

Abstract
Ataxia-telangiectasia is a multifaceted syndrome caused by null mutations in the ATM gene, which encodes the protein kinase ATM, a key participant in the DNA damage response. Retinal neurons are highly susceptible to DNA damage because they are terminally differentiated and have the highest metabolic activity in the central nervous system. In this study, we characterized the retina in young and aged Atm-deficient mice (Atm−/−). At 2 months of age, angiography revealed faint retinal vasculature in Atm−/− animals relative to wild-type controls. This finding was accompanied by increased expression of vascular endothelial growth factor protein and mRNA. Fibrinogen, generally absent from wild-type retinal tissue, was evident in Atm−/− retinas, whereas mRNA of the tight junction protein occludin was significantly decreased. Immunohistochemistry labeling for occludin in 6-month-old mice showed that this decrease persists in advanced stages of the disease. Concurrently, we noticed vascular leakage in Atm−/− retinas. Labeling for glial fibrillary acidic protein demonstrated morphological alterations in glial cells in Atm−/− retinas. Electroretinographic examination revealed amplitude aberrations in 2-month-old Atm−/− mice, which progressed to significant functional deficits in the older mice. These results suggest that impaired vascularization and astrocyte–endothelial cell interactions in the central nervous system play an important role in the etiology of ataxia-telangiectasia and that vascular abnormalities may underlie or aggravate neurodegeneration.
Some brain disorders may have a vascular origin,1,2 and vascular diseases can be directly linked to neuronal and synaptic dysfunction through changes in the blood flow, increase in blood–brain barrier permeability, and in nutrient supply.3 A healthy brain relies on the proper function and communication of all cells comprising the neurovascular unit: neurons, astrocytes, brain endothelium, and vascular smooth muscle cells.4
Impaired genomic stability interferes with cellular homeostasis and poses a constant threat to cellular viability.5 The cell combats this threat by activating the DNA damage response (DDR), a complex signaling network that detects the DNA lesions, promotes their repair, and temporarily modulates cellular metabolism while the damage is being repaired.6 The DDR is vigorously activated by DNA double-strand breaks (DSBs), a particularly cytotoxic DNA lesion induced by ionizing radiation, radiomimetic chemicals, and oxygen radicals.7,8 The DNA damage response is a hierarchical process executed by sensor/mediator proteins that accumulate at DSB sites and by protein kinases that serve as transducers of the DNA damage alarm to numerous downstream effectors.6 The primary transducer of the cellular response to DSBs is the protein kinase ATM, which phosphorylates many key players in the various branches of the DDR.9
Ataxia-telangiectasia (A-T) is an autosomal recessive disorder caused by mutations in the ATM gene that encodes the ATM protein.10 A-T is characterized by progressive neurodegeneration affecting mainly the cerebellum, which develops into severe neuromotor dysfunction; peripheral neuropathy; immunodeficiency that spans the B- and T-cell systems; thymic and gonadal atrophy; marked predisposition to lymphoreticular malignancies; and chromosomal fragility and acute sensitivity to ionizing radiation. Cultured ATM-deficient cells exhibit severe cellular sensitivity to DSB-inducing agents, with markedly defective DSB response.
DSBs are constantly induced in all body cells by metabolic byproducts such as oxygen radicals. Oxidative stress has been consistently associated with various neurodegenerative conditions.11–14 Indeed, there is substantial evidence for a role of oxidative damage in the progression of neurodegenerative disorders, including Parkinson's and Alzheimer's diseases.15 Similarly, elevated oxidative stress has been identified in several DNA repair deficiencies, including A-T.16–22 Notably, ATM has recently been shown to be activated by oxidative stress.23 Ocular tissues, especially the retina, are exposed to extremely high levels of reactive oxygen species. Nevertheless, retinal neurodegeneration has not been reported in A-T patients. The ocular manifestation of the disease reported to date is scleral telangiectasia24,25 and saccadic abnormalities.26 Moreover, no retinal pathology has been described to date in Atm-deficient mice despite their increased sensitivity to reactive oxygen species–inducing agents.27 We examined the link between retinal vascular pathology and function in young and aging Atm-deficient mice. We present evidence for vascular changes that accompany neuronal deficiencies in the retina.
Materials and Methods
Animals
Atm+/− mice28 were a generous gift from Dr. Anthony Wynshaw-Boris (University of California, San Diego, CA). Offspring of these mice were genotyped using PCR-based assays based on mouse-tail DNA prepared using the GenElute Mammalian Genomic DNA Miniprep kit (Sigma, St. Louis, MO). Two- and 6-month-old Atm−/− mice were used for this study, and age-matched Atm+/+ littermates (WT) were used as controls. Mice were housed and maintained in the animal facility of Tel Aviv University, and all experiments complied with protocols approved by the university's animal care committee.
Angiography
Mice were anesthetized by an intraperitoneal injection of ketamine (80 mg/kg) and xylazine (4 mg/kg). An incision was made into the peritoneal cavity, and the diaphragm and pericardium were incised to visualize the heart. The mice were perfused slowly through the left ventricle with saline containing 25 mg of high-molecular-mass (2 × 106) fluorescein isothiocyanate-dextran (Sigma). Mice were sacrificed by CO2 inhalation; the eyes were enucleated and fixed in 4% paraformaldehyde for 1 hour. The retinas were gently separated from the eyecups and mounted on a microscope slide with mounting medium. Four incisions were made to flatten the retina, which was then covered with a coverslip. Imaging was performed on a fluorescence microscope.
RT-PCR
To determine mRNA levels, we isolated the eyecups and extracted the RNA using MasterPure RNA Purification kit (Epicentre Biotechnologies, Madison, WI). We determined the total RNA concentration with nanodrop analysis assay (260/280 nm) and further determined the purity of the extraction (240/260 nm >1.95). The total RNA concentration was diluted with molecular water (DNase, RNase free; Sigma) to achieve approximately 500 ng/mL. Samples containing equal amounts of total RNA (0.2 to 1 μg of RNA/sample) the type of were converted into cDNA using the Verso cDNA kit (AB1453/B). The template (diluted with the water) was incubated for 5 minutes at 70°C to remove secondary structure, and the corresponding cDNA was synthesized.
Quantitative Real-Time PCR
Samples were analyzed for quantification of mRNA expression via reverse transcription followed by quantitative real-time polymerase chain reaction using TaqMan (Absolute Blue QPCR ROX Mix, AB-4138; Abgene). RT-PCR assays were designed by Applied Biosystems (Foster City, CA) and compared with β-actin (ACTB) RNA levels.29 The comparative Ct method was used for quantification of transcripts according to the manufacturer's protocol. Measurement of ΔCt was performed in duplicate or triplicate. All reactions were performed with primer concentrations of 0.25 μmol/L in a total volume of 20 μL of reaction.
