

Abstract
Increased expression of the discoidin domain receptor 2 (DDR2) results from its interaction with collagen type II. This induces expression of matrix metalloproteinase (MMP)-13, leading to osteoarthritis (OA). To investigate the impact of the pericellular matrix of chondrocytes on DDR2, we generated a mouse model with inducible overexpression of DDR2 in cartilage. Conditional overexpression of DDR2 in mature mouse articular cartilage was controlled via the cartilage oligomeric matrix protein promoter using the Tet-Off-inducible system. Doxycycline was withdrawn at 1 month of age, and knee joints were examined at 2, 3, and 4 months of age. Microsurgery was performed on 3-month-old transgenic mice overexpressing DDR2 to destabilize the medial meniscus, and serial paraffin sections were examined at 2, 4, 8, and 12 weeks after surgery. DDR2 expression increased in the knee joints of transgenic mice. However, the increased DDR2 did not induce MMP-13 expression. No OA-like changes were observed in the transgenic mice at the age of 4 months. When transgenic mice were subjected to destabilizing of the medial meniscus, we observed accelerated progression to OA, which was associated with DDR2 activation. Therefore, conditionally overexpressing DDR2 in the mature articular cartilage of mouse knee joints requires activation to induce OA, and altered biomechanical stress can accelerate the onset of cartilage loss and progression to OA in transgenic mice.
Osteoarthritis (OA) is a multifactorial disorder involving both genetic and environmental components.1,2 Environmental factors include obesity, overloading of joints, repetitive injury involving ligaments and menisci, loss of muscle strength, and joint malalignment. These conditions can result in abnormal mechanical stresses on the normal joint, leading to articular cartilage degeneration. The genetic factors such as gene mutations or variations result in defects or variability within joint structures. These conditions may lead to articular cartilage degeneration of the defective joint in the presence of normal mechanical stresses. Regardless of the nature of the factor(s) initiating the disease, it generally is accepted that the pathologic progression of articular cartilage degeneration follows a consistent pattern.3 One of the characteristic changes observed in OA progression is chondrocyte clustering within articular cartilage.4 However, the time point at which these chondrocyte clusters first appear is uncertain because the human OA samples used to study OA pathogenesis mostly are taken at the late or end stages of the disease. Recent data from animal models of OA show that chondrocyte clustering, with intensive pericellular proteoglycan staining, is one of the earliest pathologic changes seen in OA progression.5–7 Another feature characteristic of OA is the degradation of proteoglycans at the early stages. However, animal OA models indicate that increased proteoglycan staining appears before the disappearance of proteoglycans during the early stages. As OA progresses, the gradual loss of proteoglycans appears at the surface region of the articular cartilage. Interestingly, it has been shown that the loss of proteoglycans precedes or is concurrent with collagen type II degradation, as shown using an antibody that recognizes a neoepitope generated by matrix metalloproteinases (MMPs), including MMP-13.8 Cracks develop along the articular surface, producing the histologic image termed fibrillation. At later stages of the disease, fibrocartilage and osteophytes form at the periphery of the joint surface. Similarities in the pathologic progression of articular cartilage degeneration indicate the likelihood of a common molecular sequence(s) of events that eventually lead to OA.
In recent studies we have used genetic and nongenetic mouse models of OA and human OA tissues to investigate the molecular basis of articular cartilage degeneration.9–11 We found the following: i) increased expression of discoidin domain receptor 2 (DDR2), a cell membrane receptor tyrosine kinase for native collagen type II, was associated with the increased expression of MMP-13 in human OA cartilage and in the articular cartilage of the mouse models of OA; ii) reduced expression of Ddr2 attenuated OA progression in mouse models of OA; and iii) activation of DDR2 through interaction of chondrocytes with native collagen type II induced the expression of MMP-13 in chondrocytes in vitro. Based on the aforementioned results and data obtained from other investigators,12,13 we proposed a molecular chain of events responsible for articular cartilage degeneration. Our hypothesis was as follows. Altered mechanical forces, as a result of either normal loading of a defective joint or overloading of a normal joint, can incite chondrocytes to synthesize and release matrix-degrading enzymes. A consequence of this enzyme release is the degradation of the pericellular matrix of chondrocytes. This, in turn, enhances the exposure of the chondrocytes to native collagen type II. It should be duly noted that in normal mature articular cartilage, collagen type II is present within the territorial and interterritorial locations of the cartilage and that little to none is present close to the chondrocyte surface. Interaction of the chondrocytes with native collagen type II fibrils results in up-regulated activity and expression of DDR2 and induction of MMP-13. MMP-13 cleaves collagen type II and aggrecan.14,15 The resulting fragments of collagen type II and aggrecans may further increase the synthesis of MMP-13 through interaction with other cell-surface receptors.16 The result is a positive feedback amplification loop that leads to the irreversible destruction of the articular cartilage. Obviously, one of the key molecular events in our hypothesis is that the activation of DDR2, by exposure of chondrocytes to collagen type II, increases the expression of MMP-13 and the receptor itself. The prerequisite condition necessary to expose chondrocytes to collagen type II is the disappearance of the pericellular matrix of the cells. Thus, the pericellular matrix is the key component to prevent collagen type II from contacting the surface of chondrocytes. If that is the case, then the conditional induction of DDR2 in chondrocytes surrounded by a pericellular matrix should be incapable of activation because the pericellular matrix prevents DDR2 from directly contacting collagen type II. Thus, MMP-13 would not be induced under this condition.
In the present study, we used a tetracycline-controlled gene expression system to induce overexpression of DDR2 in the postnatal articular cartilage of mouse knee joints. The conditional induction was driven by a promoter of cartilage oligomeric matrix protein (COMP), which is expressed continuously in cartilage throughout life. For the first time, we were able to directly examine the biological effects of up-regulated expression of DDR2 on the normal mature articular cartilage of mouse knee joints. We also performed microsurgery to destabilize the medial meniscus (DMM) in the knee joints of mice overexpressing DDR2. We characterized the morphology of the knee joints of those mice, both with and without DMM surgery, for evidence of articular cartilage degeneration.
