

Abstract
Aberrantly hyperactivated STAT3 has been found in human liver cancers as an oncogene; however, STAT3 has also been shown to exert hepatoprotective effects during liver injury. The balancing act that STAT3 plays between hepatoprotection and liver tumorigenesis remains poorly defined. In this study, the diethylnitrosamine (DEN)-induced liver tumor model and the chronic carbon tetrachloride (CCl4)–induced liver fibrosis model were both used to investigate the role of STAT3 in liver tumorigenesis. Hepatocyte-specific STAT3 knockout mice were resistant to liver tumorigenesis induced by a single DEN injection, whose tumorigenesis was associated with minimal chronic liver inflammation, injury, and fibrosis. In contrast, long-term CCl4 treatment resulted in severe hepatic oxidative damage, inflammation, and fibrosis but rarely induced liver tumor formation in wild-type mice. Despite the oncogenic function of STAT3 in DEN-induced liver tumor, hepatocyte-specific STAT3 knockout mice were more susceptible to liver tumorigenesis after 16 weeks of CCl4 injection, which was associated with higher levels of liver injury, inflammation, fibrosis, and oxidative DNA damage compared with wild-type mice. These findings suggest that the hepatoprotective feature of STAT3 prevents hepatic damage and fibrosis under the condition of persistent inflammatory stress, consequently suppressing injury-driven liver tumor initiation. Once liver tumor cells have developed, STAT3 likely acts as an oncogenic factor to promote tumor growth.
Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related deaths worldwide and one of the fastest growing cancers in terms of incidence in developed countries.1,2 Despite recent progress in understanding its pathogenic mechanisms,2–4 HCC is still considered one of the deadliest cancers worldwide with a poor prognosis, as very few patients are eligible for potential curative treatment, such as surgical resection and liver transplantation.5–8 Various etiologies, including hepatotropic viruses (eg, hepatitis B and C viruses), chronic alcohol consumption, nonalcoholic steatohepatitis, and toxins (eg, aflatoxin B), can lead to chronic liver injury, inflammation, or fibrosis/cirrhosis, which can then culminate in liver cancer.1–3 Although the close contributory relationship between chronic liver inflammation and carcinogenesis has been well documented,2,3 the exact mechanisms that direct inflammation-induced liver cancer remain elusive.
Recent evidence suggests that liver cancer formation can be generally divided into three stages: initiation, promotion, and progression. Liver tumor initiation can be induced by many initiators, including hepatitis B virus, cirrhosis, and aflatoxin B. These factors can induce irreversible genetic mutations that subsequently cause proto-oncogene activation and/or loss of tumor suppression. For tumor development to take place, initiation must be followed by a committed tumor promotion process that requires interactions between initiated cells and their microenvironment,9 of which inflammation is an important factor.10 Although HCC usually appears after exposure to carcinogens such as aflatoxin B, viral hepatitis, and alcohol intake, it may take many years, even decades, to develop HCC from liver cirrhosis after chronic liver injury and fibrosis.2,3 Although the early etiologic events involved in liver tumorigenesis have been well documented, little is known about mechanisms underlying the progression from premalignant lesions to overt carcinomas. Without greater knowledge of these mechanisms, it is impossible to improve current unsatisfactory and suboptimal therapeutic options for HCC.
Recent results of a phase 3 clinical trial showed that sorafenib, a multikinase inhibitor that blunts multiple signaling pathways, markedly improved survival in patients with advanced HCC tumors.11,12 This pivotal study has stimulated scientific research on novel molecular therapies targeting specific signaling pathways to treat this malignancy.6 A growing body of evidence suggests that persistent activation of STAT3 is oncogenic and is prevalent in a variety of human cancers, including liver cancer.13 Hepatic activation of STAT3 occurs through various cytokines, growth factors, hormones, and hepatitis viral proteins.14 Among them, IL-6 and IL-22 are two potent inducers of STAT3 activation in hepatocytes.14 Activation of STAT3 begins on binding of IL-6 to its corresponding receptor on hepatocytes, resulting in dimerization of the signal transducer protein, gp130. The gp130-associated Janus kinases then combine to form dimers and phosphorylate each other, the gp130 protein, and cytoplasmic STAT3 monomers. IL-22 binds to IL-22R and IL-10R2 on hepatocytes, followed by inducing Janus kinase phosphorylation and subsequently STAT3 phosphorylation. Consequently, phosphorylated STAT3 (pSTAT3) monomers interact with each other and form dimers that translocate into the nuclei to induce transcription of genes involved in cell survival and proliferation.15 Activation of STAT3 by IL-6 or IL-22 has been shown to contribute to the tumorigenesis of a variety of tumors, including HCC.13,16–19 Thus, intense efforts have been made to identify suitable anti-STAT3 agents to treat human cancers such as HCC.20 However, historically, evidence from a wealth of previous studies has confirmed the hepatoprotective function of STAT3 in numerous models of liver injury.14 Thus, an understanding of the pathologic/oncogenic versus protective functions of STAT3 in liver tumorigenesis is needed to carefully assess the risks and benefits of anti-STAT3 treatment for HCC in the clinical setting.
A previous study reported that conditional deletion of STAT3 in hepatocytes prevents liver tumor development induced by a single diethylnitrosamine (DEN) injection at age 15 days,21 which was also confirmed in the present study. However, this DEN model is not associated with significant chronic liver injury, inflammation, and fibrosis, which is different from most human liver cancers that develop after chronic hepatocellular damage, inflammation, and fibrosis/cirrhosis.2,3 Thus, by using liver-specific STAT3 knockout mice, we also explored the role of hepatic STAT3 in a model of liver tumor induced by chronic carbon tetrachloride (CCl4) treatment that is associated with strong chronic liver injury, inflammation, and fibrosis. The present results suggest that hepatic STAT3 protects against CCl4-induced chronic liver injury and consequently against tumorigenesis.
Materials and Methods
Animals
Hepatocyte-specific STAT3 knockout mice (STAT3Hep−/−) (AlbCre+/− STAT3flox/flox) and littermate wild-type controls (AlbCre−STAT3flox/flox) were described previously.22 STAT1−/− and STAT1−/−STAT3Hep−/− mice were generated as described previously.23 All the animal experiments were approved by the Institutional Animal Care and Use Committee of the National Institute on Alcohol Abuse and Alcoholism.
DEN-Induced Liver Cancer Model
The DEN-induced liver tumor model was established as described previously.24,25 Briefly, 15-day-old STAT3Hep−/− mice and their littermates were injected with 5 or 20 μg/g of DEN (Sigma-Aldrich, St. Louis, MO). After 4 or 9 months on normal chow, the mice were sacrificed. Their livers were then removed, separated into individual lobes, analyzed for the presence of HCCs, and subjected to analysis of histologic and immunochemical parameters.