Western Blot Analysis
Eyecups were washed with ice-cold PBS and homogenized in ice-cold homogenization buffer containing 1:50 phosphatase inhibitor cocktail (I and II; Sigma-Aldrich, St. Louis, MO) and 1:100 protease inhibitor cocktail. Protein concentration was determined using Bio-Rad Protein Assay (Hercules, CA). Western blot analysis was performed using 10% polyacrylamide gels. Each lane was loaded with an identical amount of protein extracts, which, following electrophoresis, were transferred into an immobilon polyvinylidene disulfide membrane (Millipore, Billerica, MA) for at least 12 hours at 200 mA or for 90 minutes at 280 mA. Blots were stained with Ponceau to verify equal loading and transfer of proteins. Membranes were then probed with primary antibodies for vascular endothelial growth factor (VEGF) (1:1000; Abcam, Cambridge, UK) and fibrinogen (1:750; Dako, Glostrup, Denmark). Secondary antibody was goat anti-rabbit, IRDye infrared dye conjugated (Li-Cor antibody, 1:1000; Li-Cor Biosciences, Lincoln, NE) for 1 hour at room temperature. Blots were scanned using the Li-Cor imaging system, and band intensity was analyzed using Odyssey software (Odyssey Software, West Henrietta, NY).
Hemosiderin Labeling
Flat-mount retinas from Atm−/− and age-matched WT mice were stained for hemosiderin deposits with Prussian blue working solution (equal parts of freshly made 1% potassium ferrocyanide and 1% hydrochloric acid) for 60 minutes at room temperature, washed in deionized water, and counterstained with Nuclear Fast Red Blue-stained clusters of hemosiderin staining were qualitatively evaluated (presence/absence) from sections throughout the retina.
Immunohistochemistry
Anesthetized mice underwent intracardial perfusion with 200 mL of PBS to remove blood, and then with 25 mL of 4% paraformaldehyde. Eyes were enucleated and fixed in 4% paraformaldehyde for 1 hour at room temperature. The retinas were gently separated from the eyecups and submerged in 0.5% Triton overnight for permeabilization. The next day, the retinas were immersed in blocking medium (1% bovine serum albumin, 10% normal donkey serum, 0.25% Triton in PBS) for 2 hours, and then incubated with primary antibody in blocking medium for 1 hour at room temperature, followed by overnight at 4°C. Retinas were then incubated with the secondary antibody for 2 hours at room temperature, and finally with Sytox blue (1:1000; Invitrogen, Carlsbad, CA) in a buffer containing Tris 10 mmol/L, EDTA 1 mmol/L (pH = 7.5) for 20 minutes for nuclear staining. The stained retinas were placed in a drop of mounting medium on a microscope slide, incised for flattening, and covered with a coverslip. Primary antibodies used in this study included CD31 (rat anti-mouse, 1:100; BD Biosciences, Franklin Lakes, NJ), occludin (mouse anti-human, 1:100; Invitrogen), fibrinogen (rabbit anti-human, 1:250; Dako), and glial fibrillary acidic protein (GFAP) (rabbit anti-human, 1:500; Sigma). Secondary antibodies included AlexaFluor 488 and AlexaFluor 594 (1:250; Invitrogen). Imaging was performed on a Zeiss (Oberkochen, Germany) LSM-510 confocal microscope.
Electroretinographic Analysis
Animals were prepared under dim red light following overnight dark adaptation. Anesthesia was induced by intraperitoneal injection of ketamine (80 mg/kg) and xylazine (4 mg/kg), and body temperature was maintained with a heating pad. Electroretinographic responses (ERGs) were recorded after fully dilating the pupils with topical corneal 0.5% tropicamide and 2.5% phenylephrine HCl. A gold-wire loop electrode was placed on the cornea after topical 0.5% proparacaine HCl anesthesia. A gold-wire reference electrode was positioned to touch the sclera near the limbus of the eye, and a neutral electrode was placed on the tail. ERGs were recorded with the LKC system (LKC Technologies, Gaithersburg, MD) using ganzfeld stimulation. Dark-adapted responses were recorded across a 5 log unit range of stimulus intensity in 1 log unit steps up to maximum intensity of 1.4 log cd • s/m2. Light-adapted responses were recorded against a constant white background light of 30 cd/m2 that suppresses rod function. ERG responses were amplified and filtered (0.3 to 500 Hz).
Results
Increased Expression of VEGF in Atm−/− Retinas
A-T patients show impairment in scleral blood vessels.24,25,30 To assess similar pathological phenomena in the mouse model, we examined retinal vasculature in 2-month-old WT and Atm−/− mice using fluorescent angiography. Blood vessels in the retinas of Atm−/− mice were faintly illustrated in comparison to the WT retinas. Figure 1 shows that vessels in the Atm−/− retinas appear constricted relative to WT and suggests that there might be changes in vessel density.
Figure 1.
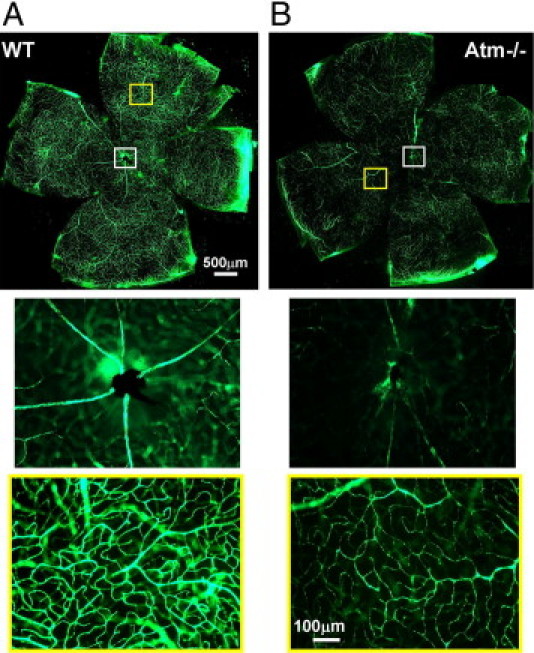
Attenuated blood vessels in retinas of Atm−/− mice. Flat-mount retinas of 2-month-old WT (A, n = 5) and Atm−/− (B, n = 5) mice imaged with a fluorescent microscope following intracardial perfusion with dextran-fluorescein. Insets show larger magnification of optic nerve region (white frame) and typical peripheral region (yellow frame).
VEGF plays a major role in angiogenesis, and under pathological conditions, changes in VEGF levels can cause vascular dysfunction.31,32 Furthermore, it was suggested that VEGF may alter vascular permeability by regulating tight junctions.33 Therefore, we investigated whether the vascular differences we observed between retinas of Atm−/− and WT mice may be related to changes in VEGF expression levels. Analysis of retinal mRNA levels revealed a significant increase of 40% in VEGF in the eyes of Atm−/− mice relative to WT controls (Figure 2A). We further analyzed the VEGF levels using Western blot analysis and found that VEGF protein expression was significantly higher (35% increase) in the retinal tissue of Atm−/− mice than in WT retinas (Figure 2B). These results suggest that there is a linkage between VEGF expression and blood vessel pathology in Atm−/− mice.