Materials and Methods
Analysis of Comp and Col2a1 mRNAs in the Mature Articular Cartilage of Mouse Knee Joints
To decide which promoter, Comp or α1(II) collagen (Col2a1), would be used to generate the transgenic mice, we measured the levels of Comp and Col2a1 mRNAs in the mature articular cartilage of mouse knee joints by real-time PCR. Articular cartilage samples were sliced from the surfaces of the knee joints from 6 mice (C57BL/6J) at 3 months of age. The cartilage samples then were pooled for isolation of RNA using the Total RNA Isolation System (Promega, Madison, WI). The cDNAs were synthesized with oligo(dT) primer using the Super-Script First-Strand Synthesis System (BD Biosciences Clontech, CA). Real-time PCR conditions were optimized for maximal PCR efficiency by adjusting the concentrations of PCR primers: Comp, forward 5′-GCTACCAAGACAGCTCCA-3′ and reverse 5′-GTGCCAAAGTGCGTTTCG-3′; Col2a1, forward 5′-ATGTAGAGATGAGGGCCG-3′ and reverse 5′-CAATAATGGGAAGGCGGGA-3′; glyceraldehyde-3-phosphate dehydrogenase, 5′-ACTGAGGACCAGGTTGTC-3′ and reverse 5′-TGCTGTAGCCGTATTCATTG-3′. The optimum concentrations of primers for each gene were as follows: forward 300 nmol/L, and reverse 300 nmol/L. The t-test analysis was used to detect differences in mRNA levels between the control and experimental groups. A 5% significance level was used. PCR was performed using 25 μL of 1X PCR buffer containing 3 μmol/L MgCl2, 200 nmol/L dNTPs, PCR primers, 1X SYBR Green, 0.5 U TaqDNA polymerase (Qiagen), and 0.5 μL cDNA. Real-time PCR was performed using the Icycler iQ detection system (Bio-Rad, Hercules, CA), and the PCR reaction was performed at 95°C for 3 minutes followed by 50 cycles at 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds, with a final extension at 72°C for 4 minutes. At the end of the PCR cycles, a melting curve, using a temperature range from 55°C to 95°C with +0.5°C intervals, was generated to test the specificity of the PCR product. A cDNA sample in each experiment was tested in triplicate, and each experiment was performed twice. We used glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the internal control. The efficiency of PCR represented by a standard curve also was tested by plotting the amount of PCR product versus the known amount of a template: 0.001 ng, 0.01 ng, 0.1 ng, 1 ng, and 10 ng. In this experiment the efficiency reached 90% or higher.
Construction of Transgenes Comp-tTA and TRE-DDR2
For the Comp-tTA transgene [the tetracycline-responsive transcriptional activator (tTA) driven by Comp promoter], a 2.3-kb (−2270 to + 14) DNA fragment was cloned by PCR using C57BL/6J genomic DNA as the template and the following primers: forward: 5′-GCACACCTTTAATCCCAGCA-3′, and reverse: 5′-GCAGCTGCAGCTCCGCCGCCGTCGAC-3′. The PCR product then was subcloned into a TA cloning vector (pCR2.1; Invitrogen) and sequenced. A DNA fragment of KpnI/XhoI containing the Comp promoter was released from the pCR2.1 vector and inserted into the pBluescript II SK. For tTA, the DNA fragment, containing amino acids 1 to 207 of the Tet repressor and the negatively charged C-terminal activation domain (130 amino acids) of the VP16 protein of herpes simplex virus, was removed by digestion of the pTet-off vector (Clontech Laboratories, CA) with EcoRI and BamHI. To construct the Comp-tTA transgene, the tTA was subcloned into the pBluescript II SK containing the Comp promoter. For TRE-DDR2 (the DDR2 driven by Tet-responsive sequence element), a 2.6-kb DNA fragment (representing an entire length of cDNA of DDR2) was removed from a previous vector by digestion with KpnI and NotI and subcloned into pTRE-HA.
Generation of Transgenic Mice
All plasmid preparations were purified using the EndoFree Plasmid Maxi-kit (Qiagen). The transgene construct, Comp-tTA, was removed from the pBluescript II by KpnI and BamHI. The transgene construct, TRE-DDR2, was removed from pTRE-HA by XhoI and NotI. The constructs were purified using the Qiagen Gel Extraction kit and eluted with the microinjection buffer, 10 mmol/L Tris-HCl, pH 8.0, and 0.25 mmol/L EDTA, pH 8.0. The constructs were sent to the Transgenic Mouse Facility at Brigham and Women's Hospital at Harvard Medical School. For generation of Tet-off transgenic mice, Comp-tTA and TRE-DDR2 mice were identified by PCR. Genomic DNA was isolated from the mouse tails (0.5 cm). Two pairs of primers were synthesized. One pair of primers was used for identification of Comp-tTA mice: forward 5′-ATGCCTTTCTCTCCTCAGACC-3′ and reverse 5′-TTGCTCCATCGCGATGACTT-3′. The other pair of primers was for identification of TRE-DDR2: forward 5′-ACGATGTTCCAGATTACGCTC-3′ and reverse 5′-ATGACGGTTCCGCCAAGAGAT-3′. Comp-tTA mice then were crossed with TRE-DDR2 mice. Pregnant mice were treated with doxycycline in the drinking water. Females with litters were treated continuously with doxycycline until weaning (about 4 weeks old). Comp-tTA, TRE-DDR2, Comp-tTA;TRE-DDR2, and their wild-type littermates were identified by PCR and maintained with regular water and food. The mice were sacrificed at the ages of 8, 12, and 16 weeks, at which time the knee joints were collected for further experiments.
Analysis of DDR2 mRNA in Comp-tTA;TRE-DDR2 Mice
Total RNA was isolated from the knee articular cartilages of 6 Comp-tTA;TRE-DDR2 mice and 6 wild-type littermates at the age of 12 weeks (2 months after doxycycline was withdrawn), using the Total RNA Isolation System (Promega). The rest of the experimental procedure was the same as that described earlier with concentrations of PCR primers for DDR2: forward 5′-GGCCACACGAAACTGTTTAGT-3′ and reverse 5′-CAAAGGCCCACACATCACT-3′.
DMM of Knee Joints in Compound Transgenic Mice
The detailed procedure for DMM has been described in our previous publications.9,11 Briefly, Comp-tTA;TRE-DDR2 and TRE-DDR2 mice (as positive control) at the age of 12 weeks (2 months after doxycycline withdrawal) were anesthetized intraperitoneally with ketamine (90 mg/kg mouse body weight) and xylazine (10 mg/kg mouse body weight). The right knees were prepared for aseptic surgery. The joint capsule immediately medial to the patellar tendon was opened to provide visualization of the medial meniscotibial ligament. The medial meniscotibial ligament was sectioned resulting in DMM. After sectioning of the medial meniscotibial ligament, the joint capsule then was closed. The mice were maintained on a daily schedule of 12 hours light/dark for further experiments. Sham surgery on the wild-type littermates was performed as a negative control.