CCl4-Induced Chronic Liver Injury, Inflammation, Fibrosis, and Liver Tumor
Eight- to 10-week-old male STAT3Hep−/− mice and their wild-type littermates were injected i.p. with 2 mL/kg body weight of 10% (v/v) CCl4 (Sigma-Aldrich) dissolved in olive oil three times a week for up to 16 weeks. Control groups were treated with vehicle (10% olive oil, 2 mL/kg). The mice were sacrificed at different time points after the last injection of chronic CCl4 treatment, and liver tissues were harvested for experiments. No mortality was observed in these mice after chronic CCl4 treatment. Ascites were reported in the mice after chronic CCl4 treatment plus phenobarbital in drinking water, especially after the inhalation protocol.26 Chronic i.p. administration of CCl4 plus phenobarbital in drinking water also induced ascites but to a lesser extent.26 No obvious ascites were observed in the mice treated with i.p. injection of CCl4 alone in the present protocol.
Liver Tumor Analysis
The whole liver was carefully removed from the euthanized animal, washed, and placed in cold PBS. The number of surface liver tumor nodules was counted for all the liver lobes in a blinded manner. Some reasonably sized tumor nodules (>2 mm in diameter) were carefully removed from the liver lobes using fine forceps and were placed in fresh cold PBS. These separated nodules were then halved using a sterile razor blade and were split into samples for RNA and protein extraction. Histologic sections, including larger tumor nodules (>2 mm in diameter), were collected and fixed in 10% formalin. Tissue section slides were stained with H&E using standard protocols. Liver nodules typically presented as basophilic foci with crowded nuclei and were classified as atypical foci (HCC) or hepatocellular adenomas. Adenomas were distinguished from atypical foci based on the presence of clearly defined margins and compression of surrounding parenchyma.
Histologic Analysis of Liver Injury and Fibrosis
Formalin-fixed liver samples were processed, and paraffin sections were stained with H&E. The score of liver injury (necrosis and inflammation) was evaluated blindly by two pathologists as described previously.27 Liver fibrosis was determined by Sirius red staining for collagens or by immunohistochemical (IHC) staining for activated hepatic stellate cells with anti–α-smooth muscle actin (Dako, Carpinteria, CA) and were quantified by digital imaging using NIH Scion Image and Adobe Photoshop (Adobe Systems Inc., San Jose, CA).28,29
Blood Chemistry
Serum alanine transaminase (ALT), aspartate aminotransferase (AST), and albumin levels were determined using a chemistry analyzer (Prochem V; Drew Scientific Co., Barrow-in-Furness, UK). Serum levels of C-reactive protein were analyzed by immunoperoxidase assay (Immunology Consultants Laboratory, Newberg, OR). Serum cytokine levels were measured by Cytometric Bead Array (BD Biosciences, San Diego, CA).
TUNEL Assay
Hepatocyte apoptosis was detected by using the ApopTag TUNEL apoptosis detection kit (Chemicon International, Temecula, CA).
Hepatocyte Proliferation in Mice after Chronic CCl4 Treatment
Hepatocyte proliferation in vivo was determined using the bromodeoxyuridine (BrdU) incorporation assay. Briefly, mice were injected (i.p) with BrdU (50 μg/g body weight) and euthanized 2 hours later, and the livers were harvested for IHC staining of BrdU using a kit (BD Biosciences). BrdU-labeled hepatocytes in each slide were counted in five low-power (×100) fields.
Real-Time PCR
Real-time PCR was performed with the indicated primers as described previously.22
Western Blotting
Western blot analyses were performed as described previously22 using STAT3, STAT1, glyceraldehyde-3-phosphate dehydrogenase (Cell Signaling Technology Inc., Danvers, MA), and anti-CYP2E1 (Chemicon International, Billerica, MA) antibodies.
Measurement of Hepatic Malondialdehyde, Glutathione, and 8-Hydroxy-2-Deoxyguanosine
Malondialdehyde (MDA) was measured using the thiobarbituric acid method and is expressed as nanomoles of MDA per milligram of protein.30,31 Glutathione (GSH) was measured as described previously.32 The hepatic GSH concentrations were adjusted according to the original wet weight of liver tissue (nanomoles per milligram of liver tissue). Hepatic 8-hydroxy-2-deoxyguanosine (8-OHdG) was measured using an enzyme-linked immunosorbent assay kit (OXIS Health Products Inc., Portland, OR) according to the manufacturer's manual.
Statistical Analysis
Data are expressed as mean ± SD (n = 5 to 12 in each group). Student's t-test was performed to compare values from two groups. To compare values obtained from three or more groups, one-factor analysis of variance was used, followed by Tukey's post hoc test. Statistical significance was taken at the P < 0.05 level.
Results
Deletion of STAT3 in Hepatocytes Ameliorates DEN-Induced HCC
To address the function of hepatic STAT3 in the development of DEN-induced HCC, STAT3Hep−/− mice and their wild-type littermates were injected with DEN at age 15 days. Figure 1A shows hepatic expression of STAT3 that was significantly lower in STAT3Hep−/− mice than in wild-type mice, confirming hepatic STAT3 deletion in STAT3Hep−/− mice at age 15 days. In the initial experimental design, we were unsure of the susceptibility of the strains of wild-type and STAT3Hep−/− mice to DEN-induced HCC development, so we injected mice with 5 or 20 μg/g of DEN. As illustrated in Figure 1B, injection of both doses induced significant liver tumor formation 9 months after injection. Injection of 20 μg/g of DEN induced a slightly higher number and larger size of tumors than did injection of 5 μg/g of DEN, but this difference did not reach statistical significance. The number and size of tumors were much smaller in STAT3Hep−/− mice than in wild-type mice after injection of either 5 or 20 μg/g of DEN.
Figure 1.
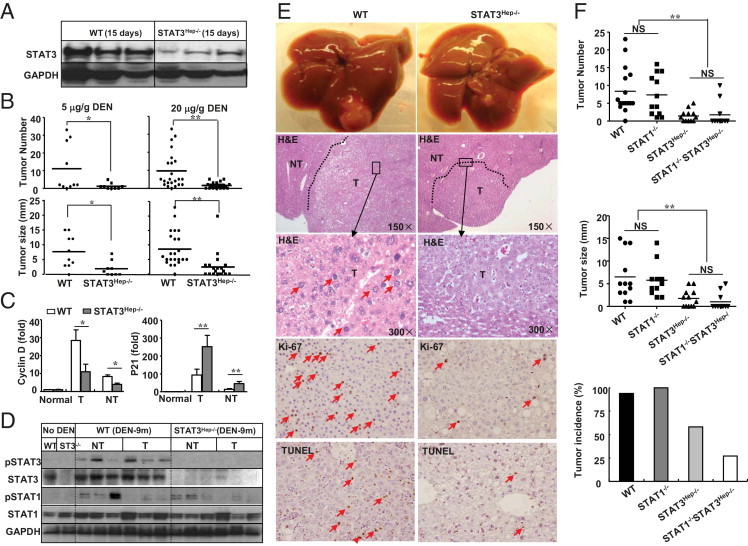
STAT3Hep−/− mice are resistant to DEN-induced liver tumorigenesis independent of STAT1. A: Western blot analyses of liver tissues isolated from 15-day-old mice. B: Wild-type (WT) and STAT3Hep−/− mice treated with 5 or 20 μg/g of DEN at age 15 days and sacrificed 9 months after DEN. The number and size of tumors on the surface were determined. C-E: Liver tumors from mice treated with 20 μg/g of DEN were further analyzed. C: Real-time PCR analysis of cyclin D and p21 mRNA in tumor (T) and nontumor (NT) tissues. Values represent mean ± SD (n = 5). D: Western blot analyses of NT and T tissues. E: Representative images of livers, H&E staining, Ki-67 immunostaining, and TUNEL staining. The arrows in H&E staining indicate the enlarged nuclei of tumor cells, and the arrows in IHC analysis indicate Ki-67+ or TUNEL+ cells. F: WT and the three lines of knockout mice were treated with a single dose of 20 μg/g of DEN at age 15 days and then were sacrificed 9 months after DEN injection. The number, size, and incidence of tumors were determined. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NS, not significant. *P < 0.05, ** P < 0.01.