Figure 2.
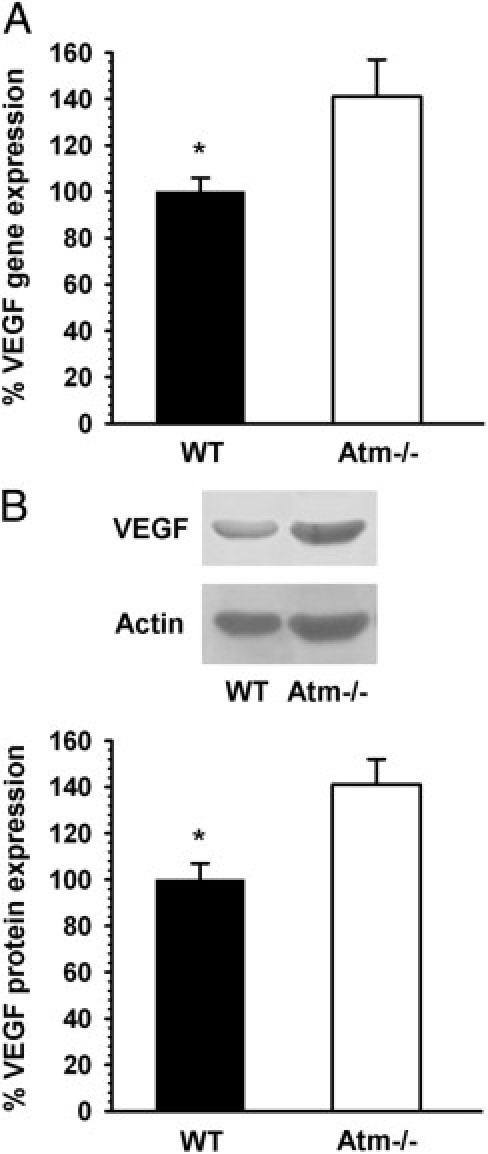
Increased VEGF levels in Atm−/− mice. Real-time RT-PCR (A) and Western blot (B) analyses reveal significantly higher levels of VEGF in eyecups of Atm−/− mice relative to VEGF levels in WT control mice. Statistical values were calculated by 2-tailed Student's t-test. *P < 0.05.
Pathological Changes in Tight Junctions in Atm−/− Mice
Increased VEGF levels have been implicated as a detrimental factor causing neovascularization and vascular permeability in age-related macular degeneration.34 Increased vascular permeability is induced through a mechanism involving decreased expression of occludin in vascular endothelial cells35; therefore, we examined occludin expression in the ocular tissues. At 2 months of age, occludin mRNA was significantly decreased in Atm−/− mice by an average of 60% compared to WT (Figure 3A). To test whether reduced occludin levels persist at older age, we examined occludin localization in the retinas of 6-month-old animals. Immunohistochemical analysis revealed distinct occludin labeling in blood vessels throughout the retina in WT mice, and only faint labeling in retinas of 6-month-old Atm−/− mice (Figure 3B), suggesting that excessive expression of VEGF alters occludin expression resulting in blood vessel pathology.
Figure 3.
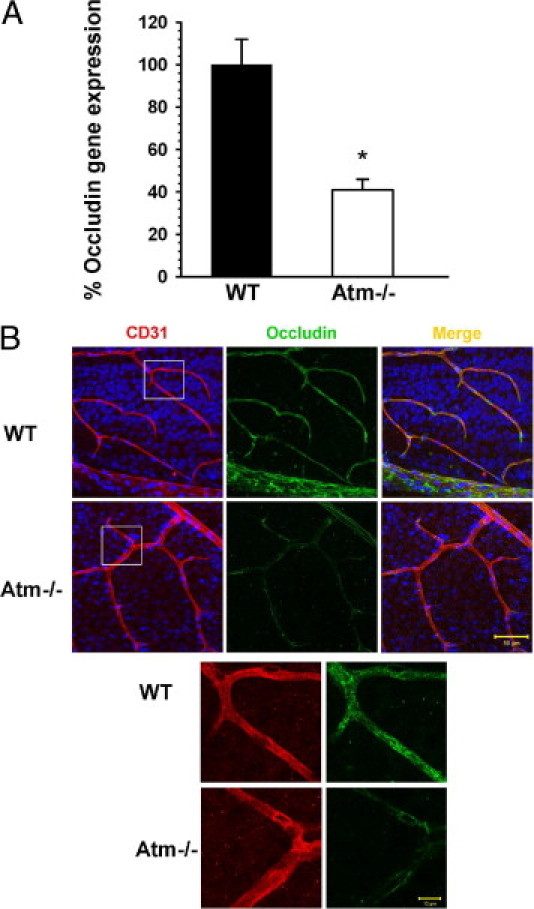
Reduced levels of occludin in Atm−/− mice. A: Real time RT-PCR analysis shows significantly lower levels of occludin in eyecups of Atm−/− mice relative to WT control mice at 2 months of age. Statistical values were calculated by 2-tailed Student's t-test. *P < 0.0025. B: Confocal images of flat-mount retinas of 6-month-old WT and Atm−/− mice immunolabeled for CD31 (red) and occludin (green). Cell nuclei are labeled with Sytox Blue (blue). Upper scale bar: 50 µm, lower scale bar: 10 µm.
Microhemorrhage Incidents in Atm−/− Mice
Fibrinogen is normally absent from healthy retinal tissue, but has been demonstrated in retinas treated with VEGF.36 It is a major player in pathologies involving damage to blood vessels. Using Western blot analysis, we found that protein levels of fibrinogen were significantly higher in retinas of 2-month-old Atm−/− mice than in age-matched controls (Figure 4A). Immunohistochemical analyses of flat-mount retinas showed fibrinogen in association with retinal blood vessels in the Atm−/− mice, whereas fibrinogen was only slightly evident in retinas of control animals (Figure 4B). We further examined whether the pathological changes apparent at 2 months are progressive. We found a similar pattern in older mice (6 months of age). Furthermore, we found evidence of vascular leakage in these animals. Using confocal microscopy, we found that fibrinogen labeling was observed in clusters outside blood vessels in Atm−/− retinas, whereas it colocalized with the endothelial cell marker in the retinas of WT mice (Figure 4C). Indeed, increases in fibrinogen levels, as well as reduced occludin content, have been previously associated with increased vascular permeability.35,37 To further examine vascular integrity by another parameter, we used the marker hemosiderin. Deposits of hemosiderin are breakdown products of hemoglobin and reflect microhemorrages that occurred previously.38 Immunohistological analysis revealed deposits of hemosiderin in Atm−/− retinas as early as 2 months of age, whereas WT retinas lacked evidence of such deposits (Figure 5).
Figure 4.