Immunohistochemistry
For double immunohistostaining, four mice from each experimental group, including a mouse model of OA (heterozygous chondrodysplasia, cho/+) at 6 months of age, mice at 8 weeks after DMM surgery, and Comp-tTA;TRE-DDR2 and TRE-DDR2 transgenic mice at 2 months after doxycycline withdrawal, were sacrificed for knee joint collection. We previously identified a mutation in Col11a1 as the genetic cause of cho in mice.17 The mutation, a single nucleotide deletion, results in premature termination of the α1 chain of type XI collagen. The protein product and mRNA of Col11a1 are not detectable in cho/cho mice, which show prenatal lethality.17 All joints were fixed and processed for paraffin embedding. For each knee joint of cho/+, Comp-tTA;TRE-DDR2, TRE-DDR2 mice, and appropriate control mice 6-μm–thick serial sections were cut from the lateral to the medial side of the joints. About 150 sections covered an entire joint. For each knee joint of mice subjected to DMM and sham surgery, 6-μm–thick serial sections were cut from the anterior to the posterior side of the joints. About 100 sections covered an entire joint. Seven to eight sections, evenly distributed throughout each joint, were collected for immunostaining. The sections were deparaffinized and quenched for endogenous peroxidase activity. The sections were incubated with primary polyclonal antibodies [rabbit polyclonal antibody against mouse Ddr2 (1:200 dilution, cat. no. H-108; Santa Cruz Biotechnology, Santa Cruz, CA), goat polyclonal antibody against mouse Mmp-13 (1:200 dilution, cat. AB8120; Chemicon, Temecula, CA), or rabbit polyclonal antibody against mouse collagen type VI (1:200 dilution, cat. no. sc-20649; Santa Cruz Biotechnology)] at 4°C overnight. After washing with PBS three times, the sections were treated with mixed secondary antibodies, Alexa Fluor 488 donkey anti-rabbit IgG, and Alexa Fluor 594 donkey anti-goat IgG at room temperature for 30 minutes. The sections then were washed three times with PBS and examined under a fluorescence microscope (Axio Imager; Zeiss).
For analysis of Tet-repressor protein in Comp-tTA;TRE-DDR2 mice, at the ages of 8, 12, and 16 weeks (1, 2, and 3 months after doxycycline withdrawal, respectively) they were sacrificed for knee joint collection (n = 4). All of the joints were fixed in 4% paraformaldehyde for 6 hours at room temperature. The samples then were processed for paraffin embedding. For each knee joint, serial sections of 6-μm thickness were taken. Every 20th section was selected for immunohistostaining. Sections were deparaffinized and quenched for endogenous peroxidase activity. The sections were incubated with rabbit polyclonal antibody (1:400 dilution) against Tet-repressor protein (cat. no. TET01, MoBiTec; Göttingen, Germany). After overnight incubation at 4°C, the sections were washed with PBS three times and subsequently treated with a biotinylated secondary antibody. Color development was performed using a peroxidase substrate (Vector NovaRED Substrate, cat. no. SK-4800; Vector Laboratories, Burlingame, CA) after treatment of the sections with a mixture of avidin and biotinylated horseradish peroxidase (Vectastain ABC Kit, cat. no. PK-4000; Vector Laboratories). Sections were counterstained with 0.2% Fast Green solution. Staining with isotype-matched normal IgG (Vector Laboratories) and staining without primary antibody also were performed as negative controls.
Histologic Analysis of the Articular Cartilage of Knee Joints of Comp-tTA;TRE-DDR2 Mice
Comp-tTA;TRE-DDR2 mice, TRE-DDR2 mice, and their wild-type littermates at 8, 12, and 16 weeks of age (corresponding to 1, 2, and 3 months after doxycycline withdrawal, respectively) were sacrificed for knee joint collection (n = 6 to 8). All of the joints were fixed and processed for paraffin embedding and stained with Safranin O/Fast green. Serial sections (6-μm thick) were cut, and every 10th section was collected for staining. The morphology of the joints was examined under light microscopy. The pathologic condition of the knee joints was evaluated by a modified Mankin scoring system.5 In this system, the condition of the joint was ranked on the basis of intensity in staining of the pericellular matrix (0 to 2), staining intensity of the territorial and interterritorial matrix (0 to 3), spatial arrangement of chondrocytes (0 to 2), and overall cartilage structure (0 to 3). The score for the articular cartilage of knee joints of wild-type mice at 2 weeks after sham surgery was 0, and the maximum score for the degenerative articular cartilage was 10.
Characterization of Knee Joints of Comp-tTA;TRE-DDR2 Mice Subjected to DMM
The experimental procedure for histology is the same as that described earlier. Six to eight knee joints were harvested from each group of Comp-tTA;TRE-DDR2 mice, TRE-DDR2 mice, their wild-type littermates, and sham surgery mice. For detection of the degraded collagen type II, knee joints from mice at 12 weeks after DMM surgery were collected. Six to eight paraffin sections from each mouse knee joint were used for immunohistostaining. The experimental procedure for immunostaining was the same as that described earlier (see Immunohistochemistry). The sections were incubated with primary rabbit polyclonal antibody against degraded collagen type II, C1-2C (1:500 dilution, cat. no. 2699; IBEX Pharmaceuticals, Inc.).
Statistical Analyses
The pathologic condition of the knee joints was evaluated by the modified Mankin scoring system.5 For statistical analyses, 8 to 10 histologic staining sections from each knee joint of an animal were scored and an average score was obtained. There were six to eight animals in each experimental group. Thus, six to eight average scores were obtained for each group. The average of the six to eight average scores was used for statistical comparison between two experimental groups with the Mann–Whitney U test. Differences were considered significant if the P < 0.01. Meniscus, subchondral bone, and osteophyte formation also were examined to evaluate the overall condition of the joints.
Results
Co-Localized Expression of Ddr2 and Mmp13 in the Degenerative Articular Cartilage of Mouse Knee Joints
Although we showed in our previous studies8–10 that increased levels of Ddr2 and Mmp13 were present in adjacent sections of OA cartilage, we did not show co-localization of these two proteins. By immunofluorescence staining, we found that the increased expression of Ddr2 indeed co-localized in the same cells with increased expression of Mmp13 in the knee joints of cho/+ mice at 6 months of age (Figure 1A) and in the knee joints of mice 8 weeks after DMM surgery (Figure 1B). Ddr2 and Mmp13 expression, therefore, corresponded with the degradation of proteoglycans and collagen type II that we previously had observed at those time points in the knee joints of the mice.5,8
Figure 1.