Tumor and nontumor tissues from wild-type and STAT3Hep−/− mice were collected and subjected to real-time PCR (Figure 1C) and Western blot analyses (Figure 1D). Figure 1C shows that expression of the typical cell cycle protein cyclin D was reduced and that expression of cell cycle regulatory protein p21 was higher in tumor and nontumor samples from STAT3Hep−/− mice than from wild-type mice. Hepatic expression of p27 was detected at low levels and was comparable in wild-type and STAT3Hep−/− mice (data not shown). Western blot analyses in Figure 1D show that pSTAT3 expression was elevated in nontumor and tumor tissues in DEN-treated wild-type mice. Such expression, as expected, was barely detected in STAT3Hep−/− mice. Expression of pSTAT1 was elevated in nontumor tissues and, to a lesser extent, in tumor tissues in DEN-treated wild-type mice compared with that in nontreated control mice. In addition, expression of pSTAT1 and STAT1 was comparable between STAT3Hep−/− and WT mice.
Figure 1E shows representative images of liver, H&E staining, Ki-67 immunostaining, and TUNEL staining from DEN-treated wild-type and knockout mice. Microscopically, the tumors in both groups formed discrete nodes surrounded by almost normal liver tissues. Histologic analyses show that the tumors from both groups had a similarly moderate degree of differentiation, with an increased nuclear-to-cytoplasmic index, enlarged and hyperchromatic nuclei, and expansive growth. In the area of tumor tissues, the normal liver architecture, such as bile duct and portal tract formation, was lost. In addition, wild-type mice had greater numbers of Ki-67+ and TUNEL+ hepatocytes than did STAT3Hep−/− mice 9 months after DEN exposure.
To explore the role of STAT1 in DEN-induced liver tumor formation, wild-type, STAT1−/−, STAT3Hep−/−, and STAT1−/−STAT3Hep−/− double knockout mice were treated with a single dose of DEN at age 15 days. Despite the well-documented antioncogenic effect of STAT1, STAT1−/− mice had a similar number and size of tumors as wild-type mice 9 months after DEN injection (Figure 1F). An additional deletion of STAT1 did not further increase liver tumor number, size, and incidence compared with STAT3Hep−/− mice (Figure 1F). These results indicate that STAT1 signaling is not enhanced and is not responsible for the reduced liver tumorigenesis in STAT3Hep−/− mice.
DEN-Induced Liver Model Is Associated with Minimal Inflammation, Injury, and Fibrosis—The Proinflammatory Effect of Hepatic STAT3
Because a causal relationship between chronic injury and inflammation and liver carcinogenesis in most patients with HCC is well established, we wondered whether the DEN-induced liver cancer mouse model is associated with chronic hepatocyte injury, inflammation, and fibrosis. As illustrated in Figure 2A, elevation of serum ALT and liver oxidative stress MDA and 8-OHdG levels and down-regulation of hepatic GSH levels were observed only 24 hours after a single dose of acute DEN injection. Such changes were comparable between wild-type and STAT3Hep−/− mice. H&E staining shows that no obvious hepatocyte necrosis and inflammatory foci were detected in the livers 4 or 9 months after DEN injection (Figure 2B). Sirius red staining revealed no obvious liver fibrosis in these DEN-treated mice (Figure 2B). Only in the very late stage of HCC, when the liver tumor grows too fast, were necrotic tumor tissue and inflammation seen in the large tumor tissues (data not shown).
Figure 2.
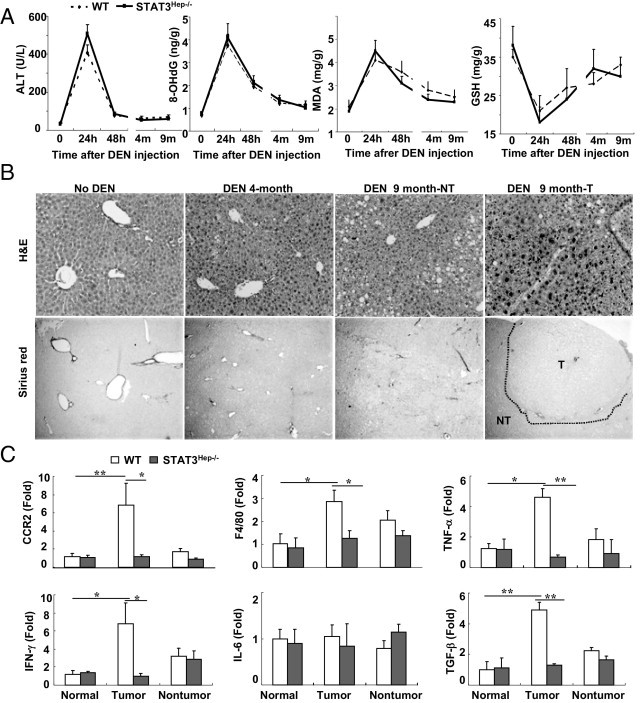
The DEN-induced liver tumor model is associated with minimal inflammation, injury, and fibrosis. Wild-type (WT) and STAT3Hep−/− mice were treated with 20 μg/g of DEN at age 15 days and then were sacrificed at different time points after DEN. A: Serum ALT, 8-OHdG, liver MDA, and GSH levels are shown (n = 6 to 8). B: Representative H&E staining and Sirius red staining are shown. NT, nontumor; T, tumor. C: Real-time PCR analyses of inflammatory markers and proinflammatory cytokines from liver tumor and nontumor tissues 9 months after DEN treatment. Values represent mean ± SD (n = 6). *P < 0.05, **P < 0.01.
Real-time PCR analyses in Figure 2C show that expression of CCR2, F4/80, tumor necrosis factor (TNF)-α, interferon (IFN)-γ, and transforming growth factor (TGF)-β was up-regulated in liver tumor tissues but not in nontumor tissues from DEN-treated wild-type mice compared with nontreated mice. Such induction was not observed in tumor tissues from DEN-treated STAT3Hep−/− mice, suggesting that hepatic STAT3 plays a role in stimulating inflammation in tumor tissues (Figure 2C).
