionic villus explants propagated in culture. Placenta. 1995;16:413–433. doi: 10.1016/0143-4004(95)90100-0. [DOI] [PubMed] [Google Scholar]
- 19.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 20.Serrano M., Lin A.W., McCurrach M.E., Beach D., Lowe S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi K., Mitsui K., Yamanaka S. Role of ERas in promoting tumour-like properties in mouse embryonic stem cells. Nature. 2003;423:541–545. doi: 10.1038/nature01646. [DOI] [PubMed] [Google Scholar]
- 22.Grümmer R., Donner A., Winterhager E. Characteristic growth of human choriocarcinoma xenografts in nude mice. Placenta. 1999;20:547–553. doi: 10.1053/plac.1999.0406. [DOI] [PubMed] [Google Scholar]
- 23.Singer G., Kurman R.J., McMaster M.T., Shih IeM. HLA-G immunoreactivity is specific for intermediate trophoblast in gestational trophoblastic disease and can serve as a useful marker in differential diagnosis. Am J Surg Pathol. 2002;26:914–920. doi: 10.1097/00000478-200207000-00010. [DOI] [PubMed] [Google Scholar]
- 24.Mühlhauser J., Crescimanno C., Kasper M., Zaccheo D., Castellucci M. Differentiation of human trophoblast populations involves alterations in cytokeratin patterns. J Histchem Cytochem. 1995;43:579–589. doi: 10.1177/43.6.7539466. [DOI] [PubMed] [Google Scholar]
- 25.Behtash N., Karimi Zarchi M. Placental site trophoblastic tumor. J Cancer Res Clin Oncol. 2008;134:1–6. doi: 10.1007/s00432-007-0208-y. [DOI] [PubMed] [Google Scholar]
- 26.Juch H., Blaschitz A., Daxböck C., Rueckert C., Kofler K., Dohr G. A novel sandwich ELISA for alpha1 domain based detection of soluble HLA-G heavy chains. J Immunol Methods. 2005;307:96–106. doi: 10.1016/j.jim.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 27.Zhau H.E., Odero-Marah V., Lue H.W., Nomura T., Wang R., Chu G., Liu Z.R., Zhou B.P., Huang W.C., Chung L.W. Epithelial to mesenchymal transition (EMT) in human prostate cancer: lessons learned from ARCaP model. Clin Exp Metastasis. 2008;25:601–610. doi: 10.1007/s10585-008-9183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pedersen I.S., Dervan P.A., Broderick D., Harrison M., Miller N., Delany E., O'Shea D., Costello P., McGoldrick A., Keating G., Tobin B., Gorey T., McCann A. Frequent loss of imprinting of PEG1/MEST in invasive breast cancer. Cancer Res. 1999;59:5449–5451. [PubMed] [Google Scholar]
- 29.Nishihara S., Hayashida T., Mitsuya K., Schulz T.C., Ikeguchi M., Kaibara N., Oshimura M. Multipoint imprinting analysis in sporadic colorectal cancers with and without microsatellite instability. Int J Oncol. 2000;17:317–322. doi: 10.3892/ijo.17.2.317. [DOI] [PubMed] [Google Scholar]
- 30.Nakanishi H., Suda T., Katoh M., Watanabe A., Igishi T., Kodani M., Matsumoto S., Nakamoto M., Shigeoka Y., Okabe T., Oshimura M., Shimizu E. Loss of imprinting of PEG1/MEST in lung cancer cell lines. Oncol Rep. 2004;12:1273–1278. [PubMed] [Google Scholar]
- 31.Paiva P., Salamonsen L.A., Manuelpillai U., Dimitriadis E. Interleukin 11 inhibits human trophoblast invasion indicating a likely role in the decidual restraint of trophoblast invasion during placentation. Biol Reprod. 2009;80:302–310. doi: 10.1095/biolreprod.108.071415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen M., Meisser A., Bischof P. Metalloproteinases and human placental invasiveness. Placenta. 2006;27:783–793. doi: 10.1016/j.placenta.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 33.Thiery J.P. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 34.Yang J., Mani S.A., Donaher J.L., Ramaswamy S., Itzykson R.A., Come C., Savagner P., Gitelman I., Richardson A., Weinberg R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 35.Craig E.A., Austin A.F., Vaillancourt R.R., Barnett J.V., Camenisch T.D. TGFβ2-mediated production of hyaluronan is important for the induction of epicardial cell differentiation and invasion. Exp Cell Res. 2010;316:3397–3405. doi: 10.1016/j.yexcr.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gubbay J., Collignon J., Koopman P., Capel B., Economou A., Münsterberg A., Vivian N., Goodfellow P., Lovell-Badge R. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature. 1990;346:245–250. doi: 10.1038/346245a0. [DOI] [PubMed] [Google Scholar]