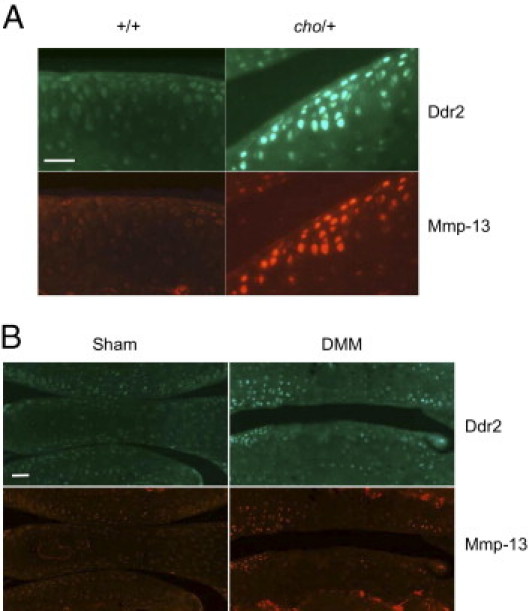
Co-localized expression of Ddr2 and Mmp-13 in the articular cartilage of mouse knee joints. The green-stained cells show positive staining for Ddr2, and the red-stained cells show positive staining for Mmp-13. A: Comparison of the top and bottom images shows that the same cells stain positively for both Ddr2 and Mmp-13 in the articular cartilage of knee joints of cho/+ mice at 6 months of age. B: The expressions of Ddr2 and Mmp-13 are also co-localized in the articular cartilage of knee joints of mice at 8 weeks after DMM surgery. Scale bar = 50 μm.
Generation of Tet-Off Transgenic Mice
We examined the levels of Comp and Col2a1 mRNAs within the knee articular cartilage of mice at 3 months of age by real-time PCR. The level of Comp mRNA was about four and a half times higher than that of Col2a1 mRNA. Based on these results, we generated two transgenic mouse strains, Comp-tTA and TRE-DDR2 (Figure 2). Four different lines of Comp-tTA and three different lines of TRE-DDR2 mice were established. No gross abnormality was observed in any of these transgenic mice. Results from skeletal staining indicated that the sizes of the transgenic mice and their wild-type littermates were similar (data not shown). Comp-tTA;TRE-DDR2 were generated by crossing Comp-tTA with TRE-DDR2 mice.
Figure 2.

The Tet-off system with Comp-tTA and TRE-DDR2 transgenic genes. The Comp-tTA transgene contains a 2.3-kb DNA fragment of mouse Comp promoter followed by a DNA fragment of the tTA gene. tTA produces a fused protein of the Tet repressor and the activation domain. The TRE-DDR2 transgene contains the Tet-responsive sequence element followed by a 2.6-kb cDNA of the human DDR2 gene. When doxycycline is present, the doxycycline binds to the fused protein to prevent it from binding to the TRE. Thus, there is no mRNA transcription of human DDR2. When doxycycline is absent, the fused protein binds to the TRE and activates transcription of human DDR2. tTA: fusion protein = Tet repressor + activation domain; TRE: Tet-responsive sequence element.
Expression of tTA, DDR2, and Collagen Type VI in Comp-tTA;TRE-DDR2 Mice
To determine whether the Comp promoter effectively induced the expression of tTA, we examined the expression of tTA protein by immunohistochemistry of Comp-tTA;TRE-DDR2 and TRE-DDR2 mice at three time points: 1, 2, and 3 months after doxycycline withdrawal. We observed the protein expression of tTA in the knee joints of Comp-tTA;TRE-DDR2 mice at all three time points throughout the cartilage but predominantly within the uncalcified layer of the articular cartilage (Figure 3A). There was no difference in the expression patterns of tTA among the mice. Expression of DDR2 was barely detectable within the knee joints of Comp-tTA;TRE-DDR2 mice at 1 month after doxycycline withdrawal (data not shown). However, DDR2 protein was present in the uncalcified layer of the knee joints of the mice at 2 months after doxycycline withdrawal (Figure 3B), as well as at 3 months after doxycycline withdrawal (data not shown). Because it is expressed exclusively in the pericellular matrix of the chondrocytes in articular cartilage, collagen type VI is a good indicator of the integrity of the pericellular matrix. We found that cells in Comp-tTA;TRE-DDR2 mice, as well as in TRE-DDR2 mice, that expressed DDR2 were surrounded by collagen type VI (Figure 3B).
Figure 3.

Expression of Tet repressor, DDR2, and collagen type VI in the TRE-DDR2 and ComptTA;TRE-DDR2 mice. Knee joints were isolated from TRE-DDR2 and ComptTA;TRE-DDR2 mice at 3 months of age (2 months after doxycycline withdrawal). A: The brown staining of cells shows positive staining for Tet repressor in ComptTA;TRE-DDR2 mice. B: The green staining of cells shows positive staining for DDR2 in ComptTA;TRE-DDR2 mice. The positive staining of cells for DDR2 was distributed predominantly in the uncalcified layer of the articular cartilage. In contrast, positive staining for tTA (A) and DDR2 (B) in TRE-DDR2 mice. The red staining of cells shows positive staining for collagen type VI (B). Collagen type VI was present in both TRE-DDR2 and ComptTA;TRE-DDR2 mice. Scale bar = 50 μm. C: mRNA was isolated from the knee articular cartilages of the TRE-DDR2 and compound transgenic mice (n = 6 in each group) at 3 months of age (2 months after doxycycline withdrawal). The levels of DDR2 mRNA in the mice were measured by real-time PCR. The level of DDR2 mRNA in the compound transgenic mice were about 2.17 times higher than that in TRE-Ddr2 mice. *2.17 fold compared to the control.
To validate our observations, we found that the level of DDR2 mRNA was increased by about 2.17-fold in the Comp-tTA;TRE-DDR2 mice at 2 months after doxycycline withdrawal, compared with the TRE-DDR2 littermates used as negative controls (Figure 3B).
Morphology of the Articular Cartilage of Knee Joints in Comp-tTA;TRE-DDR2 Mice
To determine whether the induced overexpression of DDR2 in the articular cartilage would initiate early onset degeneration, we characterized the morphology of the knee joints from Comp-tTA;TRE-DDR2 mice, TRE-DDR2 mice, and their wild-type littermates. We examined knee joints at four different time points: i) after weaning (n = 8, immediately after doxycycline withdrawal), ii) at 2 months of age (n = 8, 1 month after doxycycline withdrawal), iii) at 3 months of age (n = 8, 2 months after doxycycline withdrawal), and iv) at 4 months of age (n = 8, 3 months after doxycycline withdrawal). We did not observe any difference in morphology in the knee joints of Comp-tTA;TRE-DDR2 mice, TRE-DDR2 mice, and their wild-type littermates or any sign of early onset articular cartilage degeneration at any of the postnatal developmental time points (data not shown).
Morphology of the Articular Cartilage of Knee Joints in Comp-tTA;TRE-DDR2 Mice Subjected to DMM Surgery
To understand the effects of induced overexpression of DDR2 in conjunction with altered biomechanical stress on mouse knee joints, we performed DMM surgery on the knee joints of Comp-tTA;TRE-DDR2 mice, TRE-DDR2 mice, and their wild-type littermates at 2 months after doxycycline withdrawal. We observed the progression of articular cartilage degeneration in the knee joints of the Comp-tTA;TRE-DDR2 mice, TRE-DDR2 mice, and their wild-type littermates. No morphologic difference in the progression of degeneration was found between the TRE-DDR2 mice and their wild-type littermates. The progression of degeneration was accelerated in the knee joints of Comp-tTA;TRE-DDR2 mice after surgery, compared with TRE-DDR2 controls and their wild-type littermates (Figure 4). We also found that more degraded collagen type II was present in the Comp-tTA;TRE-DDR2 mice at 12 weeks after DMM surgery compared with the TRE-DDR2 littermates (Figure 5).