Loss of STAT3 in Hepatocytes Enhances Liver Injury and Inflammation but Reduces Liver Regeneration after CCl4 Administration
The previous data show that the DEN model is not associated with significant liver injury and inflammation; however, most human liver cancers develop after chronic liver injury, inflammation, and fibrosis.2,3 To further explore the role of STAT3 in liver tumorigenesis that is associated with chronic liver injury and fibrosis, a model of chronic liver injury and fibrosis induced by chronic CCl4 treatment was used. As illustrated in Figure 3A, the expression of STAT3 protein in the liver was barely detected in STAT3Hep−/− mice, confirming STAT3 deletion in these mice. Furthermore, expression of CYP2E1 was similar in livers from untreated wild-type and STAT3Hep−/− mice. After 4-week CCl4 challenge, expression of CYP2E1 decreased significantly in wild-type mice. A similar down-regulation was also observed in STAT3Hep−/− mice. These findings suggest that CCl4 metabolism is similar in wild-type and STAT3Hep−/− mice.
Figure 3.
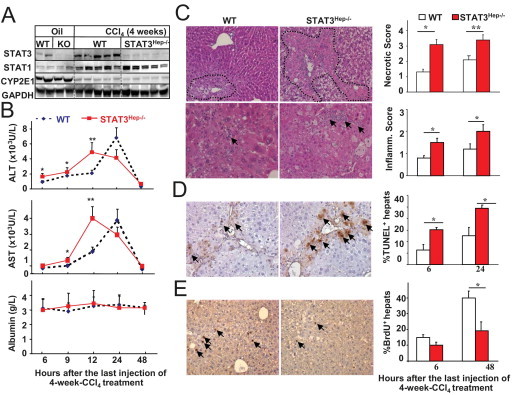
STAT3Hep−/− mice are more susceptible to liver injury and have impaired liver regeneration after 4-week CCl4 treatment. Mice were treated with CCl4 or olive oil for 4 weeks and were euthanized at various time points after the last injection. A: Western blot analyses. WT, wild type; KO, knockout; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. B: Serum ALT, AST, and albumin levels. C: H&E staining. Original magnification, ×60. Histology and inflammation scores are shown in the right panel. D: Representative TUNEL staining for hepatocyte apoptosis 24 hours after 4-week CCl4 treatment. Quantitation is shown in the right panel. E: Representative BrdU staining for hepatocyte proliferation 48 hours after 4-week CCl4 treatment. Quantitation is shown in the right panel. Values represent mean ± SD (n = 6 to 8). *P < 0.05, **P < 0.01 compared with the corresponding WT groups. Arrows indicate TUNEL+ or BrdU+ hepatocytes.
Next we compared the liver injury in wild-type and STAT3Hep−/− mice at different time points after 4-week CCl4 treatment. As illustrated in Figure 3B, STAT3Hep−/− mice had accelerated hepatocellular damage, as evidenced by elevation of higher levels of serum ALT and AST at early time points (peak at 12 hours), whereas wild-type littermates had peak serum ALT and AST levels 24 hours after 4-week CCl4 treatment. There was no significant difference in albumin levels between these 2 groups. Morphologic analyses showed prompt hemorrhagic necrosis, more extensive aberrant architecture characterized by vacuolization, and more inflammation in STAT3Hep−/− mice than in wild-type controls (Figure 3C). The necrotic and inflammatory scores were higher in STAT3Hep−/− mice 6 and 24 hours after 4-week CCl4 treatment (Figure 3C). In addition, STAT3Hep−/− mice had greater numbers of apoptotic hepatocytes (Figure 3D) but lower numbers of Brdu+ hepatocytes (Figure 3E) in the liver compared with wild-type mice after 4-week CCl4 treatment.
Figure 4 shows that compared with wild-type mice, STAT3Hep−/− mice had higher levels of myeloperoxidase, CCR2, F4/80, and TNF-α expression after 4- and 16-week CCl4 treatment (Figure 4A), whereas serum levels of inflammatory cytokines were comparable between these 2 groups (Figure 4B). Given that STAT3 plays an important role in inducing acute-phase proteins in hepatocytes, we measured levels of acute-phase proteins such as C-reactive protein, serum amyloid A, cytokine-induced neutrophil-attracting chemokine (KC), and C5. Figure 4 shows that the expression of C5, serum amyloid A, and KC mRNAs and serum levels of C-reactive protein were lower in STAT3Hep−/− mice than in wild-type mice after CCl4 treatment. Finally, serum levels of TNF-α, monocyte chemotactic protein-1, and IL-6 were comparable between these two groups after CCl4 treatment (Figure 4B).
Figure 4.
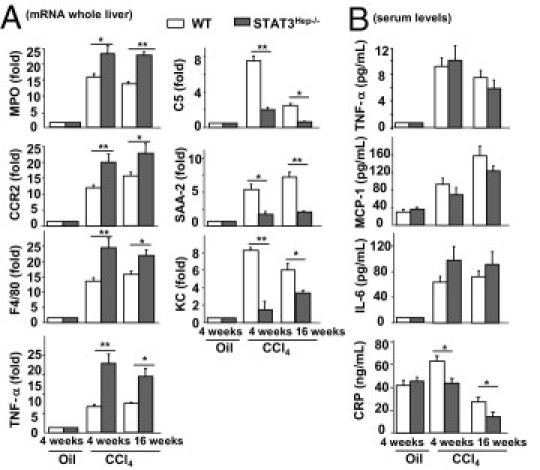
STAT3Hep−/− mice are more susceptible to chronic CCl4-induced liver inflammation. Mice were treated with CCl4 for 4 or 16 weeks and were euthanized 24 hours after the last injection. A: Liver tissue samples were collected for real-time PCR analyses. The value from wild-type (WT) mice with vehicle (olive oil) was set as onefold. B: Serum levels of cytokines and the acute-phase protein C-reactive protein (CRP). Values represent mean ± SD (n = 8). *P < 0.05, **P < 0.01. MPO, myeloperoxidase; SAA-2, serum amyloid A-2; MCP-1, monocyte chemotactic protein-1.
Loss of STAT3 in Hepatocytes Promotes Liver Fibrosis
Sirius red (Figure 5A) and α-smooth muscle actin (Figure 5B) staining show that STAT3Hep−/− mice displayed a higher degree of liver fibrosis compared with wild-type mice 4 and 16 weeks after CCl4 administration. Real-time PCR analyses also confirmed that expression of several forms of collagens and α-smooth muscle actin was higher in the liver from STAT3Hep−/− mice than in that from wild-type mice (Figure 5C).
Figure 5.
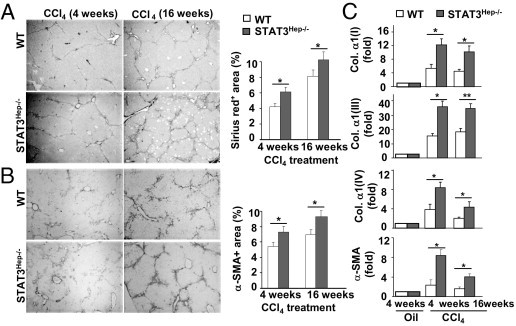
STAT3Hep−/− mice are more susceptible to CCl4-induced liver fibrosis. Mice were treated with CCl4 for 4 or 16 weeks and were euthanized 24 hours after the last injection. Liver tissue samples were collected for Sirius red staining (A) and IHC with α-smooth muscle actin (α-SMA) antibodies (B). The areas stained with Sirius red or α-SMA were quantified and are shown on the right. C: Real-time PCR analyses. The value from wild-type (WT) mice with 4-week oil treatment was set as onefold. Values represent mean ± SD (n = 6). *P < 0.05, **P < 0.01.