- 37.Kiefer J.C. Back to basics: sox genes. Dev Dyn. 2007;236:2356–2366. doi: 10.1002/dvdy.21218. [DOI] [PubMed] [Google Scholar]
- 38.Dong C., Wilhelm D., Koopman P. Sox genes and cancer. Cytogenet Genome Res. 2004;105:442–447. doi: 10.1159/000078217. [DOI] [PubMed] [Google Scholar]
- 39.Xia Y., Papalopulu N., Vogt P.K., Li J. The oncogenic potential of the high mobility group box protein Sox3. Cancer Res. 2000;60:6303–6640. [PubMed] [Google Scholar]
- 40.Kim R., Trubetskoy A., Suzuki T., Jenkins N.A., Copeland N.G., Lenz J. Genome-based identification of cancer genes by proviral tagging in mouse retrovirus-induced T-cell lymphomas. J Virol. 2003;77:2056–2062. doi: 10.1128/JVI.77.3.2056-2062.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li A.S., Siu M.K., Zhang H., Wong E.S., Chan K.Y., Ngan H.Y., Cheung A.N. Hypermethylation of SOX2 gene in hydatidiform mole and choriocarcinoma. Reprod Sci. 2008;15:735–744. doi: 10.1177/1933719108322433. [DOI] [PubMed] [Google Scholar]
- 42.Red-Horse K., Zhou Y., Genbacev O., Prakobphol A., Foulk R., McMaster M., Fisher S.J. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J Clin Invest. 2004;114:744–754. doi: 10.1172/JCI22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chou D., Ma Y., Zhang J., McGrath C., Parry S. Cytomegalovirus infection of trophoblast cells elicits an inflammatory response: a possible mechanism of placental dysfunction. Am J Obstet Gynecol. 2006;194:535–541. doi: 10.1016/j.ajog.2005.07.073. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Z., Shen T., Zhang B.H., Lv X.Y., Lin H.Y., Zhu C., Xue L.Q., Wang H. The proprotein convertase furin in human trophoblast: possible role in promoting trophoblast cell migration and invasion. Placenta. 2009;30:929–938. doi: 10.1016/j.placenta.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 45.Hu Y., Eastabrook G., Tan R., MacCalman C.D., Dutz J.P., von Dadelszen P. Decidual NK cell-derived conditioned medium enhances capillary tube and network organization in an extravillous cytotrophoblast cell line. Placenta. 2010;31:213–221. doi: 10.1016/j.placenta.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 46.Xue W.C., Feng H.C., Tsao S.W., Chan K.Y., Ngan H.Y., Chiu P.M., Maccalman C.D., Cheung A.N. Methylation status and expression of E-cadherin and cadherin-11 in gestational trophoblastic diseases. Int J Gynecol Cancer. 2003;13:879–888. doi: 10.1111/j.1525-1438.2003.13400.x. [DOI] [PubMed] [Google Scholar]
- 47.Wegner M. From head to toes: the multiple facets of Sox proteins. Nucleic Acids Res. 1999;27:1409–1420. doi: 10.1093/nar/27.6.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyagi S., Kato H., Okuda A. Role of SoxB1 transcription factors in development. Cell Mol Life Sci. 2009;66:3675–3684. doi: 10.1007/s00018-009-0097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Otsubo T., Akiyama Y., Yanagihara K., Yuasa Y. SOX2 is frequently downregulated in gastric cancers and inhibits cell growth through cell-cycle arrest and apoptosis. Br J Cancer. 2008;98:824–831. doi: 10.1038/sj.bjc.6604193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Q., He W., Lu C., Wang Z., Wang J., Giercksky K.E., Nesland J.M., Suo Z. Oct3/4 and Sox2 are significantly associated with an unfavorable clinical outcome in human esophageal squamous cell carcinoma. Anticancer Res. 2009;29:1233–1241. [PubMed] [Google Scholar]