Figure 4.
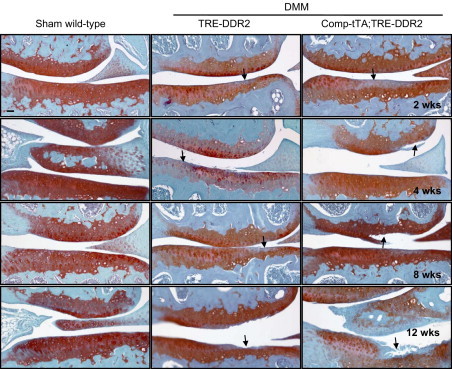
Histology of the articular cartilage of knee joints from TRE-DDR2 and Comp-tTA;TRE-DDR2 mice after DMM surgery and their wild-type littermates after sham surgery. Each image represents one section selected from 70 to 90 sections of an experimental group (about 12 sections from each knee joint and 6 to 8 knee joints in each experimental group). The representative section shows the worst conditions observed in each experimental group. There were no obvious morphologic changes observed in the knee joints of the wild-type littermates after sham surgery. However, at 2 weeks after surgery, regional reduction of Safranine O staining (arrows) was observed in both TRE-DDR2 and Comp-tTA;TRE-DDR2 mice. At 4 and 8 weeks after surgery, the regional reduction of Saranine O staining was seen through the entire articular cartilage in TRE-DDR2 mice. Furthermore, fibrillation also was seen (arrows) in Comp-tTA;TRE-DDR2 mice. At 12 weeks after surgery, the articular cartilage appears thinner (arrows) in both transgenic mice with a more severe damaged appearance of articular cartilage in Comp-tTA;TRE-DDR2 mice. All together, the degenerative condition of the articular cartilages in Comp-tTA;TRE-DDR2 mice was more advanced when compared with that in TRE-DDR2 mice at each of the time points after DMM surgery. This suggests that the rate of degenerative progression was increased in the compound transgenic mice compared with the TRE-DDR2 mice. Scale bar = 100 μm.
Figure 5.
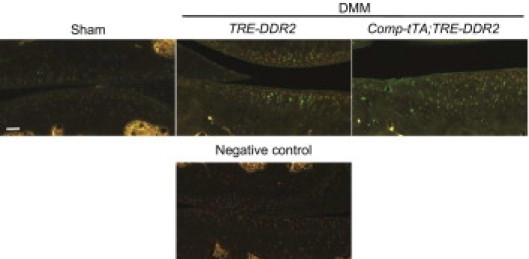
Degraded collagen type II in the articular cartilage of knee joints of mice. Degraded collagen type II was examined by use of a polyclonal antibody recognizing a collagen type II neoepitope generated by MMPs, including MMP-13. Compared with that in TRE-DDR2 mice, more degraded collagen type II (green) was detected in the articular cartilage of knee joints in the Comp-tTA;TRE-DDR2 mice at the age of 3 months (2 months after doxycycline withdrawal). Scale bar = 50 μm.
The condition of the knee joint articular cartilage also was evaluated by a modified Mankin scoring system (Figure 6). The scores were higher (indicating more damage present) in the Comp-tTA;TRE-DDR2 mice, particularly more than 4 weeks after the surgery. There were no significant differences in the scores between TRE-DDR2 mice and their wild-type littermates after DMM surgery.
Figure 6.
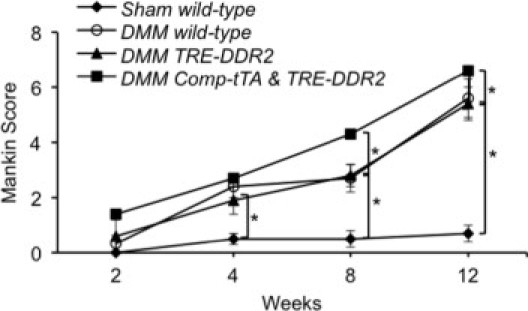
Average modified Mankin scores of the TRE-DDR2 and Comp-tTA;TRE-DDR2 mice after DMM and their wild-type littermates after sham surgery. The morphologic conditions of the articular cartilages from the knee joints were evaluated. An average score representing six to eight animals from each experimental group was obtained. A statistical comparison of the scores using the Mann–Whitney U test indicated a significant difference among the groups at each age. *P < 0.01. Wild-type mice at 2 weeks after sham surgery served as control, score 0. At 2 weeks after DMM surgery, scores for the Comp-tTA;TRE-DDR2 mice, TRE-DDR2 mice, and their wild-type littermates were 1.4, 0.6, and 0.5, respectively. At 4 weeks after surgery, scores were 2.7 for the Comp-tTA;TRE-DDR2 mice, 1.9 for TRE-DDR2 mice, 2.4 for the wild-type littermates, and 0.5 for the sham surgery mice. At 8 weeks after surgery, scores were 4.3 for the Comp-tTA;TRE-DDR2 mice, 2.8 for TRE-DDR2 mice, 2.7 for the wild-type littermates, and 0.5 for the sham surgery mice. At 12 weeks after surgery, scores were 6.6 for the Comp-tTA;TRE-DDR2 mice, 5.3 for TRE-DDR2 mice, 5.6 for their wild-type littermates, and 0.7 for the sham surgery mice. We found the degenerative progression of the Comp-tTA;TRE-DDR2 mice to be accelerated, particularly at 4 weeks after DMM surgery.
Discussion
The molecular mechanisms by which articular cartilage degeneration is initiated and progresses are largely unknown. Our objective in this study was to provide in vivo evidence to support the role of a molecular pathway, which we proposed to be the major route underlying initiation of cartilage matrix degradation. One of the molecular events in this pathway is the activation of DDR2, a receptor tyrosine kinase that interacts with native collagen type II, which results in increased expression of the receptor itself in chondrocytes and induction of Mmp13 gene expression. In our previous studies, we showed that increased expression of DDR2 was associated with increased MMP-13 protein and increased collagen type II cleavage products in the articular cartilage of human OA patients and mouse models of OA. In this study, the co-localized expression of Ddr2 and Mmp-13 in the knee joints of cho/+ mice and DMM surgical mice corresponded with our previous observations,8–10 suggesting that the presence of Ddr2 is associated directly with the induction of Mmp-13 in the chondrocytes of degenerative articular cartilages. The data also suggest that a defective extracellular matrix in cho/+ mice and mechanical injury of the joint are critical factors involved in the activation of Ddr2. Because expression of Ddr2 is barely detectable in normal articular cartilage, questions remain as to how Ddr2 is maintained in an inactive state under normal conditions and what direct effect the defective extracellular matrix of cho/+ mice or DMM surgery has on the progression of articular cartilage degeneration.