Development of Liver Tumorigenesis in STAT3Hep−/− Mice after Prolonged CCl4 Treatment
It has been well established that most HCCs develop as a result of chronic liver inflammation and cirrhosis in clinical practice.2,3 Given the enhanced hepatocyte damage, inflammation, and fibrosis in STAT3Hep−/− mice, we hypothesized that loss of STAT3 in hepatocytes facilitates tumor development after prolonged injury and repair cycles. To explore this, we prolonged the CCl4 injection course to 16 weeks in wild-type and STAT3Hep−/− mice. Approximately 60% of STAT3Hep−/− mice had developed multiple surface nodular tumors after 16 weeks of CCl4 treatment, whereas no obvious nodules were detected in the surface of livers in wild-type mice (Figure 6, A and B). Histologic analysis revealed that adenomas were found in most STAT3Hep−/− mice and that highly differentiated HCCs were found in only three STAT3Hep−/− mice (Figure 6A).
Figure 6.
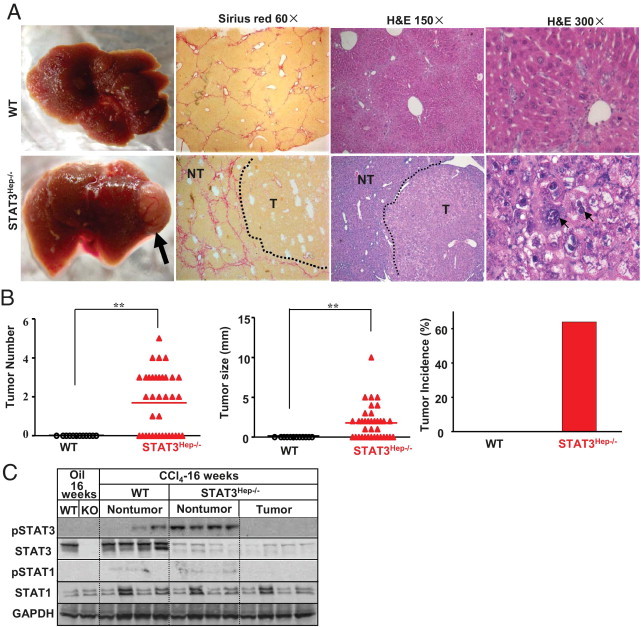
Loss of STAT3 in hepatocytes facilitates tumor development after chronic CCl4 treatment. Wild-type (WT) and STAT3Hep−/− mice were treated with CCl4 for 16 weeks and were euthanized 24 hours after the last injection. A: Liver tissue samples were collected for Sirius red staining and H&E staining. The arrow in A indicates a tumor nodule. The small arrows on the right side of A indicate enlarged and hyperchromatic nuclei. B: Tumor number, size, and incidence were determined. C: Western blot analyses. NT, nontumor tissues; T, tumor tissues; KO, knockout; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. In the WT group, no tumor tissues were available. **P < 0.01.
Western blot analyses show that STAT3 protein was detected at high levels in wild-type mouse livers but at very low levels in tumor and nontumor liver tissues in STAT3Hep−/− mice (Figure 6C), indicating that the tumors in STAT3Hep−/− mice were originated from STAT3-deleted hepatocytes. Wild-type and STAT3Hep−/− mice had comparable levels of STAT1 protein but undetectable levels of pSTAT1 protein in the liver. Weak pSTAT3 expression (STAT3 activation) was detected in nontumor liver tissue but not in tumor liver tissue of STAT3Hep−/− mice. The elevated pSTAT3 in nontumor liver tissues in STAT3Hep−/− mice may be due to infiltrated inflammatory cells.
Enhanced DNA Damage and Oxidative Stress in STAT3Hep−/− Mice
To explore the mechanisms underlying the enhanced tumorigenesis in STAT3Hep−/− mice, DNA damage and oxidative stress were measured. Levels of 8-OHdG, a marker of oxidative DNA damage, and MDA, a marker of oxidative stress, were elevated in the liver of wild-type mice after chronic CCl4 treatment (Figure 7). Such elevation was more profound in STAT3Hep−/− mice. In contrast, hepatic levels of the anti–oxidative stress enzyme GSH were decreased in wild-type and STAT3Hep−/− mice after chronic CCl4 administration, with lower levels in the latter group.
Figure 7.
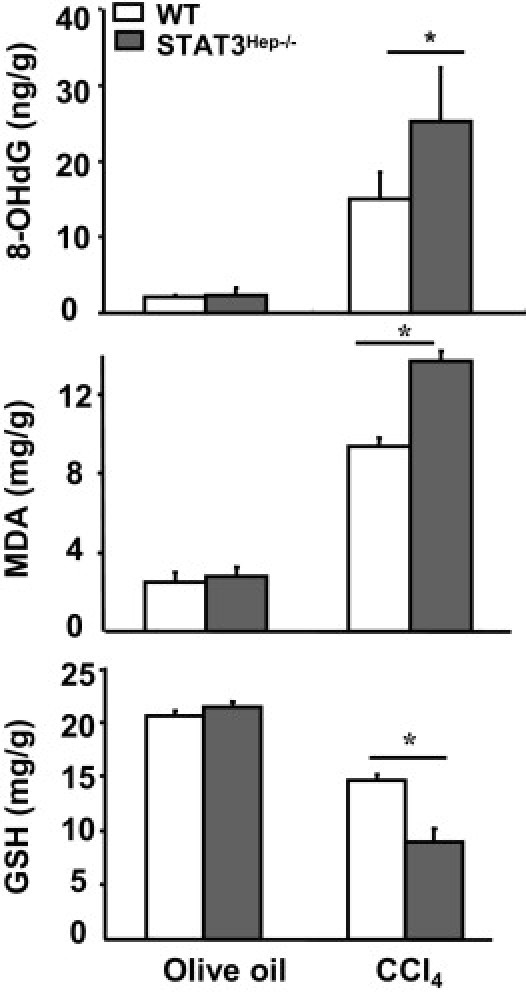
Enhanced oxidative stress and DNA damage in STAT3Hep−/− mice after CCl4 challenge. Wild-type (WT) and STAT3Hep−/− mice were treated with CCl4 for 4 weeks and were euthanized 24 hours after the last injection. Liver tissue samples were collected for measurement of 8-OHdG, MDA, and GSH levels. Values represent mean ± SD (n = 6). *P < 0.05.
Discussion
STAT3 is a transcription factor that is activated by a variety of factors, including cytokines, growth factors, hormones, and hepatitis viral proteins in the liver.14 In almost all rodent models of liver injury, hepatic STAT3 activation has been reported, playing an essential role in protecting against hepatocellular damage and promoting liver regeneration.14 It has been reported that deletion of hepatic STAT3 reduces DEN-induced tumorigenesis,21 which was also confirmed in the present study, suggesting that hepatic STAT3 promotes liver tumor development in this model. In contrast, deletion of hepatic STAT3 exacerbated CCl4-induced chronic liver inflammation and tumorigenesis, suggesting that hepatic STAT3 may also prevent hepatic tumorigenesis, especially in the condition of nonresolving inflammation.