- 51.Rodriguez-Pinilla S.M., Sarrio D., Moreno-Bueno G., Rodriguez-Gil Y., Martinez M.A., Hernandez L., Hardisson D., Reis-Filho J.S., Palacios J. Sox2: a possible driver of the basal-like phenotype in sporadic breast cancer. Mod Pathol. 2007;20:474–481. doi: 10.1038/modpathol.3800760. [DOI] [PubMed] [Google Scholar]
- 52.Schmitz M., Temme A., Senner V., Ebner R., Schwind S., Stevanovic S., Wehner R., Schackert G., Schackert H.K., Fussel M., Bachmann M., Rieber E.P., Weigle B. Identification of SOX2 as a novel glioma-associated antigen and potential target for T cell-based immunotherapy. Br J Cancer. 2007;96:1293–1301. doi: 10.1038/sj.bjc.6603696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laumonnier F., Ronce N., Hamel B.C., Thomas P., Lespinasse J., Raynaud M., Paringaux C., Van Bokhoven H., Kalscheuer V., Fryns J.P., Chelly J., Moraine C., Briault S. Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am J Hum Genet. 2002;71:1450–1455. doi: 10.1086/344661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hahn W.C., Counter C.M., Lundberg A.S., Beijersbergen R.L., Brooks M.W., Weinberg R.A. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 55.Lundberg A.S., Randell S.H., Stewart S.A., Elenbaas B., Hartwell K.A., Brooks M.W., Fleming M.D., Olsen J.C., Miller S.W., Weinberg R.A., Hahn W.C. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene. 2002;21:4577–4586. doi: 10.1038/sj.onc.1205550. [DOI] [PubMed] [Google Scholar]
- 56.Hahn W.C., Dessain S.K., Brooks M.W., King J.E., Elenbaas B., Sabatini D.M., DeCaprio J.A., Weinberg R.A. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22:2111–2123. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bilban M., Tauber S., Haslinger P., Pollheimer J., Saleh L., Pehamberger H., Wagner O., Knöfler M. Trophoblast invasion: assessment of cellular models using gene expression signatures. Placenta. 2010;31:989–996. doi: 10.1016/j.placenta.2010.08.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Histopathological findings of serial sections of tumors that were derived from HTR8/SVneo/HRASV12 and iC3-1 cells compared with isotype controls. A: Immunohistochemistry as isotype controls (mouse IgG) in HTR8/SVneo/HRASV12–derived tumors (left, ×200 magnification; right, ×600 magnification). B: Immunohistochemistry as isotype controls (rabbit IgG) in HTR8/SVneo/HRASV12–derived tumors (left, ×200 magnification; right, ×600 magnification). C: Immunohistochemistry revealed that HTR8/SVneo/HRASV12-derived tumors expressed HLA-G (left, ×200 magnification), cytokeratin (middle, ×200 magnification) and hCG (right, ×200 magnification). The identical areas of a tumor are shown in A–C. D: Immunohistochemistry as a positive control for hPL staining in human placenta tissue (×200 magnification). E: Immunohistochemistry as isotype controls (mouse IgG) in iC3-1–derived tumors (left, ×200 magnification; right, ×600 magnification). F: Immunohistochemistry as isotype controls (rabbit IgG) in iC3-1–derived tumors (left, ×200 magnification; right, ×600 magnification). G: Immunohistochemistry revealed that iC3-1–derived tumors expressed HLA-G (left, ×200 magnification), cytokeratin (middle, ×200 magnification), and hCG (right, ×200 magnification). H: Immunohistochemical staining of E-CADHERIN (left, ×200 magnification) and VIMENTIN (right, ×200 magnification) in iC3-1–derived tumors. The identical areas of a tumor are shown in E–H.
SOXB1 expression in iC3-1 cells. A: Quantitative RT-PCR showing significant upregulation of SOX1 expression in iC3-1 cells as compared with HTR8/SVneo/EGFP cells (*P < 0.05) and two choriocarcinoma cell lines (JAR and JEG3). B: Quantitative RT-PCR showing SOX2 expression in iC3-1 cells as compared with HTR8/SVneo/EGFP cells and two choriocarcinoma cell lines (JAR and JEG3).