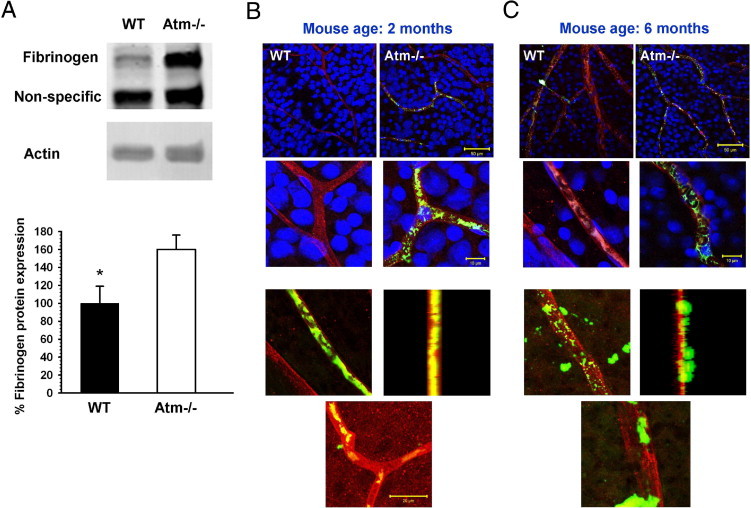
Increased fibrinogen expression in retinas of Atm−/− mice. A: Western blot analysis depicts a statistically significant increase of 60% in fibrinogen level in the Atm−/− retinas in comparison to age-matched controls. Statistical values were calculated by 2-tailed Student's t-test. *P < 0.05. B: Confocal images of flat-mount retinas of mice at 2 months of age show fibrinogen (green) in blood vessels (stained with the pan-endothelial marker CD31, red) of Atm−/− mice, but no labeling in WT controls. Cell nuclei are stained with Sytox Blue (blue). C: Similar imaging at 6 months of age reveals fibrinogen labeling external to blood vessels in a representative retina from an Atm−/− mouse. In the WT, fibrinogen labeling is absent or confined to the vessel. Upper scale bar: 50 µm, middle scale bar: 10 µm, lower scale bar: 20 µm.
Figure 5.
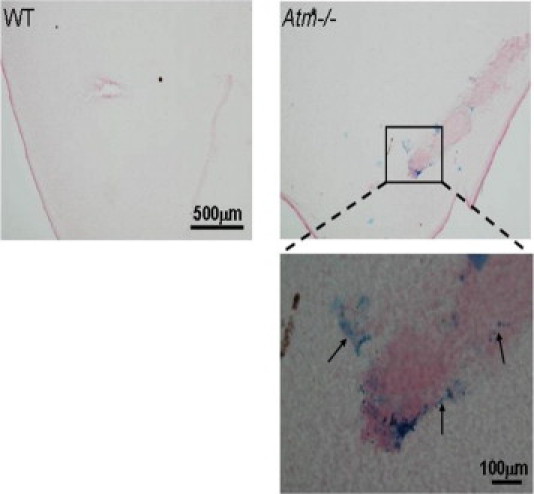
Vascular leakage in retinas of Atm−/− mice. Whole-mount retinas of WT and Atm−/− mice were stained for hemosiderin to indicate deposits resulting from microhemorrhages. The lower panel shows a larger magnification of the area marked with a rectangle in the image of Atm−/− retina. Blue deposits of hemosiderin were evident in retinas of Atm−/− mice (several indicated with arrows), but scarcely in retinas of WT mice.
Astrocyte-Vascular Pathology in Atm−/− Mice
Astrocyte–endothelial cell interaction plays a major role in the function of the vascular unit, and dysfunction in their interaction may lead to microhemorrhages.39 Furthermore, astrocytes are known to regulate the brain's endothelial barrier by releasing soluble factors such as VEGF.40 We investigated retinal astrocytes by examining the levels of GFAP in flat-mount retinas. Using confocal microscopy, we targeted the interaction of astrocytes (marked by GFAP) with endothelial cells (marked by CD31). As shown in Figure 6, in retinas of WT mice, the processes of the astrocytes appear to form an even network that encases the blood vessels, whereas in retinas of Atm−/− mice, the astrocytic processes appear short and stubby, as found in cases of gliosis, and seem to be unable to form a continuous regular net alongside the blood vessels. These results suggest that Atm deficiency leads to pathological changes in astrocytes and impairs their ability to support the blood vessels.
Figure 6.
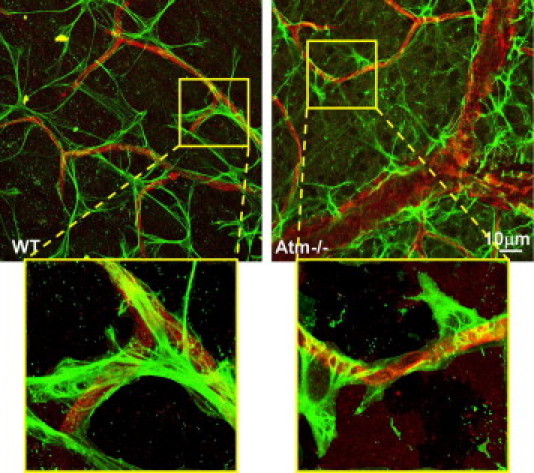
Glial cell alterations in retinas of Atm−/− mice. Confocal images of flat-mount retinas from WT and Atm−/− mice, labeled for CD31 (red) and GFAP (green). Insets depict characteristic regions marked with a yellow frame at a larger magnification.
Vascular Pathology Results in Decreased Retinal Function in Atm−/− Mice
We asked whether impairment in the astrocyte–endothelial cell interaction observed in Atm−/− mice might lead to functional deficits. Retinal function was evaluated by ERG. Dark-adapted responses of 2-month-old mice showed no differences in a- and b-wave amplitudes between Atm−/− and WT mice. In light-adapted responses, however, both a- and b-wave amplitudes of Atm−/− mice were consistently lower than in WT across the range of stimulus intensities. b-Wave amplitude at maximal stimulus intensity was 378 ± 30 μV in the wild type, and 291 ± 38 μV in the Atm−/− mice (Figure 7). To assess whether these early degenerative signs are progressive, we examined retinal function in 6-month-old mice. The a- and b-wave amplitudes of Atm−/− retinal responses were consistently lower by 40% or more than the amplitudes in control age-matched mice in the dark-adapted ERG (Figure 7). These differences were statistically significant across the range of stimulus intensities. At maximal stimulus intensity, b-wave amplitude was 73% higher in WT (1355 ± 133 μV) than in Atm−/− mice (783 ± 48 μV, P < 0.01). Similarly, b-wave amplitudes of the light-adapted responses were lower in Atm−/− mice than in the responses of the control mice. At maximal intensity, b-wave amplitude in WT mice was 40% higher than in Atm−/− mice.
Figure 7.