Results from several other research groups have indicated that the pericellular matrix of chondrocytes is the crucial structure silencing DDR2 on chondrocytes.13,18–20 The pericellular matrix is composed of numerous molecules forming a discernible extracellular matrix network, which includes matrilin 3, fibronectin, biglycan, fibromodulin, COMP, and collagens type VI and IX. There is little to no collagen type II in the pericellular matrix. Thus, DDR2 on the surface of chondrocytes is separated from collagen type II by the pericellular matrix, and thus remains inactive. Disruption of the pericellular matrix exposes chondrocytes to collagen type II, resulting in abnormal chondrocyte activities such as activation of DDR2. Poole et al21 have reported morphologic changes of the pericellular matrix of chondrocytes in the OA cartilage. Results from their study indicated that the pericellular matrix in the degenerative cartilage swells and expands. The amount of proteoglycans becomes reduced, and a fine collagen network, consisting of type VI and IX collagens, is disrupted within the pericellular matrix. Moreover, data from a number of recent studies indicated that deletions of type VI and IX collagens, matrilin 3, biglycan, and fibromodulin—all of which are components of the pericellular matrix—result in early onset articular cartilage degeneration.22–24 Although it is clear that the disruption of the pericellular matrix is implicated in the initiation of articular cartilage degeneration, the question remains as to how the pericellular matrix is degraded and then leads to progression characterized by loss of aggrecan and collagen in the interterritorial matrix.
Interestingly, we recently found that the expression of the serine protease high temperature requirement A1a (HtrA1) was increased in the articular cartilage of our four mouse models of OA,25 including cho/+ and DMM surgical models. Results from our studies indicated that increased levels of HtrA1 degraded the pericellular matrix and were associated with the increased expression of Ddr2 in the knee joints of those mouse models at early stages of OA. Expression of HTRA1 also predominantly is increased in human OA cartilage.26 More importantly, it has been shown that HTRA1 degrades most of the pericellular matrix components, including matrilin 3, fibronectin, biglycan, fibromodulin, and COMP.27–29 Thus, it is conceivable that altered mechanical stress, by either normal loading on a defective matrix (in the case of cho/+ mice) or abnormal mechanical stress on a normal joint (in the case of DMM surgery), induces up-regulation of HTRA1 in chondrocytes, which then degrades the pericellular matrix of the chondrocytes. Clearly, the pericellular matrix plays a significant role in the pathogenesis of OA.
Results from our transgenic mouse experiments, using the tetracycline-regulated gene expression system (Tet-Off), provide further in vivo evidence that the pericellular matrix is critical for maintenance of normal chondrocyte activities. We used the Tet-Off system to induce up-regulated expression of DDR2 in the mature articular cartilage of normal mouse knee joints. The advantage of using this system is the ability to investigate the consequences of increased DDR2 in mature articular cartilage without any potential abnormality resulting from increased expression of the receptor during embryonic or early postnatal developmental stages. One of the critical elements in this system is a tissue-specific promoter that controls expression of the Tet-off recombinant protein (tTA), the DNA-binding domain of Tet repression fused with herpesvirus transcriptional activation domain VP16. We chose the Comp promoter to drive tTA expression based on our previous accidental finding that Comp mRNA levels were higher than collagen type II (Col2a1) mRNA levels in a microarray analysis of mRNAs isolated from the knee joint articular cartilage of 3-month-old mice (Xu et al, unpublished data). COMP is one of the noncollagen components of the matrix of articular cartilage,30 and the promoter sequence of this gene has been identified.31 A study by Posey et al32 reported the use of the human COMP promoter for cartilage-specific expression in the mouse embryo. We investigated, therefore, the capacity of the Comp promoter to drive luciferase activity in a human chondrocyte cell line. Our unpublished data (Xu et al) suggest that the high level of Comp mRNA in mouse knee articular cartilage is caused, at least partially, by a high level of transcriptional activity of the Comp promoter. In the present study, we confirmed our previous finding by comparing the levels of Comp and Col2a1 mRNAs in articular cartilage of adult mice. More importantly, we found that the Comp promoter could drive the expression of tTA, inducing expression of TRE-driven DDR2 in the mature articular cartilage of mouse knee joints. Regarding the expression pattern of COMP, it has been suggested that its distribution is ubiquitous throughout cartilage. However, our results showed that the Comp promoter-driven tTA was expressed predominantly in the uncalcified layer of mature articular cartilage. Because there is no information about the expression pattern of the endogenous Comp promoter in mature mouse knee joints, it is unclear whether the exogenous Comp promoter reflects the in vivo situation. Although the inducible expression of DDR2 by tTA is largely limited to the uncalcified layer of articular cartilage, this, in fact, corresponds to the DDR2 expression pattern that we observed in our mouse models of OA. We noticed that the expression of DDR2 was barely detectable in Comp-tTA;TRE-DDR2 mice 1 month after doxycycline withdrawal, although the expression of tTA was evident at that time point. One plausible explanation is that 1 month was not sufficient for complete disappearance of doxycycline from the chondrocytes. Nonetheless, our findings indicate that the Comp promoter may provide a more efficient way of assessing the consequences of targeted transgene delivery or knockout in the mature articular cartilage of mouse models.
Data from knee joints of TRE-DDR2 and their wild-type littermates after DMM surgery indicate that there is no significant morphologic difference in those mice during the progression of articular cartilage degeneration. This suggests that insertion of the TRE-DDR2 transgene does not affect the biological consequences of DMM surgery. Our results show that although the overexpression of DDR2 was induced in the Comp-tTA;TRE-DDR2 mice, those cells were surrounded by collagen type VI. This suggests that the integrity of the pericellular matrix remains intact. DDR2 on the surface of chondrocytes is separated from the collagen type II by the pericellular matrix. Thus, DDR2 is not activated and MMP-13 expression is not induced in the chondrocytes. In contrast, accelerated pathologic progression of degeneration occurs in the Comp-tTA;TRE-DDR2 mice after DMM surgery. This is consistent with our previous observation that DMM surgery induces expression of HtrA1, which then disrupts the pericellular matrix. In the Comp-tTA;TRE-DDR2 mice, the disruption of the pericellular matrix, coupled with induced overexpression of DDR2, accelerates the progression of articular cartilage degeneration in the transgenic mice. These data suggest that DDR2 can accelerate OA progression but cannot initiate articular cartilage degeneration in the absence of biomechanical and/or biochemical disruption of the pericellular matrix (Figure 7). Therefore, maintaining the integrity of the pericellular matrix is one of the key factors in protecting against OA. We believe that more attention should be directed toward the pericellular matrix of chondrocytes for the identification of novel biomarkers and therapeutic targets for OA, thereby permitting intervention before significant degradation of collagen type II and aggrecan can occur.