STAT3 Is a Pro-Oncogenic Factor That Promotes Liver Tumorigenesis in a Model of DEN-Induced Liver Tumor
The pro-oncogenic role of STAT3 has been well documented in many types of tumors, including liver cancer.13 The first evidence of this was the detection of constitutively activated STAT3 in human hepatoma cells and human liver tumor tissues.13,33 In addition, it was determined that inhibition of STAT3 using chemical inhibitors or small-interfering RNA induced liver cancer cell apoptosis and cell cycle arrest in vitro and inhibited growth of transplanted liver cancer cells in vivo.33 Other evidence suggesting a role of STAT3 in tumorigenesis included findings that deletion of hepatic SOCS3, an inhibitor of STAT3, elevated STAT3 activation in the liver and accelerated DEN-induced liver tumorigenesis,34 whereas overexpression of SOCS3 inhibited HCC cell growth.35 Moreover, several cytokines (IL-6, IL-6 family cytokines, IL-22, leptin, etc) that activate STAT3 in hepatocytes have also been shown to promote HCC cell growth in vitro and in vivo.17–19,36 Finally, as we show in this study, deletion of STAT3 in hepatocytes prevents DEN-induced HCC development, which was also reported previously.21 Deletion of hepatic STAT3 also reduced hepatitis C virus core protein–mediated hepatocarcinogenesis in transgenic mice.37
In the hepatic environment, the opposing functions of STAT3 and STAT1 in controlling cell apoptosis and proliferation have been well documented.38,39 As deletion of STAT3 up-regulates STAT1 in several models of liver injury, and vice versa,38 it suggests that the proliferative, antiapoptotic, and anti-inflammatory effects of STAT3 are mediated, at least in part, via inhibition of STAT1, a transcription factor that induces cell cycle arrest and apoptosis and promotes inflammatory responses. Moreover, STAT1 is often considered a tumor suppressor.13 However, it was unexpectedly determined that STAT1 activation (pSTAT1) and protein expression were not enhanced in STAT3-deficient tumor and nontumor tissues. In addition, the added deletion of the STAT1 gene did not further reduce tumor numbers or size induced by DEN injection (Figure 1). These results suggest that the pro-oncogenic function of STAT3 in DEN-induced liver tumor is not mediated by suppression of STAT1.
STAT3 Prevents Liver Tumorigenesis in a Model of CCl4-Induced Chronic Liver Injury and Fibrosis
Tumorigenesis induced by a single DEN injection is not associated with significant inflammation, injury, and fibrosis,40,41 and, therefore, it is less representative of the type of pathogenesis occurring in most human liver cancers that develop after chronic injury, inflammation, and fibrosis/cirrhosis.2,3,6 By using a model of CCl4-induced chronic liver injury, we demonstrated that STAT3Hep−/− mice exhibited enhanced liver injury, inflammation, and fibrosis compared with wild-type mice after chronic CCl4 treatment, which is consistent with the well-documented hepatoprotective role of STAT3.14 CCl4 is a weak carcinogen and is able to induce HCC development in some strains of mice after a long-time chronic injection.42–44 For example, Fujii et al45 reported that treatment with CCl4 for 15 weeks induced liver fibrosis and well-differentiated HCC in male A/J strain mice. Furthermore, Hosui et al46 reported that chronic injection of CCl4 for 8 weeks induced HCC in 10% of liver-specific STAT5 knockout mice but not in wild-type mice. The present study shows that after 16-week CCl4 treatment, no HCC development was found in wild-type mice but approximately 60% of STAT3Hep−/− mice developed multiple surface nodular tumors (Figure 6). These findings indicate that hepatocyte STAT3 plays a protective role in preventing liver tumorigenesis induced by chronic CCl4 challenge.
Potential Mechanisms Underlying STAT3 Promotion and Inhibition of Liver Tumorigenesis in DEN- and CCl4-Induced Liver Injury Models, Respectively
As mentioned previously herein, activation of STAT3 in hepatocytes plays an important role in promoting liver tumorigenesis in many models, including DEN-induced liver tumor. The pro-oncogenic function of STAT3 is likely mediated via up-regulation of a variety of genes involved in cell cycle progression (cyclin D1, c-myc), cell survival (Bcl-xL, Bcl-2), angiogenesis (VEGF), and tissue remodeling (Mmp-1, Mmp-3).13 In addition, a previous study showed that treatment of mice with an antioxidant before a single DEN injection at age 18 days prevented liver tumor development,47 suggesting that DEN induction of oxidative stress contributes to hepatic tumorigenesis. Herein, we showed that a single DEN injection induces a transient elevation in 8-OHdG and MDA levels in the liver, which returned to basal levels 4 and 9 months after injection, respectively (Figure 2A). There was no difference in hepatic levels of MDA and 8-OHdG between wild-type and STAT3Hep−/− mice at all time points examined (Figure 2A). This suggests that the decreased DEN-induced liver tumorigenesis in STAT3Hep−/− mice is not caused by a decrease in oxidative stress.
In contrast to weak and transient induction of oxidative stress in the liver tumor model induced by a single DEN injection, chronic CCl4 challenge induced sixfold and twofold higher levels of 8-OHdG and MDA, respectively, in the liver compared with a single DEN injection (Figure 7 versus Figure 2A). In addition, hepatic levels of 8-OHdG and MDA were higher in STAT3Hep−/− mice than in wild-type mice. Elevation of 8-OHdG, a mutation prone to inducing G-C to T-A transversion during DNA replication, has been implicated in tumorigenesis in many types of tumors, including liver tumors.48 Furthermore, compared with wild-type mice, STAT3Hep−/− mice had greater liver injury, inflammation, and fibrosis after chronic CCl4 administration. Each of these changes in the liver is a risk factor for developing liver cancer.2,3,6 Therefore, enhanced liver tumorigenesis in STAT3Hep−/− mice may be caused by higher levels of liver injury, inflammation, oxidative stress, and DNA damage in these mice compared with wild-type mice. It would be interesting to test whether treatment with antioxidants, such as N-acetylcysteine, prevents CCl4-induced chronic liver injury and tumorigenesis in STAT3Hep−/− mice in a future study.
STAT3 and Inflammation in the DEN and CCl4 Models
It has been documented that STAT3 signaling is a major intrinsic pathway whereby cancers induce transcription of a variety of inflammation-related genes.13,49 Such cancer cell STAT3-mediated inflammation was observed in the DEN-induced liver tumor model. As shown in Figure 2C, expression of CCR2, F4/80, TNF-α, IFN-γ, and TGF-β was only slightly elevated in the nontumor tissues of wild-type mice, confirming a lack of overt injury and inflammation in this model. In contrast, these inflammatory mediators were markedly up-regulated in the liver tumor tissues of wild-type mice. The observed induction was not found in STAT3-deficient tumor tissues, indicating that the activation of STAT3 observed in these tumor tissues (Figure 2C) contributed to induction of inflammation in the tumor tissues. At present, how this STAT3-associated inflammation affects the pathogenesis of liver tumor is unknown.