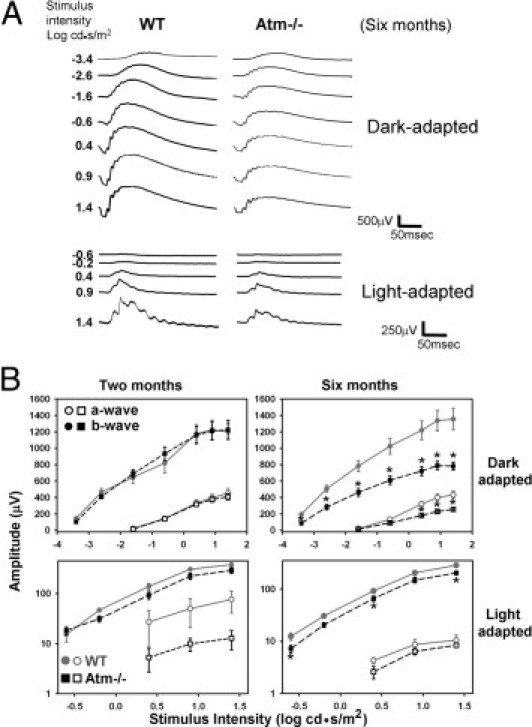
Retinal functional deficits in Atm−/− mice. A: Representative dark- and light-adapted responses of 6-month-old WT and Atm−/− mice. B: Average responses of WT (full gray line, circles, n = 5) and Atm−/− (broken black line, squares, n = 8) mice at 2 months and 6 months (WT n = 5, Atm−/−n = 5) of age. Average and SE bars are shown for a-wave (open symbols) and b-wave (filled symbols). The a- and b-wave amplitudes were significantly higher in dark-adapted responses of WT mice compared to Atm−/− mice at all stimulus intensities at 6 months. An asterisk denotes statistically significant amplitude difference at individual stimulus intensities in the graph of light-adapted responses. Statistical values were calculated by 2-tailed Student's t-test. *P < 0.05.
Discussion
Here, we show that Atm deficiency in mice leads to impaired retinal vasculature. Furthermore, we found significant elevation in astrocyte-secreted VEGF, which might play a role in vascular pathology. Collectively, our data suggest an important role for astrocyte–vascular interaction in Atm-deficient mice and probably A-T patients.
There is growing attention to the role of glial cells in neuropathologies.41 Transgenic and knockout mouse models for certain astrocyte-specific proteins demonstrated neuropathologies.42,43 Astrocyte abnormalities and their influence on Purkinje cells have also been reported in Atm−/− cells.41,44,45 Electron microscopy observations have detected structural activation of glial cells in Atm knockout mice.46 Ultrastructural alterations in astrocytes have been demonstrated alongside neuronal degenerative processes in Atm−/− mice in cerebellar tissue.46 Furthermore, astrocytes from Atm−/− mice exhibited growth arrest and growth defects, which may be due to oxidative stress.41,47 Retinal neurons are terminally differentiated and are therefore, like other neurons in the brain, irreplaceable. The ongoing visual processes require large amounts of energy, which cause the retina to have the highest oxygen consumption in the mammalian brain.27,48,49 This creates a stressful environment, with the metabolic byproducts that are formed, primarily reactive oxygen species, constantly attacking neuroretinal DNA.27,50,51 Like the rest of the central nervous system, the retina depends on glia, mostly Muller cells and astrocytes, for proper function. We have discovered that the pathological appearance of astrocytes along the retinal blood vessel may result in microhemorrhage incidents.
ATM controls DDR, which is, in turn, critically important for normal retinal development and maintenance. Retinal and choroidal vascular abnormalities were associated with Atm missense variants.52,53 Our findings support this and show vascular abnormalities in the mouse model of Atm. These blood vessel abnormalities are likely to result in impaired oxygen diffusion to the retina. The resulting hypoxia can strongly up-regulate VEGF.31,32,54 VEGF is a major factor in different types of angiogenic disorders including those associated with cancer, ischemia, and inflammation.55 It is a member of a group of growth factors and hormones that alter vascular permeability by regulating tight junctions.33
In the eye, excessive VEGF has been implicated as a detrimental factor causing neovascularization and vascular leakage in age-related macular degeneration34 and in proliferative retinopathies.56 VEGF is not only up-regulated in response to hypoxia and nonperfusion, but may also be an underlying cause of them.57 In addition, VEGF can induce hypertrophy of endothelial cells of retinal capillaries, at the expense of lumen diameter.57,58 We provide evidence for an increase in VEGF in the retinas of Atm−/− mice. This can account for the vasoconstriction and vascular leakage that we found in the retinas of Atm−/− mice.
Fibrinogen, a factor that is intertwined with VEGF, may also have multiple effects on the retinal abnormalities in the Atm−/− mice that we report here. We report a significant increase in fibrinogen levels, as early as 2 months of age and ongoing to advanced stages of the disease. Increased fibrinogen content leads to its binding to endothelial receptors, intercellular adhesion molecule-1 (ICAM-159,60) and α5β1 integrin.60,61 This induces increased formation of F-actin, leading to stiffening of cells and actin filament retraction.37,62–66 Furthermore, increased levels of fibrinogen have been shown to down-regulate endothelial tight junction proteins.67 Both of these processes lead to widening of interendothelial junctions and increased vascular leakage in the paracellular route, as demonstrated by fibrinogen and hemosiderin labeling in this study. Fibrinogen can also induce vasoconstriction through the production of endothelin-1.63 This, too, could be manifested in the vessel narrowing evident in the angiography imaging.
One of the tight junction proteins that is affected by fibrinogen as well as by VEGF and that contributes to vascular permeability is occludin.67 We demonstrated here that occludin is significantly reduced in the retinas of Atm−/− mice at 2 months of age. In parallel, we found indications for interstitial accumulation of blood by hemosiderin staining. However, the angiographic and fibrinogen evaluation at 2 months did not reveal clear evidence of vascular leakage as one might expect. A study on occludin-null mice showed that occludin is not required for maintaining functional tight junctions, and that cells of these mice form structurally intact tight junctions despite the absence of occludin.68,69 It is possible that in our case, too, the reduction in occludin is compensated for by the presence of other tight junction proteins at this relatively early stage of the disease. The symptomatic picture is aggravated with time, as shown by fibrinogen labeling at 6 months, perhaps as other components of the tight junction are also impaired.
High levels of the activated phosphorylated form of ATM were detected in the cytoplasm of mouse photoreceptors.27 Cerebellar cells of Atm−/− mice spontaneously develop elevated levels of oxidative DNA damage,15,70,71 and it is reasonable that photoreceptor cells will suffer similar damage and show reduced performance. The functional changes that we report here demonstrate progressive conspicuous neuroretinal deficiencies. We found that ERG at 2 months points to early signs of degeneration. In a progressive stage of the disease, at 6 months of age, significant functional defects are evident that involve both rod and cone systems. Amplitude reductions in the b-wave suggest that the damage may be more extensive and include the inner layers of the retina beyond the photoreceptors as well. These findings concur with subtle neurological abnormalities reported previously, including electrophysiological deficiencies.15,22,46,72–77
The symptomatic picture presented in A-T patients is complex and multifaceted, partly progressive, and occasionally fixed over a limited period of time. The diversity of systems affected by the disease and their interconnections makes it difficult to determine which of the disease manifestations is primary and which reflects added systemic complications. The diagnosis of A-T based on its clinical manifestations is challenging.78 Ocular symptoms of telangiectasia and saccadic abnormalities in A-T patients generally appear 4 to 5 years after first signs of ataxia.26
The abnormalities that we present here encompass neuronal, glial, and vascular changes. We suggest that the changes in the retinal glio-neurovascular unit are underlying factors that aggravate the functional deficits that we report in advanced stages of the disease. We suggest that the retina could serve as a useful window to central nervous system pathology in A-T models in future studies, as signs are apparent at the young age of 2 months. The additional ocular alterations described here, if replicated in human patients, could assist in earlier diagnosis of the disease.