Figure 7.
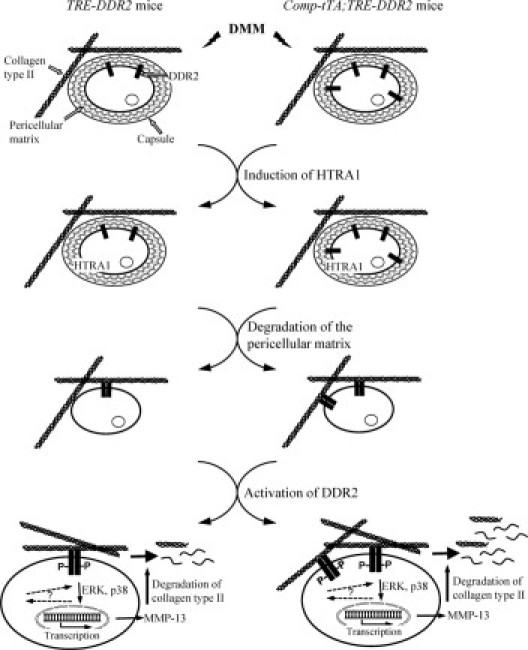
A molecular pathway underlying articular cartilage degeneration. Altered mechanical stress, caused by DMM, activates chondrocytes to synthesize and release HTRA1. The left side represents the condition of TRE-DDR2 mice and the right side represents the condition of the Comp-tTA;TRE-DDR2 mice, which contain a higher level of DDR2. HTRA1 degrades the pericellular matrix of chondrocytes. This, in turn, enhances the exposure of the chondrocytes to collagen type II. Interaction of the chondrocytes with collagen type II–containing fibrils results in active DDR2 and increased expression of the receptor itself. The activation of DDR2 then induces the expression of MMP-13, which cleaves collagen type II and aggrecan. Accelerated articular cartilage degeneration seen in the Comp-tTA;TRE-DDR2 mice is caused by the interaction of collagen type II with a higher level of DDR2.
Footnotes
Supported by National Institutes of Health grants R01-AR051989 (Y.L. and L.X.) and grant R01-AG022021 (M.B.G.).
Contributor Information
Lin Xu, Email: lin_xu@hms.harvard.edu.
Yefu Li, Email: yefu_li@hms.harvard.edu.
References
- 1.Felson D.T., Lawrence R.C., Dieppe P.A., Hirsch R., Helmick C.G., Jordan J.M., Kington R.S., Lane N.E., Nevitt M.C., Zhang Y., Sowers M., McAlindon T., Spector T.D., Poole A.R., Yanovski S.Z., Ateshian G., Sharma L., Buckwalter J.A., Brandt K.D., Fries J.F. Osteoarthritis: new insights: Part 1: the disease and its risk factors. Ann Intern Med. 2000;133:635–646. doi: 10.7326/0003-4819-133-8-200010170-00016. [DOI] [PubMed] [Google Scholar]
- 2.Li Y., Xu L., Olsen B.R. Lessons from genetic forms of osteoarthritis for the pathogenesis of the disease. Osteoarth Cartil. 2007;15:1101–1105. doi: 10.1016/j.joca.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamerman D. The biology of osteoarthritis. N Engl J Med. 1989;320:1322–1330. doi: 10.1056/NEJM198905183202006. [DOI] [PubMed] [Google Scholar]
- 4.Lotz M.K., Otsuki S., Grogan S.P., Sah R., Terkeltaub R., D'Lima D. Cartilage cell clusters. Arthritis Rheum. 2010;62:2206–2218. doi: 10.1002/art.27528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu L., Flahiff C.M., Waldman B.A., Wu D., Olsen B.R., Setton L.A., Li Y. Osteoarthritis-like changes and decreased mechanical function of articular cartilage in the joints of chondrodysplasia (cho) mice. Arthritis Rheum. 2003;48:2509–2518. doi: 10.1002/art.11233. [DOI] [PubMed] [Google Scholar]
- 6.Hu K., Xu L., Cao L., Flahiff C.M., Setton L.A., Youn I., Brussiau J., Ho K., Guilak F., Olsen B.R., Li Y. Pathogenesis of osteoarthritis-like changes in joints of type IX collagen-deficient mice. Arthritis Rheum. 2006;9:2891–2900. doi: 10.1002/art.22040. [DOI] [PubMed] [Google Scholar]
- 7.Xu L., Polur I., Lim C., Servais J.M., Dobeck J., Li Y., Olsen B.R. Early-onset osteoarthritis of mouse temporomandibular joint induced by partial discectomy. Osteoarth Cartil. 2009;17:903–908. doi: 10.1016/j.joca.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu L., Peng H., Wu D., Hu K., Goldring M.B., Olsen B.R., Li Y. Activation of the discoidin domain receptor 2 induces expression of matrix metalloproteinase 13 associated with osteoarthritis in cho/+ mice. J Biol Chem. 2005;280:548–555. doi: 10.1074/jbc.M411036200. [DOI] [PubMed] [Google Scholar]
- 9.Xu L., Peng H., Glosson S., Lee P.L., Hu K., Ijiri K., Olsen B.R., Goldring M.B., Li Y. Increased expression of a collagen receptor discoidin domain receptor 2 in articular cartilage as a key event in the pathogenesis of osteoarthritis. Arthritis Rheum. 2007;56:2663–2673. doi: 10.1002/art.22761. [DOI] [PubMed] [Google Scholar]
- 10.Sunk I., Bobacz K., Hofstaetter J.G., Amoyo L., Soleiman A., Smolen J., Xu L., Li Y. Increased expression of discoidin domain receptor 2 is linked to the degree of cartilage damage in human knee joints: a potential role in osteoarthritis pathogenesis. Arthritis Rheum. 2007;56:3685–3692. doi: 10.1002/art.22970. [DOI] [PubMed] [Google Scholar]
- 11.Xu L., Servais J., Polur I., Kim D., Lee P.L., Chung K., Li Y. Attenuation of osteoarthritis progression by reduction of the discoidin domain receptor 2 in mice. Arthritis Rheum. 2010;62:2736–2744. doi: 10.1002/art.27582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klatt A.R., Zech D., Kühn G., Paul-Klausch B., Klinger G., Renno J.H., Schmidt J., Malchau G., Wielckens K. Discoidin domain receptor 2 mediates the collagen II-dependent release of interleukin-6 in primary human chondrocytes. J Pathol. 2009;218:241–247. doi: 10.1002/path.2529. [DOI] [PubMed] [Google Scholar]
- 13.Vonk L.A., Doulabi B.Z., Huang C., Helder M.N., Everts V., Bank R.A. Collagen-induced expression of collagenase-3 by primary chondrocytes is mediated by integrin {alpha}1 and discoidin domain receptor 2: a protein kinase C-dependent pathway. Rheumatology (Oxford) 2011;50:463–472. doi: 10.1093/rheumatology/keq305. [DOI] [PubMed] [Google Scholar]
- 14.Fosang A.J., Last K., Knauper V., Murphy G., Neame P.J. Degradation of cartilage aggrecan by collagenase-3 (MMP-13) FEBS Lett. 1996;380:17–20. doi: 10.1016/0014-5793(95)01539-6. [DOI] [PubMed] [Google Scholar]