Previously, we showed that compared with wild-type mice, STAT3Hep−/− mice had enhanced liver injury but reduced liver inflammation after acute CCl4 injection, suggesting that hepatic STAT3 protects against hepatocellular damage but promotes liver inflammation in CCl4-induced acute liver injury.50 In contrast, chronic CCl4 injection induced higher levels of liver inflammation in STAT3Hep−/− mice than in wild-type mice (Figures 3 and 4), which might be indirectly caused by the greater hepatocellular damage in STAT3Hep−/− mice than in wild-type mice. We did not compare the role of STAT3 in inflammation in tumor tissues between wild-type and STAT3Hep−/− mice in this model because of a lack of liver tumors in wild-type mice.
Conclusions and Clinical Implications
Most human liver cancers develop after chronic liver injury, inflammation, and cirrhosis. Activation of hepatic STAT3 in the early stage of liver injury likely plays an important role in protecting against hepatocyte death and DNA damage, thereby acting as a tumor suppressor to prevent liver tumorigenesis. However, once tumor cells have developed, STAT3 likely acts as an oncogenic factor to promote liver tumor cell proliferation and survival.
In recent years, intense efforts have been made to develop STAT3 inhibitors for treating various types of tumors. It has been reported that treatment with STAT3 inhibitors effectively blocks the growth of transplanted liver tumor cells in nude mice.33 However, the effects of STAT3 inhibitors on nontransplanted solid liver tumor and liver injury remain unknown and should be further investigated. Clinical application of STAT3 inhibitors in treating HCC should be cautious with monitoring hepatocellular damage.
Footnotes
Supported in part by the intramural program of the National Institute on Alcohol Abuse and Alcoholism, NIH (B.G.), and in part by the Natural Science Foundation of China (30973467/H1611 to H.W.).
None of the authors disclosed any relevant financial relationships.
Current address of F.L., Laboratory of Liver Pathophysiology, INSERM U955, Hopital Henri Mondor, Université Paris-Est, Creteil Cedex, France.
References
- 1.Ferenci P., Fried M., Labrecque D., Bruix J., Sherman M., Omata M., Heathcote J., Piratsivuth T., Kew M., Otegbayo J.A., Zheng S.S., Sarin S., Hamid S.S., Modawi S.B., Fleig W., Fedail S., Thomson A., Khan A., Malfertheiner P., Lau G., Carillo F.J., Krabshuis J., Le Mair A. Hepatocellular carcinoma (HCC): a global perspective. J Clin Gastroenterol. 2010;44:239–245. doi: 10.1097/MCG.0b013e3181d46ef2. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag H.B., Rudolph K.L. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 3.Aravalli R.N., Steer C.J., Cressman E.N. Molecular mechanisms of hepatocellular carcinoma. Hepatology. 2008;48:2047–2063. doi: 10.1002/hep.22580. [DOI] [PubMed] [Google Scholar]
- 4.Mishra L., Banker T., Murray J., Byers S., Thenappan A., He A.R., Shetty K., Johnson L., Reddy E.P. Liver stem cells and hepatocellular carcinoma. Hepatology. 2009;49:318–329. doi: 10.1002/hep.22704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Serag H.B., Marrero J.A., Rudolph L., Reddy K.R. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology. 2008;134:1752–1763. doi: 10.1053/j.gastro.2008.02.090. [DOI] [PubMed] [Google Scholar]
- 6.Llovet J.M., Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–1327. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruix J., Llovet J.M. Major achievements in hepatocellular carcinoma. Lancet. 2009;373:614–616. doi: 10.1016/S0140-6736(09)60381-0. [DOI] [PubMed] [Google Scholar]
- 8.Villanueva A., Llovet J.M. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011;140:1410–1426. doi: 10.1053/j.gastro.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Albini A., Sporn M.B. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;7:139–147. doi: 10.1038/nrc2067. [DOI] [PubMed] [Google Scholar]
- 10.Grivennikov S.I., Greten F.R., Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Llovet J.M., Ricci S., Mazzaferro V., Hilgard P., Gane E., Blanc J.F., de Oliveira A.C., Santoro A., Raoul J.L., Forner A., Schwartz M., Porta C., Zeuzem S., Bolondi L., Greten T.F., Galle P.R., Seitz J.F., Borbath I., Haussinger D., Giannaris T., Shan M., Moscovici M., Voliotis D., Bruix J. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 12.Siegel A.B., Olsen S.K., Magun A., Brown R.S., Jr Sorafenib: where do we go from here? Hepatology. 2010;52:360–369. doi: 10.1002/hep.23633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu H., Pardoll D., Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H., Lafdil F., Kong X., Gao B. Signal transducer and activator of transcription 3 in liver diseases: a novel therapeutic target. Int J Biol Sci. 2011;7:536–550. doi: 10.7150/ijbs.7.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kishimoto T. IL-6: from its discovery to clinical applications. Int Immunol. 2010;22:347–352. doi: 10.1093/intimm/dxq030. [DOI] [PubMed] [Google Scholar]
- 16.Bromberg J., Wang T.C. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell. 2009;15:79–80. doi: 10.1016/j.ccr.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naugler W.E., Sakurai T., Kim S., Maeda S., Kim K., Elsharkawy A.M., Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 18.Radaeva S., Sun R., Pan H.N., Hong F., Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332–1342. doi: 10.1002/hep.20184. [DOI] [PubMed] [Google Scholar]
- 19.Park O., Wang H., Weng H., Feigenbaum L., Li H., Yin S., Ki S., Yoo S.H., Dooley S., Wang F.S., Young H.A., Gao B. In vivo consequences of liver-specific interleukin-22 expression: implications for human liver disease progression. Hepatology. 2011 doi: 10.1002/hep.24339. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yue P., Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs. 2009;18:45–56. doi: 10.1517/13543780802565791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He G., Yu G.Y., Temkin V., Ogata H., Kuntzen C., Sakurai T., Sieghart W., Peck-Radosavljevic M., Leffert H.L., Karin M. Hepatocyte IKKβ/NF-κB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17:286–297. doi: 10.1016/j.ccr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horiguchi N., Wang L., Mukhopadhyay P., Park O., Jeong W.I., Lafdil F., Osei-Hyiaman D., Moh A., Fu X.Y., Pacher P., Kunos G., Gao B. Cell type-dependent pro- and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology. 2008;134:1148–1158. doi: 10.1053/j.gastro.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H., Park O., Lafdil F., Shen K., Horiguchi N., Yin S., Fu X.Y., Kunos G., Gao B. Interplay of hepatic and myeloid signal transducer and activator of transcription 3 in facilitating liver regeneration via tempering innate immunity. Hepatology. 2010;51:1354–1362. doi: 10.1002/hep.23430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vesselinovitch S.D., Mihailovich N. Kinetics of diethylnitrosamine hepatocarcinogenesis in the infant mouse. Cancer Res. 1983;43:4253–4259. [PubMed] [Google Scholar]