Footnotes
Supported by grants from the Human Frontier Science Program organization: CDA – 0019/2008-C; and Legacy Heritage Biomedical Science Partnership: 862/09. Work in the laboratory of A.B. was supported by research grants from the A-T Children's Project: 160240; the Israeli Ministry of Health: 3-00000-6068; Israel Science Foundation: 365/08; US-Israel Binational Science Foundation: 2005046; and the German-Israeli Foundation (GIF): I-935-270.1/2006. Work in the laboratory of Y.S. was supported by the A-T Medical Research Foundation, the A-T Ease Foundation, and the Israel Cancer Research Fund. Y.S. is a Research Professor of the Israel Cancer Research Fund.
References
- 1.Forstl H., Howard R. Recent studies on dementia senilis and brain disorders caused by atheromatous vascular disease: by A. Alzheimer, 1898. Alzheimer Dis Assoc Disord. 1991;5:257–264. doi: 10.1097/00002093-199100540-00005. [DOI] [PubMed] [Google Scholar]
- 2.Baldwin R.C., O'Brien J. Vascular basis of late-onset depressive disorder. Br J Psychiatry. 2002;180:157–160. doi: 10.1192/bjp.180.2.157. [DOI] [PubMed] [Google Scholar]
- 3.Zlokovic B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Bell R.D., Zlokovic B.V. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropath. 2009;118:103–113. doi: 10.1007/s00401-009-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciccia A., Elledge S.J. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bassing C.H., Alt F.W. The cellular response to general and programmed DNA double strand breaks. DNA Repair (Amst) 2004;3:781–796. doi: 10.1016/j.dnarep.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 8.Hartlerode A.J., Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–168. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lavin M.F., Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6:931–942. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- 10.Lavin M.F. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 11.Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 12.Perry J.J., Fan L., Tainer J.A. Developing master keys to brain pathology, cancer and aging from the structural biology of proteins controlling reactive oxygen species and DNA repair. Neuroscience. 2007;145:1280–1299. doi: 10.1016/j.neuroscience.2006.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryter S.W., Kim H.P., Hoetzel A., Park J.W., Nakahira K., Wang X., Choi A.M. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal. 2007;9:49–89. doi: 10.1089/ars.2007.9.49. [DOI] [PubMed] [Google Scholar]
- 14.Trushina E., McMurray C.T. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience. 2007;145:1233–1248. doi: 10.1016/j.neuroscience.2006.10.056. [DOI] [PubMed] [Google Scholar]
- 15.Barlow C., Dennery P.A., Shigenaga M.K., Smith M.A., Morrow J.D., Roberts L.J., 2nd, Wynshaw-Boris A., Levine R.L. Loss of the ataxia-telangiectasia gene product causes oxidative damage in target organs. Proc Natl Acad Sci U S A. 1999;96:9915–9919. doi: 10.1073/pnas.96.17.9915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barzilai A., Biton S., Shiloh Y. The role of the DNA damage response in neuronal development, organization and maintenance. DNA Repair (Amst) 2008;7:1010–1027. doi: 10.1016/j.dnarep.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Barzilai A., Rotman G., Shiloh Y. ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA Repair (Amst) 2002;1:3–25. doi: 10.1016/s1568-7864(01)00007-6. [DOI] [PubMed] [Google Scholar]
- 18.Chen P., Peng C., Luff J., Spring K., Watters D., Bottle S., Furuya S., Lavin M.F. Oxidative stress is responsible for deficient survival and dendritogenesis in purkinje neurons from ataxia-telangiectasia mutated mutant mice. J Neurosci. 2003;23:11453–11460. doi: 10.1523/JNEUROSCI.23-36-11453.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito K., Hirao A., Arai F., Matsuoka S., Takubo K., Hamaguchi I., Nomiyama K., Hosokawa K., Sakurada K., Nakagata N., Ikeda Y., Mak T.W., Suda T. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 20.Karanjawala Z.E., Murphy N., Hinton D.R., Hsieh C.L., Lieber M.R. Oxygen metabolism causes chromosome breaks and is associated with the neuronal apoptosis observed in DNA double-strand break repair mutants. Curr Biol. 2002;12:397–402. doi: 10.1016/s0960-9822(02)00684-x. [DOI] [PubMed] [Google Scholar]
- 21.Suraweera A., Becherel O.J., Chen P., Rundle N., Woods R., Nakamura J., Gatei M., Criscuolo C., Filla A., Chessa L., Fusser M., Epe B., Gueven N., Lavin M.F. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol. 2007;177:969–979. doi: 10.1083/jcb.200701042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watters D.J. Oxidative stress in ataxia telangiectasia. Redox Rep. 2003;8:23–29. doi: 10.1179/135100003125001206. [DOI] [PubMed] [Google Scholar]
- 23.Guo Z., Kozlov S., Lavin M.F., Person M.D., Paull T.T. ATM activation by oxidative stress. Science. 2010;330:517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 24.Crawford T.O., Mandir A.S., Lefton-Greif M.A., Goodman S.N., Goodman B.K., Sengul H., Lederman H.M. Quantitative neurologic assessment of ataxia-telangiectasia. Neurology. 2000;54:1505–1509. doi: 10.1212/wnl.54.7.1505. [DOI] [PubMed] [Google Scholar]
- 25.Lavin M.F., Gueven N., Bottle S., Gatti R.A. Current and potential therapeutic strategies for the treatment of ataxia-telangiectasia. Br Med Bull. 2007;81 doi: 10.1093/bmb/ldm012. [DOI] [PubMed] [Google Scholar]; –82:129–147
- 26.Crawford T.O. Ataxia telangiectasia. Semin Pediatr Neurol. 1998;5:287–294. doi: 10.1016/s1071-9091(98)80007-7. [DOI] [PubMed] [Google Scholar]
- 27.Leemput J., Masson C., Bigot K., Errachid A., Dansault A., Provost A., Gadin S., Aoufouchi S., Menasche M., Abitbol M. ATM localization and gene expression in the adult mouse eye. Mol Vis. 2009;15:393–416. [PMC free article] [PubMed] [Google Scholar]