- 15.Knäuper V., López-Otin C., Smith B., Knight G., Murphy G. Biochemical characterization of human collagenase-3. J Biol Chem. 1996;271:1544–1550. doi: 10.1074/jbc.271.3.1544. [DOI] [PubMed] [Google Scholar]
- 16.Fichter M., Körner U., Schömburg J., Jennings L., Cole A.A., Mollenhauer J. Collagen degradation products modulate matrix metalloproteinase expression in cultured articular chondrocytes. J Orthop Res. 2006;24:63–70. doi: 10.1002/jor.20001. [DOI] [PubMed] [Google Scholar]
- 17.Li Y., Lacerda D.L., Warman M.L., Beier D.R., Oxford J.T., Morris N.P., Andrikopoulos K., Ramirez F., Wardell B.B., Lifferth G.D., Teuscher C., Woodward S.R., Taylor B.A., Seegmiller R.E., Olsen B.R. A fibrillar collagen gene: Col11a1, is essential for skeletal morphogenesis. Cell. 1995;80:423–430. doi: 10.1016/0092-8674(95)90492-1. [DOI] [PubMed] [Google Scholar]
- 18.Poole C.A., Flint M.H., Beaumont B.W. Chondrons extracted from canine tibial cartilage: preliminary report on their isolation and structure. J Orthop Res. 1988;6:408–419. doi: 10.1002/jor.1100060312. [DOI] [PubMed] [Google Scholar]
- 19.Poole C.A., Flint M.H., Beaumont B.W. Chondrons in cartilage: ultrastructural analysis of the pericellular microenvironment in adult human articular cartilages. J Orthop Res. 1987;5:509–522. doi: 10.1002/jor.1100050406. [DOI] [PubMed] [Google Scholar]
- 20.Hunziker E.B., Michel M., Studer D. Ultrastructure of adult human articular cartilage matrix after cryotechnical processing. Microsc Res Tech. 1997;37:271–284. doi: 10.1002/(SICI)1097-0029(19970515)37:4<271::AID-JEMT3>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 21.Poole C.A., Matsuoka A., Schofield J.R. Chondrons from articular cartilage: III. Morphologic changes in the cellular microenvironment of chondrons isolated from osteoarthritic cartilage. Arthritis Rheum. 1991;34:22–35. doi: 10.1002/art.1780340105. [DOI] [PubMed] [Google Scholar]
- 22.Wadhwa S., Embree M., Ameye L., Young M.F. Mice deficient in biglycan and fibromodulin as a model for temporomandibular joint osteoarthritis. Cells Tissues Organs. 2005;18:136–143. doi: 10.1159/000091375. [DOI] [PubMed] [Google Scholar]
- 23.Alexopoulos L.G., Inchan Youn I., Paolo Bonaldo P., Guilak F. Developmental and osteoarthritic changes in Col6a1-knockout mice: biomechanics of type VI collagen in the cartilage pericellular matrix. Arthritis Rheum. 2009;60:771–779. doi: 10.1002/art.24293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Weyden L., Wei L., Luo J., Yang X., Birk D.E., Adams D.J., Bradley A., Chen Q. Functional knockout of the matrilin-3 gene causes premature chondrocyte maturation to hypertrophy and increases bone mineral density and osteoarthritis. Am J Pathol. 2006;169:515–527. doi: 10.2353/ajpath.2006.050981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polur I., Lee P.L., Servais J.M., Xu L., Li Y. Role of HTRA1, a serine protease, in the progression of articular cartilage degeneration. Histol Histopathol. 2010;25:599–608. doi: 10.14670/hh-25.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu J., Liu W., Bemis A., Wang E., Qiu Y., Morris E.A., Flannery C.R., Yang Z. Comparative proteomic characterization of articular cartilage tissue from normal donors and patients with osteoarthritis. Arthritis Rheum. 2007;56:3675–3684. doi: 10.1002/art.22876. [DOI] [PubMed] [Google Scholar]
- 27.Grau S., Richards P.J., Kerr B., Hughes C., Caterson B., Williams A.S., Junker U., Jones S.A., Clausen T., Ehrmann M. The role of human HtrA1 in arthritic disease. J Biol Chem. 2006;281:6124–6129. doi: 10.1074/jbc.M500361200. [DOI] [PubMed] [Google Scholar]
- 28.Tsuchiya A., Yano M., Tocharus J., Kojima H., Fukumoto M., Kawaichi M., Oka C. Expression of mouse HtrA1 serine protease in normal bone and cartilage and its upregulation in joint cartilage damaged by experimental arthritis. Bone. 2005;37:323–336. doi: 10.1016/j.bone.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 29.Hu S.I., Carozza M., Klein M., Nantermet P., Luk D., Crowl R.M. Human HtrA, an evolutionarily conserved serine protease identified as a differentially expressed gene product in osteoarthritic cartilage. J Biol Chem. 1998;273:34406–34412. doi: 10.1074/jbc.273.51.34406. [DOI] [PubMed] [Google Scholar]
- 30.DiCesare P.E., Morgelin M., Carlson C.S., Pasumarti S., Paulsson M. Cartilage oligomeric matrix protein: isolation and characterization from human articular cartilage. J Orthop Res. 1995;13:422–428. doi: 10.1002/jor.1100130316. [DOI] [PubMed] [Google Scholar]
- 31.Issack P.S., Liu C.J., Prazak L., Di Cesare P.E. A silencer element in the cartilage oligomeric matrix protein gene regulates chondrocyte-specific expression. J Orthop Res. 2004;22:751–758. doi: 10.1016/j.orthres.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 32.Posey K.L., Davies S., Bales E.S., Haynes R., Sandell L.J., Hecht J.T. In vivo human cartilage oligomeric matrix protein (COMP) promoter activity. Matrix Biol. 2005;24:539–549. doi: 10.1016/j.matbio.2005.07.007. [DOI] [PubMed] [Google Scholar]