- 25.Fan Y., Boivin G.P., Knudsen E.S., Nebert D.W., Xia Y., Puga A. The aryl hydrocarbon receptor functions as a tumor suppressor of liver carcinogenesis. Cancer Res. 2010;70:212–220. doi: 10.1158/0008-5472.CAN-09-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Domenicali M., Caraceni P., Giannone F., Baldassarre M., Lucchetti G., Quarta C., Patti C., Catani L., Nanni C., Lemoli R.M., Bernardi M. A novel model of CCl4-induced cirrhosis with ascites in the mouse. J Hepatol. 2009;51:991–999. doi: 10.1016/j.jhep.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 27.Goodman Z.D. Grading and staging systems for inflammation and fibrosis in chronic liver diseases. J Hepatol. 2007;47:598–607. doi: 10.1016/j.jhep.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Radaeva S., Sun R., Jaruga B., Nguyen V.T., Tian Z., Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130:435–452. doi: 10.1053/j.gastro.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 29.Paik Y.H., Kim J.K., Lee J.I., Kang S.H., Kim D.Y., An S.H., Lee S.J., Lee D.K., Han K.H., Chon C.Y., Lee S.I., Lee K.S., Brenner D.A. Celecoxib induces hepatic stellate cell apoptosis through inhibition of Akt activation and suppresses hepatic fibrosis in rats. Gut. 2009;58:1517–1527. doi: 10.1136/gut.2008.157420. [DOI] [PubMed] [Google Scholar]
- 30.Gavino V.C., Miller J.S., Ikharebha S.O., Milo G.E., Cornwell D.G. Effect of polyunsaturated fatty acids and antioxidants on lipid peroxidation in tissue cultures. J Lipid Res. 1981;22:763–769. [PubMed] [Google Scholar]
- 31.Ohkawa H., Ohishi N., Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 32.Baker M.A., Cerniglia G.J., Zaman A. Microtiter plate assay for the measurement of glutathione and glutathione disulfide in large numbers of biological samples. Anal Biochem. 1990;190:360–365. doi: 10.1016/0003-2697(90)90208-q. [DOI] [PubMed] [Google Scholar]
- 33.Lin L., Amin R., Gallicano G.I., Glasgow E., Jogunoori W., Jessup J.M., Zasloff M., Marshall J.L., Shetty K., Johnson L., Mishra L., He A.R. The STAT3 inhibitor NSC 74859 is effective in hepatocellular cancers with disrupted TGF-β signaling. Oncogene. 2009;28:961–972. doi: 10.1038/onc.2008.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riehle K.J., Campbell J.S., McMahan R.S., Johnson M.M., Beyer R.P., Bammler T.K., Fausto N. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. J Exp Med. 2008;205:91–103. doi: 10.1084/jem.20070820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cui Q., Jiang W., Wang Y., Lv C., Luo J., Zhang W., Lin F., Yin Y., Cai R., Wei P., Qian C. Transfer of suppressor of cytokine signaling 3 by an oncolytic adenovirus induces potential antitumor activities in hepatocellular carcinoma. Hepatology. 2008;47:105–112. doi: 10.1002/hep.21951. [DOI] [PubMed] [Google Scholar]
- 36.Sharma D., Wang J., Fu P.P., Sharma S., Nagalingam A., Mells J., Handy J., Page A.J., Cohen C., Anania F.A., Saxena N.K. Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology. 2010;52:1713–1722. doi: 10.1002/hep.23892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Machida K., Tsukamoto H., Liu J.C., Han Y.P., Govindarajan S., Lai M.M., Akira S., Ou J.H. c-Jun mediates hepatitis C virus hepatocarcinogenesis through signal transducer and activator of transcription 3 and nitric oxide-dependent impairment of oxidative DNA repair. Hepatology. 2010;52:480–492. doi: 10.1002/hep.23697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong F., Jaruga B., Kim W.H., Radaeva S., El-Assal O.N., Tian Z., Nguyen V.A., Gao B. Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. J Clin Invest. 2002;110:1503–1513. doi: 10.1172/JCI15841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Regis G., Pensa S., Boselli D., Novelli F., Poli V. Ups and downs: the STAT1:STAT3 seesaw of Interferon and gp130 receptor signalling. Semin Cell Dev Biol. 2008;19:351–359. doi: 10.1016/j.semcdb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 40.Verna L., Whysner J., Williams G.M. N-nitrosodiethylamine mechanistic data and risk assessment: bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol Ther. 1996;71:57–81. doi: 10.1016/0163-7258(96)00062-9. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X.F., Tan X., Zeng G., Misse A., Singh S., Kim Y., Klaunig J.E., Monga S.P. Conditional β-catenin loss in mice promotes chemical hepatocarcinogenesis: role of oxidative stress and platelet-derived growth factor receptor α/phosphoinositide 3-kinase signaling. Hepatology. 2010;52:954–965. doi: 10.1002/hep.23747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farazi P.A., Glickman J., Horner J., Depinho R.A. Cooperative interactions of p53 mutation, telomere dysfunction, and chronic liver damage in hepatocellular carcinoma progression. Cancer Res. 2006;66:4766–4773. doi: 10.1158/0008-5472.CAN-05-4608. [DOI] [PubMed] [Google Scholar]
- 43.Fausto N., Campbell J.S. Mouse models of hepatocellular carcinoma. Semin Liver Dis. 2010;30:87–98. doi: 10.1055/s-0030-1247135. [DOI] [PubMed] [Google Scholar]
- 44.Newell P., Villanueva A., Friedman S.L., Koike K., Llovet J.M. Experimental models of hepatocellular carcinoma. J Hepatol. 2008;48:858–879. doi: 10.1016/j.jhep.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujii T., Fuchs B.C., Yamada S., Lauwers G.Y., Kulu Y., Goodwin J.M., Lanuti M., Tanabe K.K. Mouse model of carbon tetrachloride induced liver fibrosis: histopathological changes and expression of CD133 and epidermal growth factor. BMC Gastroenterol. 2010;10:79. doi: 10.1186/1471-230X-10-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hosui A., Kimura A., Yamaji D., Zhu B.M., Na R., Hennighausen L. Loss of STAT5 causes liver fibrosis and cancer development through increased TGF-β and STAT3 activation. J Exp Med. 2009;206:819–831. doi: 10.1084/jem.20080003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maeda S., Kamata H., Luo J.L., Leffert H., Karin M. IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 48.Chuma M., Hige S., Nakanishi M., Ogawa K., Natsuizaka M., Yamamoto Y., Asaka M. 8-Hydroxy-2′-deoxy-guanosine is a risk factor for development of hepatocellular carcinoma in patients with chronic hepatitis C virus infection. J Gastroenterol Hepatol. 2008;23:1431–1436. doi: 10.1111/j.1440-1746.2008.05502.x. [DOI] [PubMed] [Google Scholar]
- 49.Kortylewski M., Yu H. Role of Stat3 in suppressing anti-tumor immunity. Curr Opin Immunol. 2008;20:228–233. doi: 10.1016/j.coi.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horiguchi N., Lafdil F., Miller A.M., Park O., Wang H., Rajesh M., Mukhopadhyay P., Fu X.Y., Pacher P., Gao B. Dissociation between liver inflammation and hepatocellular damage induced by carbon tetrachloride in myeloid cell-specific signal transducer and activator of transcription 3 gene knockout mice. Hepatology. 2010;51:1724–1734. doi: 10.1002/hep.23532. [DOI] [PMC free article] [PubMed] [Google Scholar]