- 28.Barlow C., Hirotsune S., Paylor R., Liyanage M., Eckhaus M., Collins F., Shiloh Y., Crawley J.N., Ried T., Tagle D., Wynshaw-Boris A. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 29.Frenkel D., Puckett L., Petrovic S., Xia W., Chen G., Vega J., Dembinsky-Vaknin A., Shen J., Plante M., Burt D.S., Weiner H.L. A nasal proteosome adjuvant activates microglia and prevents amyloid deposition. Ann Neurol. 2008;63:591–601. doi: 10.1002/ana.21340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biton S., Barzilai A., Shiloh Y. The neurological phenotype of ataxia-telangiectasia: solving a persistent puzzle. DNA Repair (Amst) 2008;7:1028–1038. doi: 10.1016/j.dnarep.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 31.Detmar M., Brown L.F., Berse B., Jackman R.W., Elicker B.M., Dvorak H.F., Claffey K.P. Hypoxia regulates the expression of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) and its receptors in human skin. J Invest Dermatol. 1997;108:263–268. doi: 10.1111/1523-1747.ep12286453. [DOI] [PubMed] [Google Scholar]
- 32.Shweiki D., Itin A., Soffer D., Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 33.Harhaj N.S., Antonetti D.A. Regulation of tight junctions and loss of barrier function in pathophysiology. Int J Biochem Cell Biol. 2004;36:1206–1237. doi: 10.1016/j.biocel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 34.Blaauwgeers H.G., Holtkamp G.M., Rutten H., Witmer A.N., Koolwijk P., Partanen T.A., Alitalo K., Kroon M.E., Kijlstra A., van Hinsbergh V.W., Schlingemann R.O. Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris: Evidence for a trophic paracrine relation. Am J Pathol. 1999;155:421–428. doi: 10.1016/S0002-9440(10)65138-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Antonetti D.A., Barber A.J., Khin S., Lieth E., Tarbell J.M., Gardner T.W. Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content: vascular endothelial growth factor decreases occludin in retinal endothelial cells: Penn State Retina Research Group. Diabetes. 1998;47:1953–1959. doi: 10.2337/diabetes.47.12.1953. [DOI] [PubMed] [Google Scholar]
- 36.Hofman P., Blaauwgeers H.G., Vrensen G.F., Schlingemann R.O. Role of VEGF-A in endothelial phenotypic shift in human diabetic retinopathy and VEGF-A-induced retinopathy in monkeys. Ophthalmic Res. 2001;33:156–162. doi: 10.1159/000055663. [DOI] [PubMed] [Google Scholar]
- 37.Tyagi N., Roberts A.M., Dean W.L., Tyagi S.C., Lominadze D. Fibrinogen induces endothelial cell permeability. Mol Cell Biochem. 2008;307:13–22. doi: 10.1007/s11010-007-9579-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Viswanathan A., Chabriat H. Cerebral microhemorrhage. Stroke. 2006;37:550–555. doi: 10.1161/01.STR.0000199847.96188.12. [DOI] [PubMed] [Google Scholar]
- 39.Lifshitz V., Frenkel D. Animal models for cerebrovascular impairment and its relevance in vascular dementia. In: Landow M.L., editor. Cognitive Impairment: Causes, Diagnosis and Treatments. Nova Biomedical Books; New York: 2009. pp. 1–20. [Google Scholar]
- 40.Fruttiger M. Development of the retinal vasculature. Angiogenesis. 2007;10:77–88. doi: 10.1007/s10456-007-9065-1. [DOI] [PubMed] [Google Scholar]
- 41.Kim J., Wong P.K. Oxidative stress is linked to ERK1/2-p16 signaling-mediated growth defect in ATM-deficient astrocytes. J Biol Chem. 2009;284:14396–14404. doi: 10.1074/jbc.M808116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otani N., Nawashiro H., Fukui S., Ooigawa H., Ohsumi A., Toyooka T., Shima K., Gomi H., Brenner M. Enhanced hippocampal neurodegeneration after traumatic or kainate excitotoxicity in GFAP-null mice. J Clin Neurosci. 2006;13:934–938. doi: 10.1016/j.jocn.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 43.Pardo A.C., Wong V., Benson L.M., Dykes M., Tanaka K., Rothstein J.D., Maragakis N.J. Loss of the astrocyte glutamate transporter GLT1 modifies disease in SOD1(G93A) mice. Exp Neurol. 2006;201:120–130. doi: 10.1016/j.expneurol.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 44.Liu N., Stoica G., Yan M., Scofield V.L., Qiang W., Lynn W.S., Wong P.K. ATM deficiency induces oxidative stress and endoplasmic reticulum stress in astrocytes. Lab Invest. 2005;85:1471–1480. doi: 10.1038/labinvest.3700354. [DOI] [PubMed] [Google Scholar]
- 45.Wang X.F., Cynader M.S. Astrocytes provide cysteine to neurons by releasing glutathione. J Neurochem. 2000;74:1434–1442. doi: 10.1046/j.1471-4159.2000.0741434.x. [DOI] [PubMed] [Google Scholar]
- 46.Kuljis R.O., Xu Y., Aguila M.C., Baltimore D. Degeneration of neurons, synapses, and neuropil and glial activation in a murine Atm knockout model of ataxia-telangiectasia. Proc Natl Acad Sci U S A. 1997;94:12688–12693. doi: 10.1073/pnas.94.23.12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gosink E.C., Chong M.J., McKinnon P.J. Ataxia telangiectasia mutated deficiency affects astrocyte growth but not radiosensitivity. Cancer Res. 1999;59:5294–5298. [PubMed] [Google Scholar]
- 48.Yu D.Y., Cringle S.J. Oxygen distribution and consumption within the retina in vascularised and avascular retinas and in animal models of retinal disease. Prog Retin Eye Res. 2001;20:175–208. doi: 10.1016/s1350-9462(00)00027-6. [DOI] [PubMed] [Google Scholar]
- 49.Yu D.Y., Cringle S.J. Retinal degeneration and local oxygen metabolism. Exp Eye Res. 2005;80:745–751. doi: 10.1016/j.exer.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 50.Reme C.E., Braschler U.F., Roberts J., Dillon J. Light damage in the rat retina: effect of a radioprotective agent (WR-77913) on acute rod outer segment disk disruptions. Photochem Photobiol. 1991;54:137–142. doi: 10.1111/j.1751-1097.1991.tb01997.x. [DOI] [PubMed] [Google Scholar]
- 51.Specht S., Leffak M., Darrow R.M., Organisciak D.T. Damage to rat retinal DNA induced in vivo by visible light. Photochem Photobiol. 1999;69:91–98. [PubMed] [Google Scholar]
- 52.Barbazetto I.A., Room M., Yannuzzi N.A., Barile G.R., Merriam J.E., Bardal A.M., Freund K.B., Yannuzzi L.A., Allikmets R. ATM gene variants in patients with idiopathic perifoveal telangiectasia. Invest Ophthalmol Vis Sci. 2008;49:3806–3811. doi: 10.1167/iovs.07-1357. [DOI] [PubMed] [Google Scholar]
- 53.Mauget-Faysse M., Vuillaume M., Quaranta M., Moullan N., Angele S., Friesen M.D., Hall J. Idiopathic and radiation-induced ocular telangiectasia: the involvement of the ATM gene. Invest Ophthalmol Vis Sci. 2003;44:3257–3262. doi: 10.1167/iovs.02-1269. [DOI] [PubMed] [Google Scholar]
- 54.Kerbel R.S. Tumor angiogenesis. N Engl J Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 56.Aiello L.P., Avery R.L., Arrigg P.G., Keyt B.A., Jampel H.D., Shah S.T., Pasquale L.R., Thieme H., Iwamoto M.A., Park J.E. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- 57.Tolentino M.J., Miller J.W., Gragoudas E.S., Jakobiec F.A., Flynn E., Chatzistefanou K., Ferrara N., Adamis A.P. Intravitreous injections of vascular endothelial growth factor produce retinal ischemia and microangiopathy in an adult primate. Ophthalmology. 1996;103:1820–1828. doi: 10.1016/s0161-6420(96)30420-x. [DOI] [PubMed] [Google Scholar]
- 58.Hofman P., van Blijswijk B.C., Gaillard P.J., Vrensen G.F., Schlingemann R.O. Endothelial cell hypertrophy induced by vascular endothelial growth factor in the retina: new insights into the pathogenesis of capillary nonperfusion. Arch Ophthalmol. 2001;119:861–866. doi: 10.1001/archopht.119.6.861. [DOI] [PubMed] [Google Scholar]
- 59.D'Souza S.E., Byers-Ward V.J., Gardiner E.E., Wang H., Sung S.S. Identification of an active sequence within the first immunoglobulin domain of intercellular cell adhesion molecule-1 (ICAM-1) that interacts with fibrinogen. J Biol Chem. 1996;271:24270–24277. doi: 10.1074/jbc.271.39.24270. [DOI] [PubMed] [Google Scholar]
- 60.Plow E.F., Haas T.A., Zhang L., Loftus J., Smith J.W. Ligand binding to integrins. J Biol Chem. 2000;275:21785–21788. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- 61.Luscinskas F.W., Lawler J. Integrins as dynamic regulators of vascular function. FASEB J. 1994;8:929–938. doi: 10.1096/fasebj.8.12.7522194. [DOI] [PubMed] [Google Scholar]
- 62.Ehringer W.D., Yamany S., Steier K., Farag A., Roisen F.J., Dozier A., Miller F.N. Quantitative image analysis of F-actin in endothelial cells. Microcirculation. 1999;6:291–303. [PubMed] [Google Scholar]
- 63.Lominadze D., Saari J.T., Percival S.S., Schuschke D.A. Proinflammatory effects of copper deficiency on neutrophils and lung endothelial cells. Immunol Cell Biol. 2004;82:231–238. doi: 10.1046/j.1440-1711.2004.01231.x. [DOI] [PubMed] [Google Scholar]
- 64.Mehta D., Malik A.B. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 65.Qiao R.L., Yan W., Lum H., Malik A.B. Arg-Gly-Asp peptide increases endothelial hydraulic conductivity: comparison with thrombin response. Am J Physiol. 1995;269:C110–C117. doi: 10.1152/ajpcell.1995.269.1.C110. [DOI] [PubMed] [Google Scholar]
- 66.Trepat X., Grabulosa M., Buscemi L., Rico F., Farre R., Navajas D. Thrombin and histamine induce stiffening of alveolar epithelial cells. J Appl Physiol. 2005;98:1567–1574. doi: 10.1152/japplphysiol.00925.2004. [DOI] [PubMed] [Google Scholar]
- 67.Patibandla P.K., Tyagi N., Dean W.L., Tyagi S.C., Roberts A.M., Lominadze D. Fibrinogen induces alterations of endothelial cell tight junction proteins. J Cell Physiol. 2009;221:195–203. doi: 10.1002/jcp.21845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saitou M., Fujimoto K., Doi Y., Itoh M., Fujimoto T., Furuse M., Takano H., Noda T., Tsukita S. Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J Cell Biol. 1998;141:397–408. doi: 10.1083/jcb.141.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saitou M., Furuse M., Sasaki H., Schulzke J.D., Fromm M., Takano H., Noda T., Tsukita S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. 2000;11:4131–4142. doi: 10.1091/mbc.11.12.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abraham R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 71.Reliene R., Fischer E., Schiestl R.H. Effect of N-acetyl cysteine on oxidative DNA damage and the frequency of DNA deletions in atm-deficient mice. Cancer Res. 2004;64:5148–5153. doi: 10.1158/0008-5472.CAN-04-0442. [DOI] [PubMed] [Google Scholar]
- 72.Allen D.M., van Praag H., Ray J., Weaver Z., Winrow C.J., Carter T.A., Braquet R., Harrington E., Ried T., Brown K.D., Gage F.H., Barlow C. Ataxia telangiectasia mutated is essential during adult neurogenesis. Genes Dev. 2001;15:554–566. doi: 10.1101/gad.869001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Borghesani P.R., Alt F.W., Bottaro A., Davidson L., Aksoy S., Rathbun G.A., Roberts T.M., Swat W., Segal R.A., Gu Y. Abnormal development of Purkinje cells and lymphocytes in Atm mutant mice. Proc Natl Acad Sci U S A. 2000;97:3336–3341. doi: 10.1073/pnas.050584897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chiesa N., Barlow C., Wynshaw-Boris A., Strata P., Tempia F. Atm-deficient mice Purkinje cells show age-dependent defects in calcium spike bursts and calcium currents. Neuroscience. 2000;96:575–583. doi: 10.1016/s0306-4522(99)00581-3. [DOI] [PubMed] [Google Scholar]
- 75.Eilam R., Peter Y., Elson A., Rotman G., Shiloh Y., Groner Y., Segal M. Selective loss of dopaminergic nigro-striatal neurons in brains of Atm-deficient mice. Proc Natl Acad Sci U S A. 1998;95:12653–12656. doi: 10.1073/pnas.95.21.12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eilam R., Peter Y., Groner Y., Segal M. Late degeneration of nigro-striatal neurons in ATM-/- mice. Neuroscience. 2003;121:83–98. doi: 10.1016/s0306-4522(03)00322-1. [DOI] [PubMed] [Google Scholar]
- 77.Mount H.T., Martel J.C., Fluit P., Wu Y., Gallo-Hendrikx E., Cosi C., Marien M.R. Progressive sensorimotor impairment is not associated with reduced dopamine and high energy phosphate donors in a model of ataxia-telangiectasia. J Neurochem. 2004;88:1449–1454. doi: 10.1046/j.1471-4159.2003.02278.x. [DOI] [PubMed] [Google Scholar]
- 78.Cabana M.D., Crawford T.O., Winkelstein J.A., Christensen J.R., Lederman H.M. Consequences of the delayed diagnosis of ataxia-telangiectasia. Pediatrics. 1998;102:98–100. doi: 10.1542/peds.102.1.98. [DOI] [PubMed] [Google Scholar]